

Effect of N-Acetylcysteine in Mitochondrial Function, Redox Signaling, and Sirtuin 3 Levels in the Heart During Cardiorenal Syndrome Type 4 Development

 ,
, 
 , , , , ,
, , , , ,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethics Statement
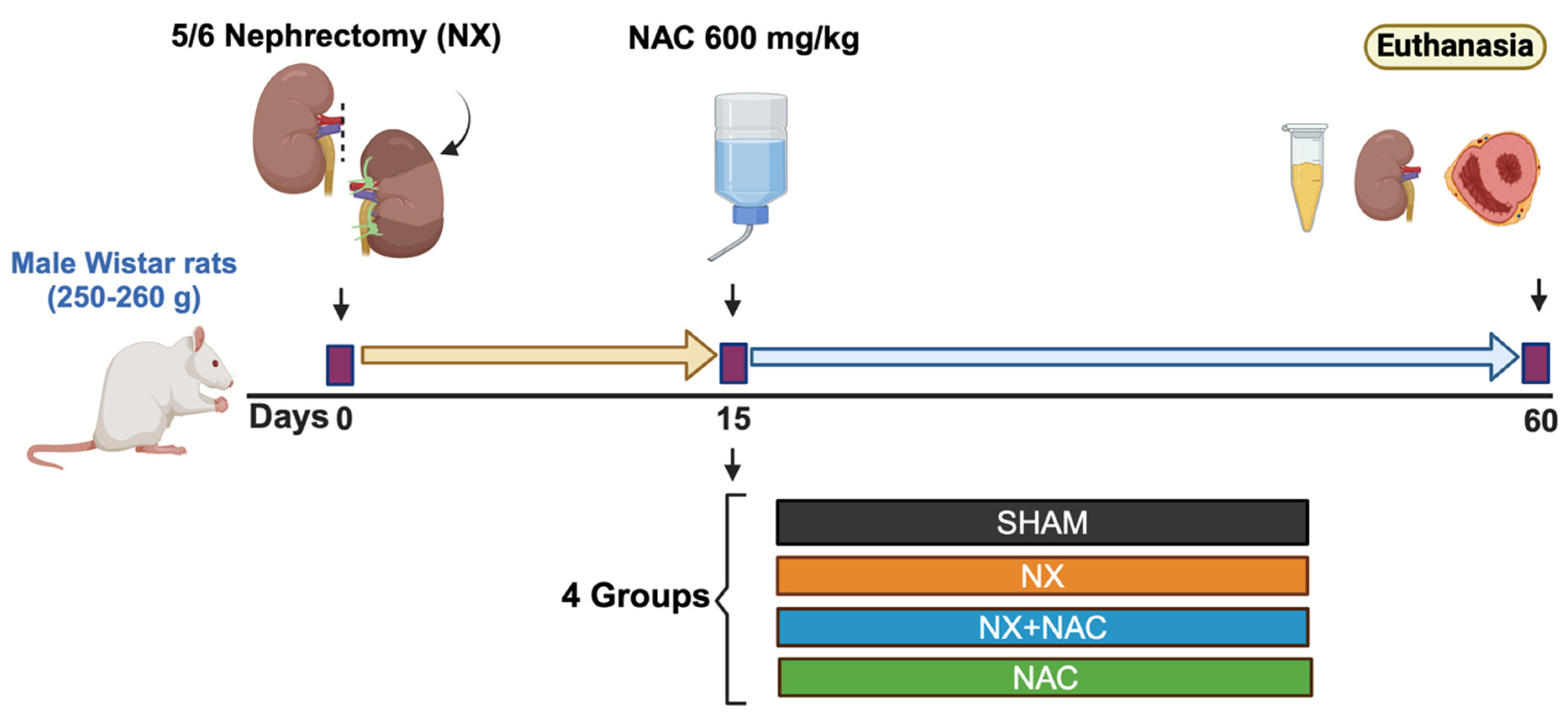
2.3. Experimental Design
2.4. Renal Damage Markers (BUN, Creatinine, and Proteinuria)
2.5. Systolic Blood Pressure (SBP)
2.6. Histology Evaluation
2.7. Protein Extraction and Western Blot (WB) Assay
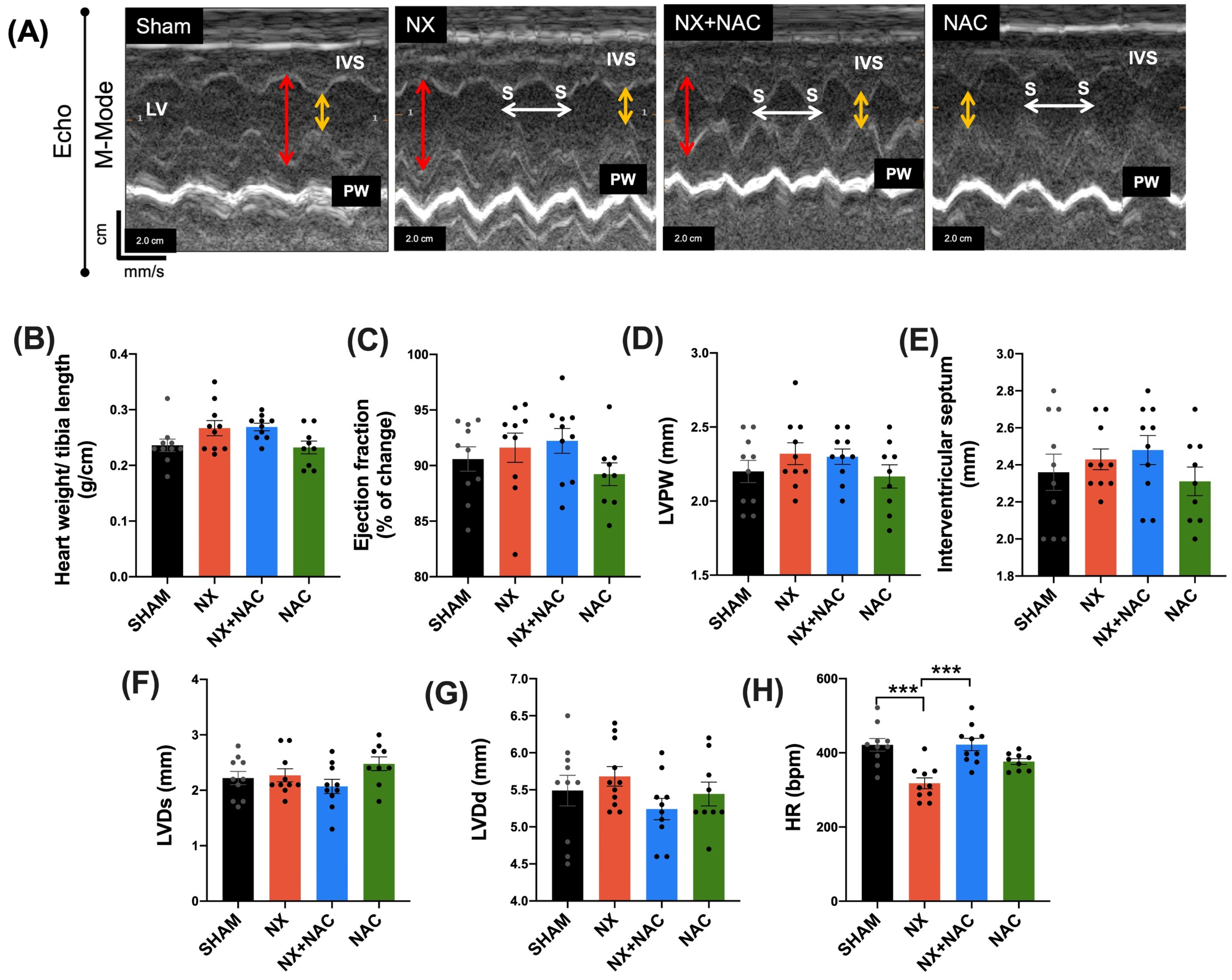
2.8. Evaluation of Cardiac Function by Echocardiography (Echo)
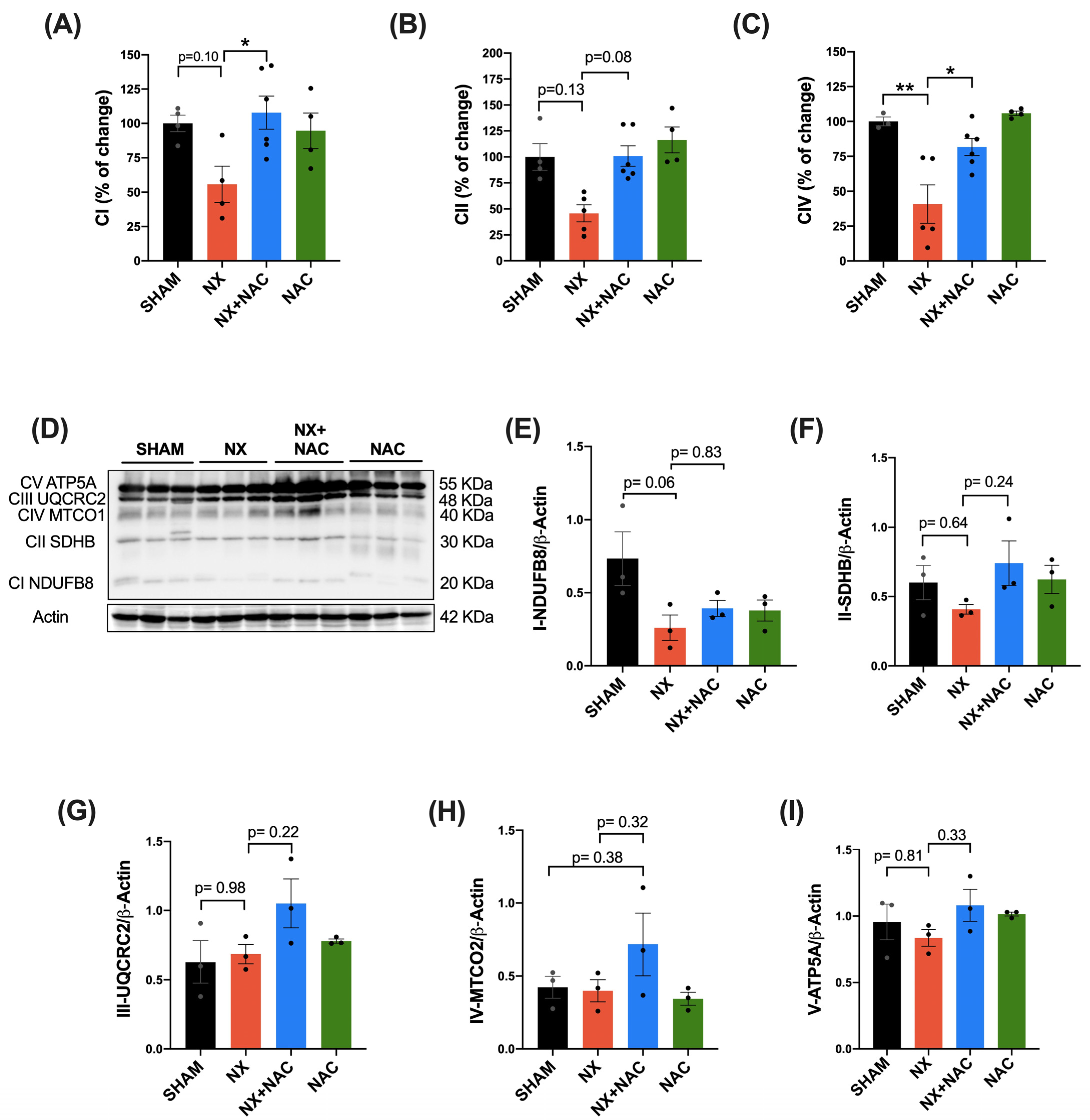
2.9. OXPHOS Subunit Protein Content in Total Heart Tissue
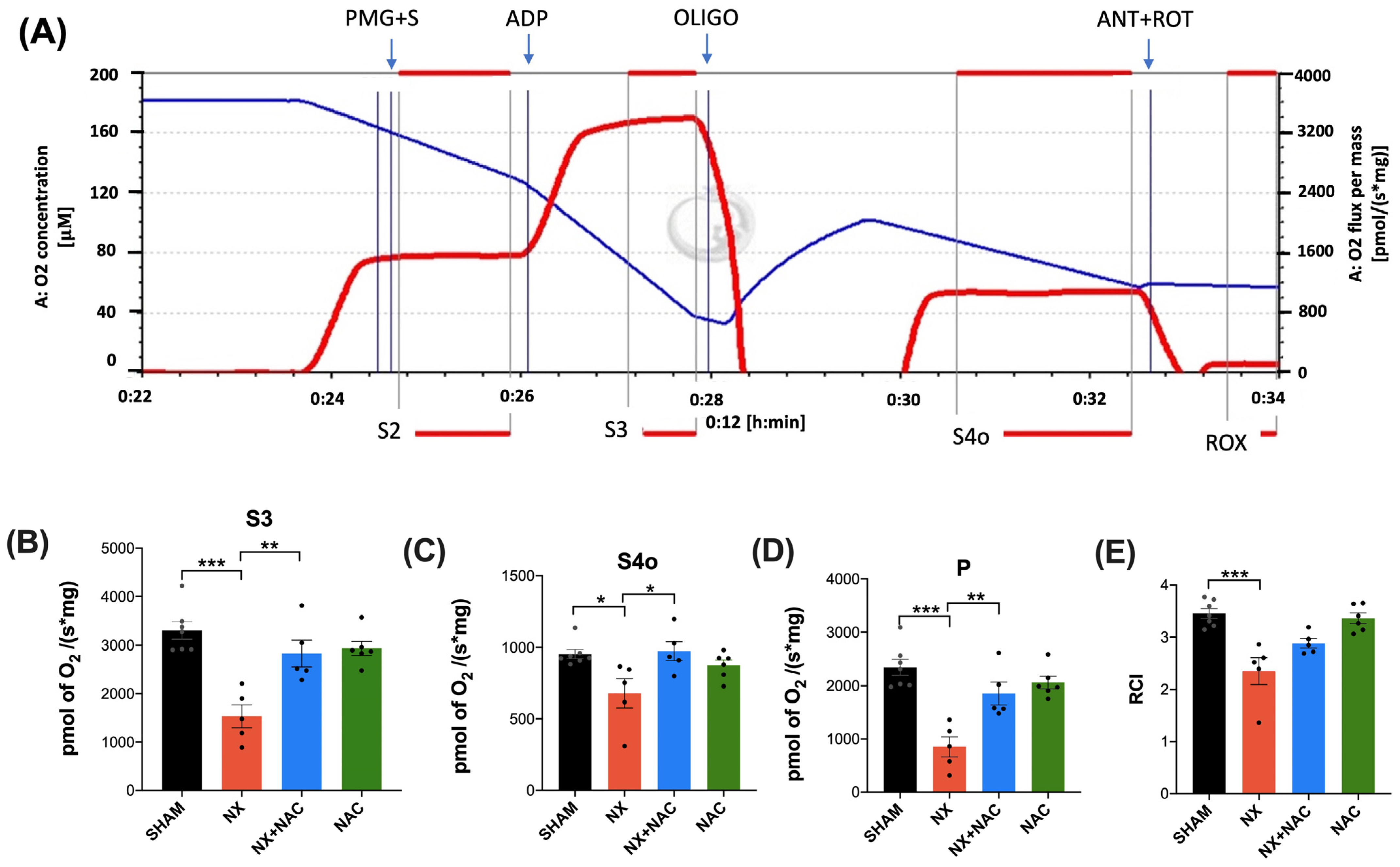
2.10. Mitochondrial Isolation and Oxygen Consumption
2.11. Mitochondrial Complexes’ Activity in Total Heart Tissue
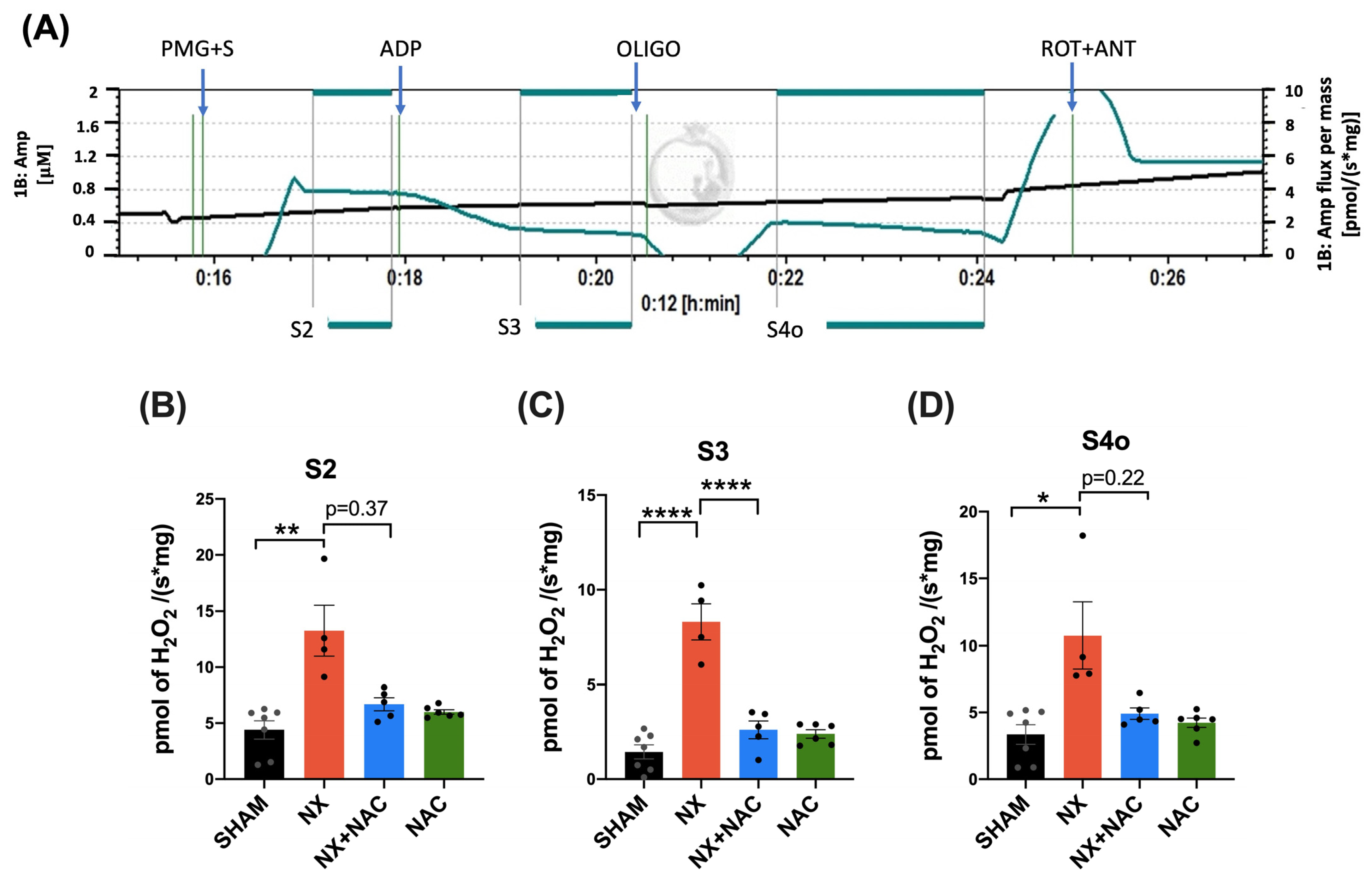
2.12. Mitochondrial H2O2 Production
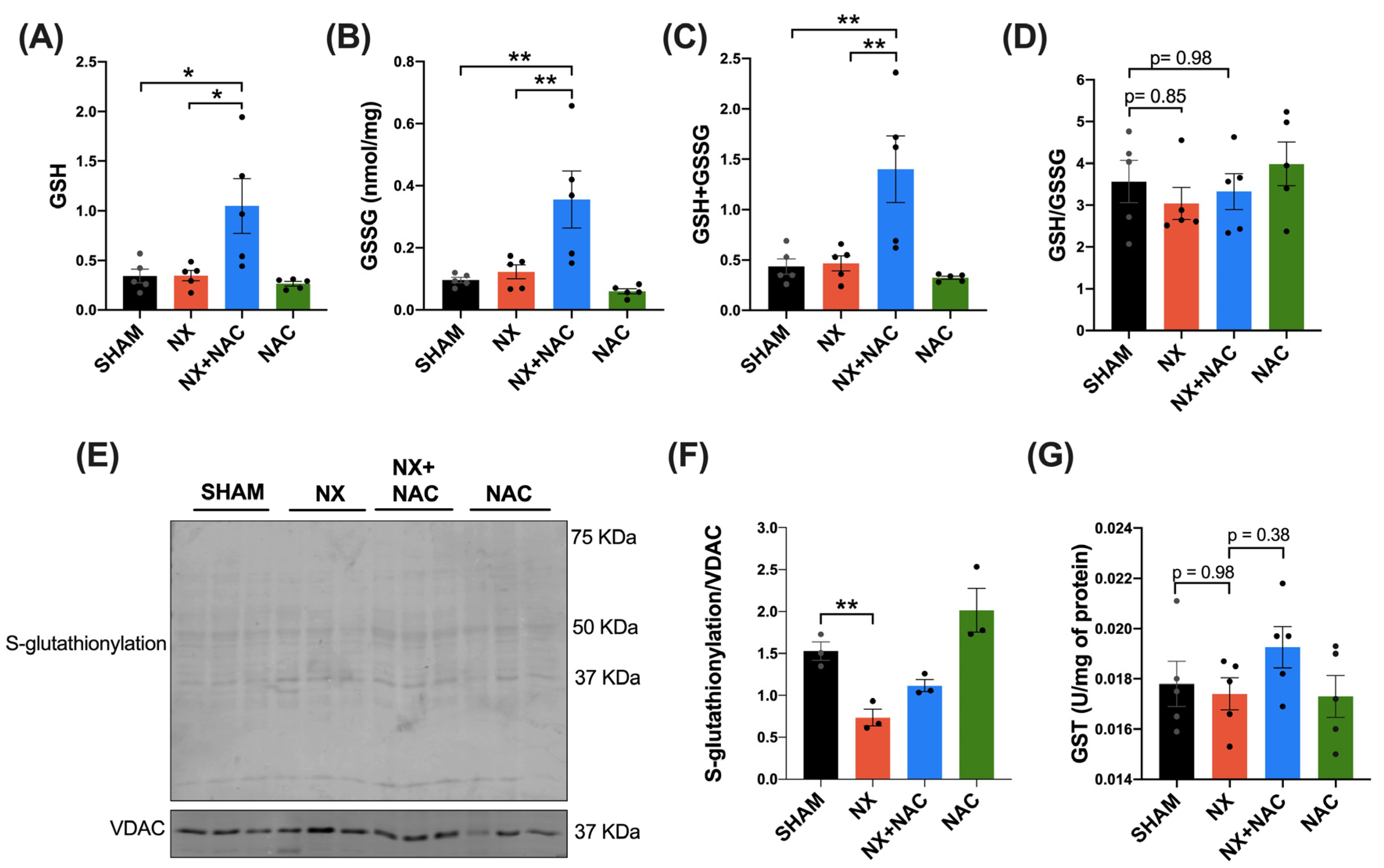
2.13. Glutathione Content in the Total Homogenate, Isolated Mitochondria, and Mitochondrial Protein RS-SG in the LV of the Heart
2.14. Transmission Electron Microscopy
2.15. NAD+/NADH Measurements in Heart Tissue
2.16. Activity of Antioxidant Enzymes in Heart Tissue
2.17. Statistical Analysis
3. Results
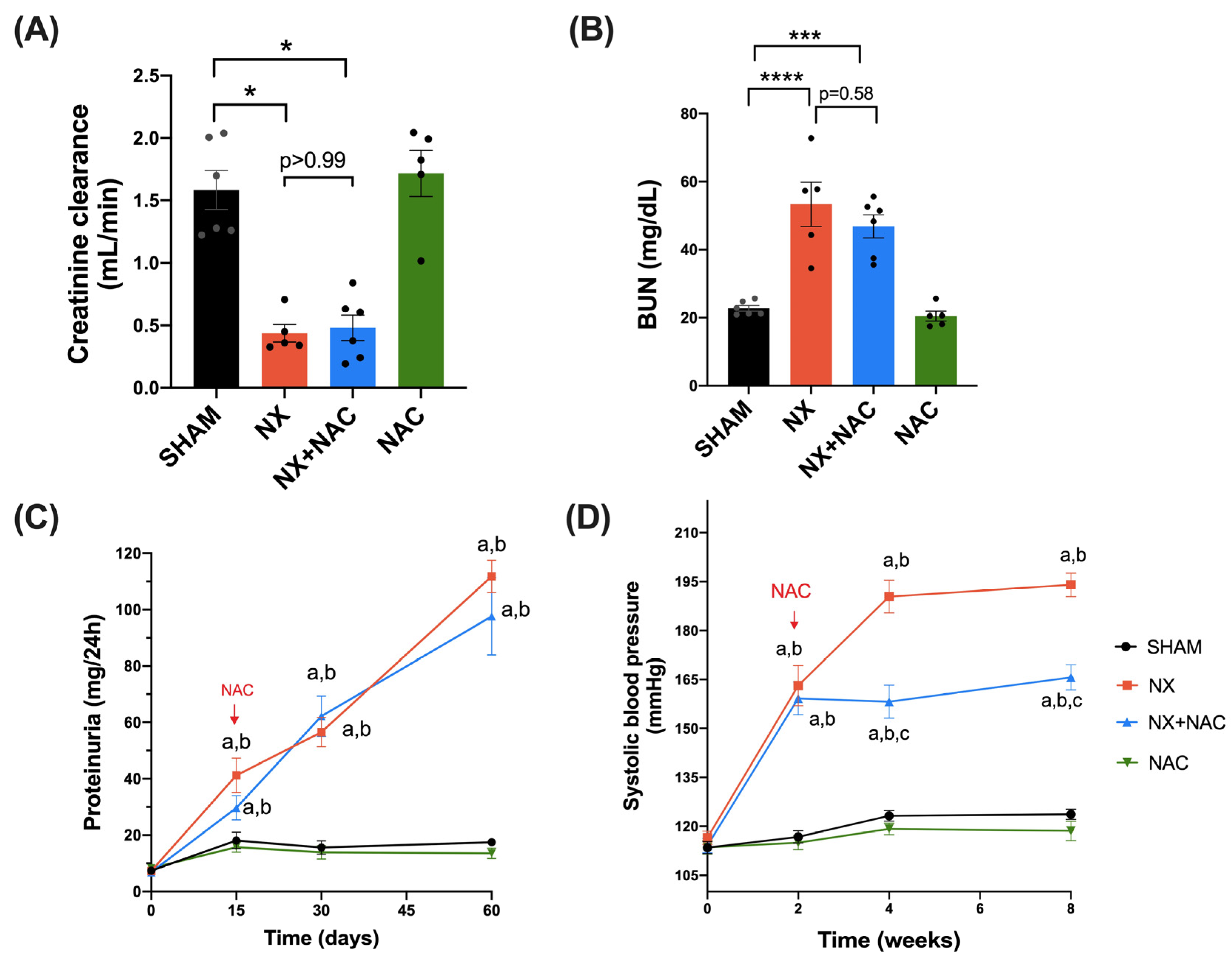
3.1. Administration of NAC-Reduced CKD-Induced Cardiac Alterations at Two-Month Follow-Up
3.2. NAC Recovered the CKD-Induced Decrease in Heart ETS Activity, OXPHOS Capacity, and Mitochondrial Decoupling During CRS-4
3.3. NAC Prevented Increased Mitochondrial H2O2 Production in Cardiac Mitochondria by Regulating GSH Levels and RS-SG During CRS-4
3.4. NAC Protected Cardiac Mitochondria by Restoring the Redox Status, Increasing SIRT3 Levels, and Improving the SOD-2 Activity in the LV of the Heart in Animals with CRS-4 Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Δψm | Mitochondrial membrane potential |
| 2-VP | 2-vinylpyridine |
| ADP | Adenosine 5′-diphosphate |
| ANOVA | Analysis of variance |
| ATP | Adenosine 5′-triphosphate |
| BPM | Beats per minute |
| BSA | Bovine serum albumin |
| BUN | Blood urea nitrogen |
| CCCP | Carbonyl cyanide m-chlorophenylhydrazone |
| CDNB | 1-Chloro-2,4-dinitrobenzene |
| CI | Complex I |
| CII | Complex II |
| CIII | Complex III |
| CIV | Complex IV |
| CI-NDUFB8 | Reduced form of nicotinamide adenine dinucleotide ubiquinone oxidoreductase subunit B8 |
| CII-SDHB | Succinate dehydrogenase B |
| CIII-UQCRC2 | Ubiquinol–cytochrome c reductase core protein 2 |
| CIV-MTCO1 | Cytochrome c oxidase subunit I |
| CV-ATP5A | Adenine triphosphate synthase-α subunit |
| CKD | Chronic kidney disease |
| CoA | Coenzyme A |
| CRS-4 | Cardiorenal syndrome type 4 |
| DCPIP | 2,6-dichlorophenolindophenol sodium salt hydrate |
| DTNB | 5,5′-dithio-bis-(2-nitrobenzoic acid) |
| DTT | Dithiothreitol |
| DUB | Decyl ubiquinone |
| DUBH2 | Decyl ubiquinol |
| Echo | Echocardiography |
| EDTA | Ethylenediaminetetraacetic acid |
| EF | Ejection fraction |
| EGTA | Ethylene glycol-bis (2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| ETS | Electron transport system |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GCL | Glutamate cysteine ligase |
| GCLC | Glutamate–cysteine ligase catalytic subunit |
| GCLM | Glutamate–cysteine ligase modifier subunit |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Glutathione |
| GSH/GSSG | Glutathione ratio |
| GSH + GSSG | Total glutathione |
| GSSG | Oxidized glutathione |
| GST | Glutathione S-transferase |
| H&E | Hematoxylin and eosin |
| H2O2 | Hydrogen peroxide |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HR | Heart rate |
| HRP | Horseradish peroxidase |
| IL-1β | Interleukin-1β |
| IVS | Interventricular septum |
| KCN | Potassium cyanide |
| KH2PO4 | Potassium Phosphate monobasic |
| KOH | Potassium hydroxide |
| LDH | Lactate dehydrogenase |
| LV | Left ventricle |
| LVDd | Left ventricle dimensions at end-diastole |
| LVDs | Left ventricle dimensions at end-systole |
| LVPW | Left ventricular posterior wall |
| MgCl2 | Manganese (II) chloride |
| Na2HPO4 | Sodium phosphate dibasic |
| Na3VO4 | Sodium orthovanadate |
| NAC | N-acetylcysteine |
| NaCl | Sodium chloride |
| NAD+ | β-Nicotinamide adenine dinucleotide oxidized |
| NADH | β-Nicotinamide adenine dinucleotide hydrogen |
| NADP+ | β-Nicotinamide adenine dinucleotide phosphate oxidized |
| NADPH | β-Nicotinamide adenine dinucleotide phosphate reduced |
| NaF | Sodium fluoride |
| NaH2PO4 | Sodium phosphate monobasic |
| NaHCO3 | Sodium bicarbonate |
| NaOH | Sodium hydroxide |
| NaN3 | Sodium azide |
| NBT | Nitro blue tetrazolium |
| NEM | N-ethyl maleimide |
| NH4Cl | Ammonium chloride |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NX | 5/6 Nephrectomy |
| NX + NAC | 5/6 Nephrectomy treated with NAC |
| O2 | Oxygen |
| P | Oxidative phosphorylation-associated respiration |
| PAGE | SDS–polyacrylamide gel electrophoresis |
| p-AMPK | Phosphorylated-adenosine monophosphate-activated protein kinase |
| PEP | Phosphoenolpyruvate |
| PGC-1α | Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha |
| PMSF | Phenylmethanesulfonyl fluoride |
| PVDF | Polyvinylidene fluoride |
| PW | Posterior wall |
| PYK | Pyruvate kinase |
| RCI | Respirometry control index |
| RIPA | Radioimmunoprecipitation buffer |
| ROX | Residual respiration |
| RS- | Thiolate anions |
| RS-SG | Protein-S-glutathionylation |
| S3 | State 3 |
| S4o | State 4 induced by oligomycin |
| SBP | Systolic blood pressure |
| SDHB | Succinate dehydrogenase B |
| SDS | Sodium dodecyl sulfate |
| Sham | Simulated surgery |
| SIRT3 | Sirtuin 3 |
| SOD | Superoxide dismutase |
| SOD-2 | Superoxide dismutase-2 |
| TBS-T | Tris-buffered saline with 0.1% Tween 20 detergent |
| TFAM | Transcription factor associated mitochondria |
| TMPD | Tetramethyl-p-phenylenediamine |
| TNB | 5-thio-2-nitrobenzoic acid |
| TnT | Troponin T |
| Tris | Trizma |
| Tris-HCl | Trizma-hydrochloride |
| UQCRC2 | Ubiquinol–cytochrome c reductase core protein 2 |
| VDAC | Voltage-dependent anion channel |
| WB | Western blot |
References
- Ren, J.; Dai, C. Pathophysiology of Chronic Kidney Disease. In Chronic Kidney Disease; Yang, J., He, W., Eds.; Springer: Singapore, 2020. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A Single Number for Advocacy and Communication-Worldwide More than 850 Million Individuals Have Kidney Diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic Kidney Disease and Cardiovascular Risk: Epidemiology, Mechanisms, and Prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Xue, Y.; Xu, B.; Su, C.; Han, Q.; Wang, T.; Tang, W. Cardiorenal syndrome in incident peritoneal dialysis patients: What is its effect on patients’ outcomes? PLoS ONE 2019, 14, e0218082. [Google Scholar] [CrossRef] [PubMed]
- Prothasis, M.; Varma, A.; Gaidhane, S.; Kumar, S.; Khatib, N.; Zahiruddin, Q.; Gaidhane, A. Prevalence, types, risk factors, and outcomes of cardiorenal syndrome in a rural population of central India: A cross-sectional study. J. Fam. Med. Prim. Care 2020, 9, 4127. [Google Scholar] [CrossRef]
- Suresh, H.; Arun, B.S.; Moger, V.; Swamy, M. Cardiorenal syndrome type 4: A study of cardiovascular diseases in chronic kidney disease. Indian Heart J. 2017, 69, 11–16. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Rapsomanikis, K.-P.; Dounousi, E. Chronic Kidney Disease and Disproportionally Increased Cardiovascular Damage: Does Oxidative Stress Explain the Burden? Oxidative Med. Cell. Longev. 2017, 2017, 9036450. [Google Scholar] [CrossRef]
- Caio-Silva, W.; Da Silva Dias, D.; Junho, C.V.C.; Panico, K.; Neres-Santos, R.S.; Pelegrino, M.T.; Pieretti, J.C.; Seabra, A.B.; De Angelis, K.; Carneiro-Ramos, M.S. Characterization of the Oxidative Stress in Renal Ischemia/Reperfusion-Induced Cardiorenal Syndrome Type 3. BioMed Res. Int. 2020, 2020, 1605358. [Google Scholar] [CrossRef]
- Wollenhaupt, J.; Frisch, J.; Harlacher, E.; Wong, D.W.L.; Jin, H.; Schulte, C.; Vondenhoff, S.; Moellmann, J.; Klinkhammer, B.M.; Zhang, L.; et al. Pro-Oxidative Priming but Maintained Cardiac Function in a Broad Spectrum of Murine Models of Chronic Kidney Disease. Redox Biol. 2022, 56, 102459. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; Sowers, J.R. Oxidative Stress in the Cardiorenal Metabolic Syndrome. Curr. Hypertens. Rep. 2012, 14, 360–365. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Treberg, J.R. Protein S-Glutathionlyation Links Energy Metabolism to Redox Signaling in Mitochondria. Redox Biol. 2016, 8, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Marti, C.N.; Sabbah, H.N.; Roessig, L.; Greene, S.J.; Böhm, M.; Burnett, J.C.; Campia, U.; Cleland, J.G.F.; Collins, S.P.; et al. Soluble Guanylate Cyclase: A Potential Therapeutic Target for Heart Failure. Heart Fail. Rev. 2013, 18, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Rashdan, N.A.; Shrestha, B.; Pattillo, C.B. S-Glutathionylation, Friend or Foe in Cardiovascular Health and Disease. Redox Biol. 2020, 37, 101693. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Picot, I.; Witko-Sarsat, V.; Merad-Boudia, M.; Nguyen, A.T.; Thévenin, M.; Jaudon, M.C.; Zingraff, J.; Verger, C.; Jingers, P.; Descamps-Latscha, B. Glutathione Antioxidant System as a Marker of Oxidative Stress in Chronic Renal Failure. Free Radic. Biol. Med. 1996, 21, 845–853. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Sadowska, A.M.; Manuel-y-Keenoy, B.; De Backer, W.A. Antioxidant and Anti-Inflammatory Efficacy of NAC in the Treatment of COPD: Discordant In Vitro and In Vivo Dose-Effects: A Review. Pulm. Pharmacol. Ther. 2007, 20, 9–22. [Google Scholar] [CrossRef]
- Ershad, M.; Naji, A.; Patel, P.; Vearrier, D. N-Acetylcysteine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537183/ (accessed on 1 March 2025).
- Cuevas-López, B.; Romero-Ramirez, E.I.; García-Arroyo, F.E.; Tapia, E.; León-Contreras, J.C.; Silva-Palacios, A.; Roldán, F.-J.; Campos, O.N.M.; Hernandez-Esquivel, L.; Marín-Hernández, A.; et al. NAC Pre-Administration Prevents Cardiac Mitochondrial Bioenergetics, Dynamics, Biogenesis, and Redox Alteration in Folic Acid-AKI-Induced Cardio-Renal Syndrome Type 3. Antioxidants 2023, 12, 1592. [Google Scholar] [CrossRef]
- Li, Q.; Liao, J.; Chen, W.; Zhang, K.; Li, H.; Ma, F.; Zhang, H.; Han, Q.; Guo, J.; Li, Y.; et al. Corrigendum to “NAC Alleviative Ferroptosis in Diabetic Nephropathy via Maintaining Mitochondrial Redox Homeostasis through Activating SIRT3-SOD2/Gpx4 Pathway”. Free Radic. Biol. Med. 2022, 189, 1. [Google Scholar] [CrossRef]
- Peerapanyasut, W.; Kobroob, A.; Palee, S.; Chattipakorn, N.; Wongmekiat, O. Activation of Sirtuin 3 and Maintenance of Mitochondrial Integrity by N-Acetylcysteine Protects Against Bisphenol A-Induced Kidney and Liver Toxicity in Rats. Int. J. Mol. Sci. 2019, 20, 267. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, L.G.; Braam, B.; Gaillard, C.A.; Cramer, M.J.; Goldschmeding, R.; Verhaar, M.C.; Doevendans, P.A.; Joles, J.A. Target Organ Cross Talk in Cardiorenal Syndrome: Animal Models. Am. J. Physiol. Ren. Physiol. 2012, 303, F1253–F1263. [Google Scholar] [CrossRef]
- Chang, D.; Wang, Y.-C.; Zhang, S.-J.; Bai, Y.-Y.; Liu, D.-F.; Zang, F.-C.; Wang, G.; Wang, B.; Ju, S. Visualizing Myocardial Inflammation in a Rat Model of Type 4 Cardiorenal Syndrome by Dual-Modality Molecular Imaging. Biomaterials 2015, 68, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; García-Niño, W.R.; Roldán, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Curcumin Maintains Cardiac and Mitochondrial Function in Chronic Kidney Disease. Free Radic. Biol. Med. 2013, 61, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Tapia, E.; Zatarain-Barrón, Z.L.; Hernández-Pando, R.; Zarco-Márquez, G.; Molina-Jijón, E.; Cristóbal-García, M.; Santamaría, J.; Pedraza-Chaverri, J. Curcumin Reverses Glomerular Hemodynamic Alterations and Oxidant Stress in 5/6 Nephrectomized Rats. Phytomedicine 2013, 20, 359–366. [Google Scholar] [CrossRef]
- Giam, B.; Kuruppu, S.; Chu, P.-Y.; Smith, A.I.; Marques, F.Z.; Fiedler, A.; Horlock, D.; Kiriazis, H.; Du, X.-J.; Kaye, D.M.; et al. N-Acetylcysteine Attenuates the Development of Renal Fibrosis in Transgenic Mice with Dilated Cardiomyopathy. Sci. Rep. 2017, 7, 17718. [Google Scholar] [CrossRef]
- Heloisa, M.; Shimizu, M.; Coimbra, T.M.; De Araujo, M.; Menezes, L.F.; Seguro, A.C. N-Acetylcysteine Attenuates the Progression of Chronic Renal Failure. Kidney Int. 2005, 68, 2208–2217. [Google Scholar] [CrossRef]
- García-Arroyo, F.E.; Gonzaga-Sánchez, G.; Tapia, E.; Muñoz-Jiménez, I.; Manterola-Romero, L.; Osorio-Alonso, H.; Arellano-Buendía, A.S.; Pedraza-Chaverri, J.; Roncal-Jiménez, C.A.; Lanaspa, M.A.; et al. Osthol Ameliorates Kidney Damage and Metabolic Syndrome Induced by a High-Fat/High-Sugar Diet. Int. J. Mol. Sci. 2021, 22, 2431. [Google Scholar] [CrossRef]
- Prieto-Carrasco, R.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; Rojas-Morales, P.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Tapia, E.; Pedraza-Chaverri, J. Progressive Reduction in Mitochondrial Mass Is Triggered by Alterations in Mitochondrial Biogenesis and Dynamics in Chronic Kidney Disease Induced by 5/6 Nephrectomy. Biology 2021, 10, 349. [Google Scholar] [CrossRef]
- Bazzano, T.; Restel, T.I.; Porfirio, L.C.; de Souza, A.S.; Silva, I.S. Renal biomarkers of male and female Wistar rats (Rattus norvegicus) undergoing renal ischemia and reperfusion. Acta Cir. Bras. 2015, 30, 277–288. [Google Scholar] [CrossRef]
- Hammond, J.B.W.; Kruger, N.J. The Bradford Method for Protein Quantitation. In New Protein Techniques; Humana Press: Totowa, NJ, USA, 1988; Volume 3, pp. 25–32. ISBN 978-0-89603-126-5. [Google Scholar]
- Prieto-Carrasco, R.; Silva-Palacios, A.; Rojas-Morales, P.; Aparicio-Trejo, O.E.; Medina-Reyes, E.I.; Hernández-Cruz, E.Y.; Sánchez-Garibay, C.; Salinas-Lara, C.; Pavón, N.; Roldán, F.J.; et al. Unilateral Ureteral Obstruction for 28 Days in Rats Is Not Associated with Changes in Cardiac Function or Alterations in Mitochondrial Function. Biology 2021, 10, 671. [Google Scholar] [CrossRef]
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez- Lozada, L.G.; Pedraza-Chaverri, J. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, J.H.; Matthews, H.R. The Lowry Method for Protein Quantitation. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 1984; Volume 1, pp. 1–3. [Google Scholar]
- Orozco-Ibarra, M.; Aparicio-Trejo, O.E.; Jiménez-Uribe, A.P.; Hernández-Cruz, E.Y.; Aranda-Rivera, A.K.; Amador-Martínez, I.; Fernández-Valverde, F.; Pedraza-Chaverri, J. Assessment of Kidney Mitochondrial Function by High-Resolution Respirometry, Transmission Electron Microscopy, and Histological Techniques. In Kidney Research; Hewitson, T.D., Toussaint, N.D., Smith, E.R., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; Volume 2664, pp. 283–308. ISBN 978-1-07-163178-2. [Google Scholar]
- Aparicio-Trejo, O.E.; Rojas-Morales, P.; Avila-Rojas, S.H.; León-Contreras, J.C.; Hernández-Pando, R.; Jiménez-Uribe, A.P.; Prieto-Carrasco, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. Int. J. Mol. Sci. 2020, 21, 6512. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for Quantitative Determination of Glutathione and Glutathione Disulfide Levels Using Enzymatic Recycling Method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Tomin, T.; Schittmayer, M.; Birner-Gruenberger, R. Addressing Glutathione Redox Status in Clinical Samples by Two-Step Alkylation with N-Ethylmaleimide Isotopologues. Metabolites 2020, 10, 71. [Google Scholar] [CrossRef]
- Durak, I.; Yurtarslanl, Z.; Canbolat, O.; Akyol, O. A Methodological Approach to Superoxide Dismutase (SOD) Activity Assay Based on Inhibition of Nitroblue Tetrazolium (NBT) Reduction. Clin. Chim. Acta Int. J. Clin. Chem. 1993, 214, 103–104. [Google Scholar] [CrossRef]
- Tapia, E.; Soto, V.; Ortiz-Vega, K.M.; Zarco-Márquez, G.; Molina-Jijón, E.; Cristóbal-García, M.; Santamaría, J.; García-Niño, W.R.; Correa, F.; Zazueta, C.; et al. Curcumin Induces Nrf2 Nuclear Translocation and Prevents Glomerular Hypertension, Hyperfiltration, Oxidant Stress, and the Decrease in Antioxidant Enzymes in 5/6 Nephrectomized Rats. Oxidative Med. Cell. Longev. 2012, 2012, e269039. [Google Scholar] [CrossRef]
- Aebi, H. [13] Catalase in Vitro. In Methods in Enzymology; Oxygen Radicals in Biological Systems; Academic Press: Cambridge, MA, USA, 1984; Volume 105, pp. 121–126. [Google Scholar]
- Lawrence, R.A.; Burk, R.F. Glutathione Peroxidase Activity in Selenium-Deficient Rat Liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Purification and Characterization of the Flavoenzyme Glutathione Reductase from Rat Liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Belliard, A.; Mulder, P.; Chwastyniak, M.; Beseme, O.; Henry, J.; Thuillez, C.; Amouyel, P.; Richard, V.; Pinet, F. Circulating Plasma Serine 208-phosphorylated Troponin T Levels Are Indicator of Cardiac Dysfunction. J. Cell. Mol. Med. 2013, 17, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.-Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224. [Google Scholar] [CrossRef] [PubMed]
- Scholze, A.; Rinder, C.; Beige, J.; Riezler, R.; Zidek, W.; Tepel, M. Acetylcysteine Reduces Plasma Homocysteine Concentration and Improves Pulse Pressure and Endothelial Function in Patients With End-Stage Renal Failure. Circulation 2004, 109, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD + /NADH Redox State and Diabetic Cardiomyopathy. Antioxid. Redox Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef]
- Muoio, D.M.; Noland, R.C.; Kovalik, J.-P.; Seiler, S.E.; Davies, M.N.; DeBalsi, K.L.; Ilkayeva, O.R.; Stevens, R.D.; Kheterpal, I.; Zhang, J.; et al. Muscle-Specific Deletion of Carnitine Acetyltransferase Compromises Glucose Tolerance and Metabolic Flexibility. Cell Metab. 2012, 15, 764–777. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a New Target of PGC-1α, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef]
- Xin, T.; Lu, C. SirT3 Activates AMPK-Related Mitochondrial Biogenesis and Ameliorates Sepsis-Induced Myocardial Injury. Aging 2020, 12, 16224–16237. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating Oxidative Stress in Heart Failure: Past, Present and Future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Reule, S.; Drawz, P.E. Heart rate and blood pressure: Any possible implications for management of hypertension? Curr. Hypertens. Rep. 2012, 14, 478–484. [Google Scholar] [CrossRef]
- Nagata, D.; Hishida, E. Elucidating the complex interplay between chronic kidney disease and hypertension. Hypertens. Res. 2024, 47, 3409–3422. [Google Scholar] [CrossRef]
- Borire, A.A.; Arnold, R.; Pussell, B.A.; Kwai, N.C.; Visser, L.H.; Padua, L.; Simon, N.G.; Kiernan, M.C.; Krishnan, A.V. Haemodialysis alters peripheral nerve morphology in end-stage kidney disease. Clin. Neurophysiol. 2017, 128, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Manzella, D.; Paolisso, G.; Caliendo, A.; Varricchio, M.; Giordano, C. Differences in heart rate variability parameters during the post-dialytic period in type II diabetic and non-diabetic ESRD patients. Nephrol. Dial. Transplant. 2001, 16, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, S.; Singal, P.; Gill, V. The Antihypertensive Effect of Cysteine. Int. J. Angiol. 2009, 18, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Camuglia, A.C.; Maeder, M.T.; Starr, J.; Farrington, C.; Kaye, D.M. Impact of N-Acetylcysteine on Endothelial Function, B-Type Natriuretic Peptide and Renal Function in Patients with the Cardiorenal Syndrome: A Pilot Cross Over Randomised Controlled Trial. Heart Lung Circ. 2013, 22, 256–259. [Google Scholar] [CrossRef]
- Whillier, S.; Raftos, J.E.; Chapman, B.; Kuchel, P.W. Role of N-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. 2009, 14, 115–124. [Google Scholar] [CrossRef]
- Forgacsova, A.; Galba, J.; Mojzisova, J.; Mikus, P.; Piestansky, J.; Kovac, A. Ultra-high performance hydrophilic interaction liquid chromatography—Triple quadrupole tandem mass spectrometry method for determination of cysteine, homocysteine, cysteinyl-glycine and glutathione in rat plasma. J. Pharm. Biomed. Anal. 2019, 164, 442–451. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Hartman, J.; Brannon, P.C.; Reeves-Daniel, A.; Satko, S.G.; Russell, G. Characteristics of sudden death in hemodialysis patients. Kidney Int. 2006, 69, 2268–2273. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of Oxidative Stress in Cardiac Hypertrophy and Remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Amador-Martínez, I.; García-Ballhaus, J.; Buelna-Chontal, M.; Cortés-González, C.; Massó, F.; Jaisser, F.; Barrera-Chimal, J. Early Inflammatory Changes and CC Chemokine Ligand-8 Upregulation in the Heart Contribute to Uremic Cardiomyopathy. FASEB J. 2021, 35, e21761. [Google Scholar] [CrossRef]
- Kingma, J.G.; Simard, D.; Rouleau, J.R.; Drolet, B.; Simard, C. The Physiopathology of Cardiorenal Syndrome: A Review of the Potential Contributions of Inflammation. J. Cardiovasc. Dev. Dis. 2017, 4, 21. [Google Scholar] [CrossRef]
- Edelstein, C.L.; Akcay, A.; Nguyen, Q. Mediators of Inflammation in Acute Kidney Injury. Mediat. Inflamm. 2009, 2009, 137072. [Google Scholar]
- de Deyn, P.P.; Vanholder, R.; D’Hooge, R. Nitric Oxide in Uremia: Effects of Several Potentially Toxic Guanidino Compounds. Kidney Int. Suppl. 2003, 63 (Suppl. S84), S25–S28. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of IL-1 in the pathogenesis of heart disease. Arch. Immunol. Ther. Exp. 2009, 57, 165–176. [Google Scholar] [CrossRef]
- Bevere, M.; Morabito, C.; Mariggiò, M.A.; Guarnieri, S. The Oxidative Balance Orchestrates the Main Keystones of the Functional Activity of Cardiomyocytes. Oxidative Med. Cell. Longev. 2022, 2022, 7714542. [Google Scholar] [CrossRef]
- Junho, C.V.C.; Trentin-Sonoda, M.; Panico, K.; dos Santos, R.S.N.; Abrahão, M.V.; Vernier, I.C.S.; Fürstenau, C.R.; Carneiro-Ramos, M.S. Cardiorenal syndrome: Long road between kidney and heart. Heart Fail. Rev. 2022, 27, 2137–2153. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 Catalyzes the Reversible Oxidation and Glutathionylation of Mitochondrial Membrane Thiol Proteins: Implications for Mitochondrial Redox Regulation and Antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.D.; França, K.C.; Dallin, F.T.; Fujihara, C.K.; Nascimento, A.J.; Pecoits-Filho, R.; Nakao, L.S. N-Acetylcysteine as a Potential Strategy to Attenuate the Oxidative Stress Induced by Uremic Serum in the Vascular System. Life Sci. 2015, 121, 110–116. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Lee, J.-I.; Kang, J.; Stipanuk, M.H. Differential Regulation of Glutamate–Cysteine Ligase Subunit Expression and Increased Holoenzyme Formation in Response to Cysteine Deprivation. Biochem. J. 2006, 393, 181–190. [Google Scholar] [CrossRef]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial Characterization of the Glutamate-Cysteine Ligase Modifier Subunit Gclm(−/−) Knockout Mouse. J. Biol. Chem. 2002, 277, 49446–49452. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Mitochondrial Glutathione: Features, Regulation and Role in Disease. Biochim. Biophys. Acta BBA—Gen. Subj. 2013, 1830, 3317–3328. [Google Scholar] [CrossRef]
- Chen, X.; Gan, B. SLC25A39 Links Mitochondrial GSH Sensing with Iron Metabolism. Mol. Cell 2024, 84, 616–618. [Google Scholar] [CrossRef]
- Wengrowski, A.M.; Kuzmiak-Glancy, S.; Jaimes, R.; Kay, M.W. NADH Changes during Hypoxia, Ischemia, and Increased Work Differ between Isolated Heart Preparations. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H529–H537. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Ganote, C.E. Mitochondrial Structure and Function in Acute Myocardial Ischemic Injury. Circ. Res. 1976, 38, 1256 I80-91. [Google Scholar]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; MacK, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Guarente, L. The SirT3 Divining Rod Points to Oxidative Stress. Mol. Cell 2011, 42, 561–568. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, J.; Li, X.; Zhu, H.; Sun, J.; Jiang, L.; Xue, C.; Zhang, L.; Xu, C.; Xing, S.; et al. Tetrahydrocurcumin Ameliorates Postinfarction Cardiac Dysfunction and Remodeling by Inhibiting Oxidative Stress and Preserving Mitochondrial Function via SIRT3 Signaling Pathway. Phytomedicine 2023, 121, 155127. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The Diabetes Medication Canagliflozin Promotes Mitochondrial Remodelling of Adipocyte via the AMPK-Sirt1-Pgc-1α Signalling Pathway. Adipocyte 2020, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, C.; Xie, M.; Zhen, Y.; Lai, B.; Liu, J.; Qiao, L.; Liu, S.; Shi, D. Compartmentally Scavenging Hepatic Oxidants through AMPK/SIRT3-PGC1α Axis Improves Mitochondrial Biogenesis and Glucose Catabolism. Free Radic. Biol. Med. 2021, 168, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Sanchez, W.Y.; Roy, S.F.; Morais, C.; Brooks, H.L.; Coombes, J.S.; Johnson, D.W.; Gobe, G.C. N-Acetyl-Cysteine Increases Cellular Dysfunction in Progressive Chronic Kidney Damage after Acute Kidney Injury by Dampening Endogenous Antioxidant Responses. Am. J. Physiol. Ren. Physiol. 2018, 314, F956–F968. [Google Scholar] [CrossRef]
- Sandilands, E.A.; Bateman, D.N. Adverse reactions associated with acetylcysteine. Clin. Toxicol. 2009, 47, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The Mechanism of Action of N-Acetylcysteine (NAC): The Emerging Role of H2S and Sulfane Sulfur Species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Renke, M.; Tylicki, L.; Rutkowski, P.; Larczyński, W.; Aleksandrowicz, E.; Łysiak-Szydłowska, W.; Rutkowski, B. The Effect of N-Acetylcysteine on Proteinuria and Markers of Tubular Injury in Non-Diabetic Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2008, 31, 404–410. [Google Scholar] [CrossRef]
- Wittstock, A.; Burkert, M.; Zidek, W.; Tepel, M.; Scholze, A. N-acetylcysteine improves arterial vascular reactivity in patients with chronic kidney disease. Nephron J. 2009, 12, c184–c189. [Google Scholar]
- Hamzaoui, M.; Djerada, Z.; Brunel, V.; Mulder, P.; Richard, V.; Bellien, J.; Guerrot, D. 5/6 nephrectomy induces different renal, cardiac and vascular consequences in 129/Sv and C57BL/6JRj mice. Sci Rep. 2020, 10, 1524. [Google Scholar] [CrossRef]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAD+/NADH Levels (pmol/mg of Protein) | SHAM | NX | NX + NAC |
|---|---|---|---|
| NAD+ + NADH | 752 ± 65 | 666 ± 68 | 620 ± 94 |
| NADH | 93 ± 12 | 192 ± 45 * | 69 ± 17 |
| NAD+/NADH | 7 ± 0.2 | 3 ± 1.2 * | 8 ± 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amador-Martínez, I.; Aparicio-Trejo, O.E.; Aranda-Rivera, A.K.; Bernabe-Yepes, B.; Medina-Campos, O.N.; Tapia, E.; Cortés-González, C.C.; Silva-Palacios, A.; Roldán, F.J.; León-Contreras, J.C.; et al. Effect of N-Acetylcysteine in Mitochondrial Function, Redox Signaling, and Sirtuin 3 Levels in the Heart During Cardiorenal Syndrome Type 4 Development. Antioxidants 2025, 14, 367. https://doi.org/10.3390/antiox14030367
Amador-Martínez I, Aparicio-Trejo OE, Aranda-Rivera AK, Bernabe-Yepes B, Medina-Campos ON, Tapia E, Cortés-González CC, Silva-Palacios A, Roldán FJ, León-Contreras JC, et al. Effect of N-Acetylcysteine in Mitochondrial Function, Redox Signaling, and Sirtuin 3 Levels in the Heart During Cardiorenal Syndrome Type 4 Development. Antioxidants. 2025; 14(3):367. https://doi.org/10.3390/antiox14030367
Chicago/Turabian StyleAmador-Martínez, Isabel, Omar Emiliano Aparicio-Trejo, Ana Karina Aranda-Rivera, Bismarck Bernabe-Yepes, Omar Noel Medina-Campos, Edilia Tapia, Carlo César Cortés-González, Alejandro Silva-Palacios, Francisco Javier Roldán, Juan Carlos León-Contreras, and et al. 2025. "Effect of N-Acetylcysteine in Mitochondrial Function, Redox Signaling, and Sirtuin 3 Levels in the Heart During Cardiorenal Syndrome Type 4 Development" Antioxidants 14, no. 3: 367. https://doi.org/10.3390/antiox14030367
APA StyleAmador-Martínez, I., Aparicio-Trejo, O. E., Aranda-Rivera, A. K., Bernabe-Yepes, B., Medina-Campos, O. N., Tapia, E., Cortés-González, C. C., Silva-Palacios, A., Roldán, F. J., León-Contreras, J. C., Hernández-Pando, R., Saavedra, E., Gonzaga-Sánchez, J. G., Ceja-Galicia, Z. A., Sánchez-Lozada, L. G., & Pedraza-Chaverri, J. (2025). Effect of N-Acetylcysteine in Mitochondrial Function, Redox Signaling, and Sirtuin 3 Levels in the Heart During Cardiorenal Syndrome Type 4 Development. Antioxidants, 14(3), 367. https://doi.org/10.3390/antiox14030367