
Mitochondrial Dysfunction in Endothelial Cells: A Key Driver of Organ Disorders and Aging



Abstract
1. Introduction
2. Mitochondria’s Structure and Origin
2.1. Origin
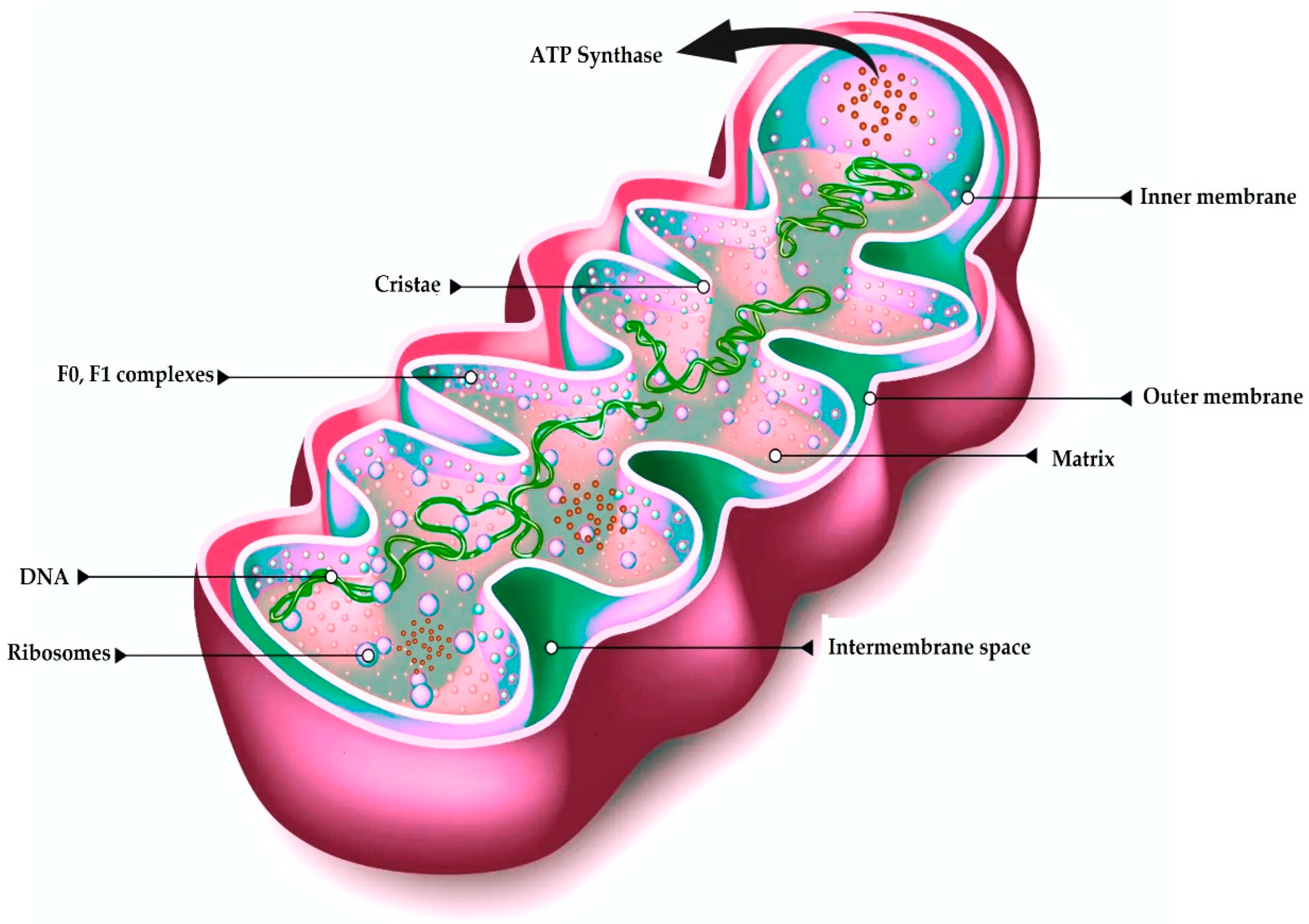
2.2. Structure
3. Mitochondrial Genomic Structure
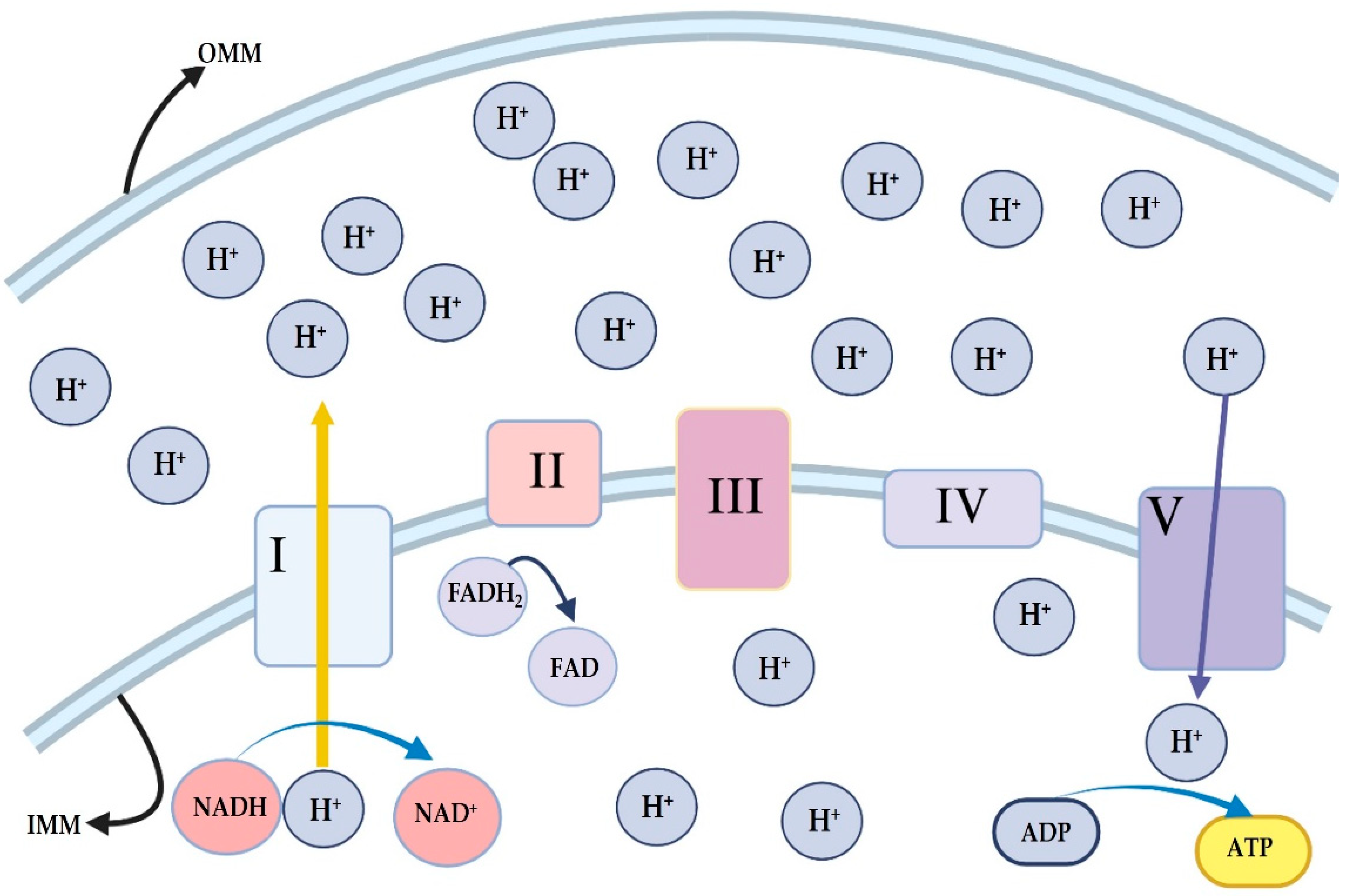
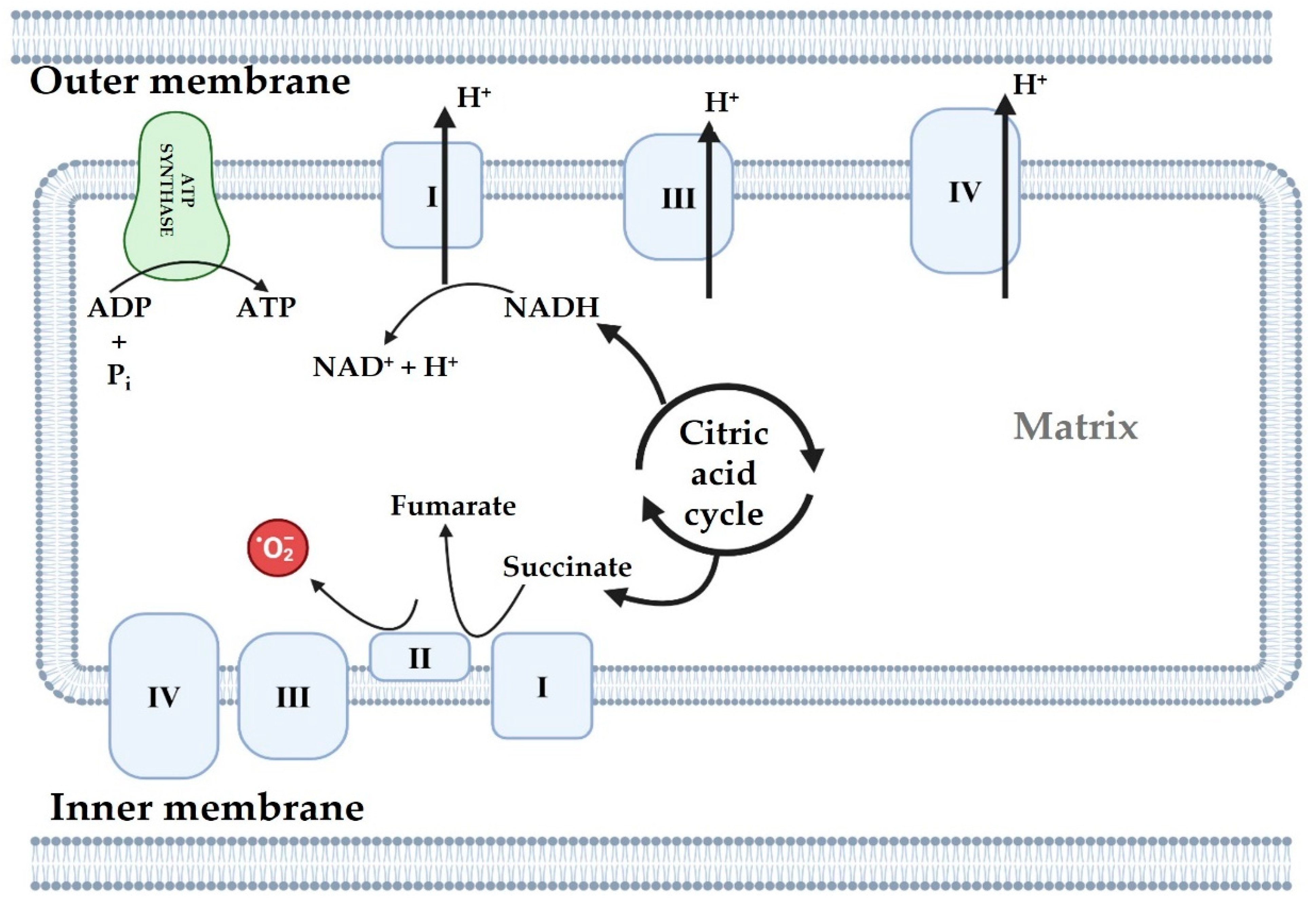
4. ATP Generation in Mitochondria
4.1. Oxidative Phosphorylation System
4.2. Mitochondrial ROS
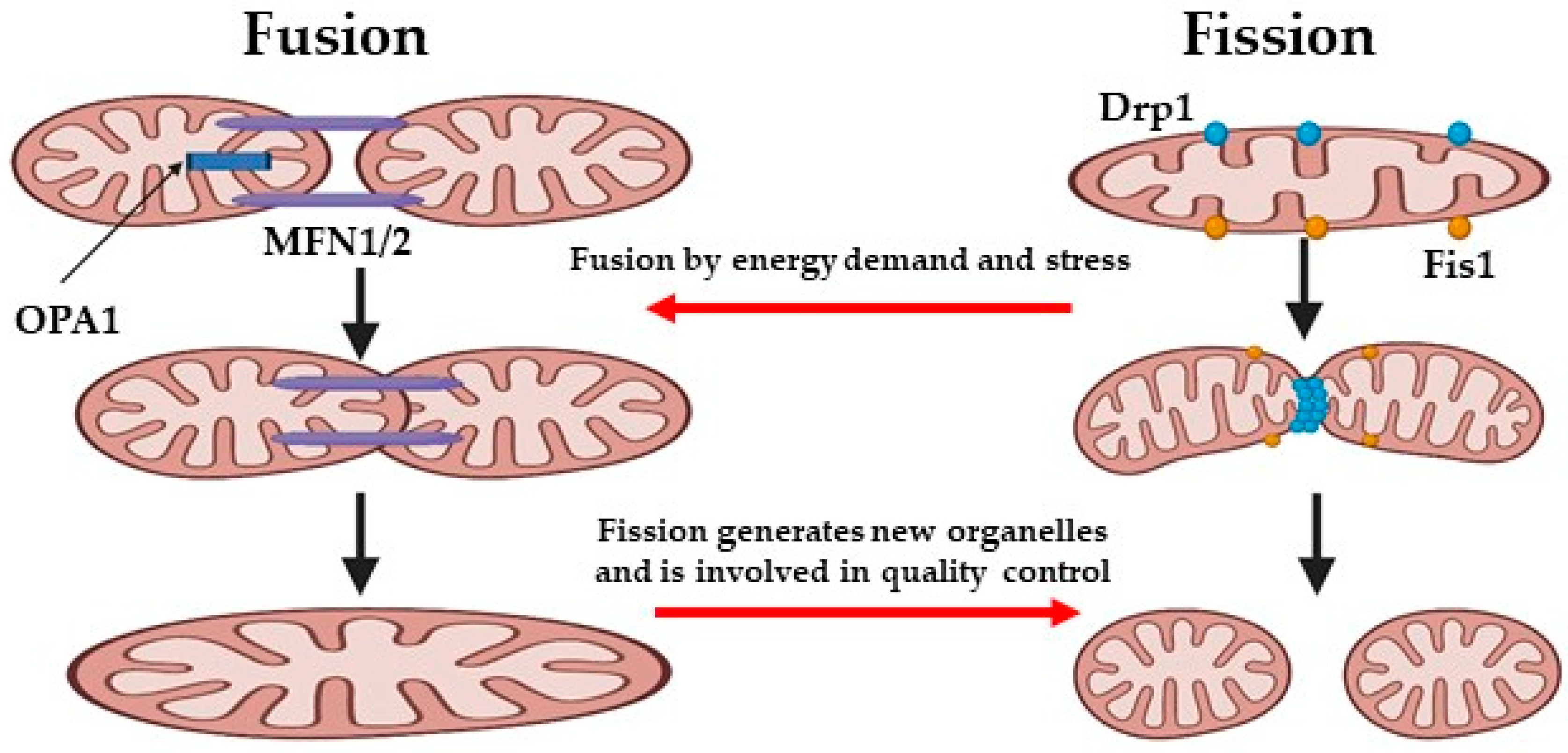
5. Mitochondrial Dynamics
6. Mitochondria and Endothelial Cells Under Physiological Conditions
7. Endothelial Cells and Mitochondria Dysfunction and Aging
8. Endothelial Cells and Mitochondria in Cardio-Metabolic Dysfunction
9. Mitochondrial Dysfunction and Renal Diseases
10. Neurodegenerative Diseases
10.1. Blood Brain Barrier, Mitochondria, and Endothelial Cells
10.2. Alzheimer’s Disease (AD)
10.3. Huntington’s Disease (HD)
10.4. Parkinson’s Disease (PD)
10.5. Amyotrophic Lateral Sclerosis (ALS)
10.6. Plant Antioxidant Supplementation for Improving Mitochondrial Function in Neurodegenerative Disorders
11. Mitochondrial Dysfunction and Liver Diseases
12. Mitochondria as a Potential Therapeutic Target in Diseases
12.1. Exposure to Hypoxia
12.2. Mitochondrial Function Targeted Drugs
12.3. Gene Therapy
12.4. Mitochondrial Transplantation
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pang, B.; Dong, G.; Pang, T.; Sun, X.; Liu, X.; Nie, Y.; Chang, X. Emerging insights into the pathogenesis and therapeutic strategies for vascular endothelial injury-associated diseases: Focus on mitochondrial dysfunction. Angiogenesis 2024, 27, 623–639. [Google Scholar] [CrossRef]
- Wang, X.; He, B. Endothelial dysfunction: Molecular mechanisms and clinical implications. MedComm 2024, 5, e651. [Google Scholar] [CrossRef]
- Campagna, R.; Mazzanti, L.; Pompei, V.; Alia, S.; Vignini, A.; Emanuelli, M. The Multifaceted Role of Endothelial Sirt1 in Vascular Aging: An Update. Cells 2024, 13, 1469. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Gupta, S.; Bakshi, A.; Kaur, H.; Jain, A.; Senapati, S.; Baghel, M.S. Targeting mitochondria in the regulation of neurodegenerative diseases: A comprehensive review. J. Neurosci. Res. 2022, 100, 1845–1861. [Google Scholar] [CrossRef] [PubMed]
- Parente, A.; Flores Carvalho, M.; Schlegel, A. Endothelial Cells and Mitochondria: Two Key Players in Liver Transplantation. Int. J. Mol. Sci. 2023, 24, 10091. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Dong, G.; Pang, T.; Sun, X.; Liu, X.; Nie, Y.; Chang, X. Advances in pathogenesis and treatment of vascular endothelial injury-related diseases mediated by mitochondrial abnormality. Front. Pharmacol. 2024, 15, 1422686. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Dunn, C.D. Some Liked It Hot: A Hypothesis Regarding Establishment of the Proto-Mitochondrial Endosymbiont During Eukaryogenesis. J. Mol. Evol. 2017, 85, 99–106. [Google Scholar] [CrossRef]
- Zachar, I.; Boza, G. Endosymbiosis before eukaryotes: Mitochondrial establishment in protoeukaryotes. Cell. Mol. Life Sci. 2020, 77, 3503–3523. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb. Cell 2015, 2, 105–125. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Mehrotra, A.; Rigoni, G.; Soriano, M.E. Who and how in the regulation of mitochondrial cristae shape and function. Biochem. Biophys. Res. Commun. 2018, 500, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, A.A. The Mitochondrial Genome. The Nucleoid. Biochemistry 2016, 81, 1057–1065. [Google Scholar] [CrossRef]
- Decker, S.T.; Funai, K. Mitochondrial membrane lipids in the regulation of bioenergetic flux. Cell Metab. 2024, 36, 1963–1978. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Q.; Liao, X.; Fan, M.; Liu, X.; Liu, Q.; Wang, M.; Wu, X.; Huang, C.K.; Tan, R.; et al. Mitochondrial Dysfunction in Arrhythmia and Cardiac Hypertrophy. Rev. Cardiovasc. Med. 2023, 24, 364. [Google Scholar] [CrossRef]
- Castellanos, E.; Lanning, N.J. Phosphorylation of OXPHOS Machinery Subunits: Functional Implications in Cell Biology and Disease. Yale J. Biol. Med. 2019, 92, 523–531. [Google Scholar]
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 52. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Rahmani, S.; Roohbakhsh, A.; Pourbarkhordar, V.; Hayes, A.W.; Karimi, G. Melatonin regulates mitochondrial dynamics and mitophagy: Cardiovascular protection. J. Cell. Mol. Med. 2024, 28, e70074. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, T.; Yanagi, S. Role of Mitochondrial Dynamics in Heart Diseases. Genes 2023, 14, 1876. [Google Scholar] [CrossRef]
- Goglia, I.; Węglarz-Tomczak, E.; Gioia, C.; Liu, Y.; Virtuoso, A.; Bonanomi, M.; Gaglio, D.; Salmistraro, N.; De Luca, C.; Papa, M.; et al. Fusion-fission-mitophagy cycling and metabolic reprogramming coordinate nerve growth factor (NGF)-dependent neuronal differentiation. FEBS J. 2024, 291, 2811–2835. [Google Scholar] [CrossRef]
- Ranieri, M.; Brajkovic, S.; Riboldi, G.; Ronchi, D.; Rizzo, F.; Bresolin, N.; Corti, S.; Comi, G.P. Mitochondrial fusion proteins and human diseases. Neurol. Res. Int. 2013, 2013, 293893. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef]
- Prajapat, S.K.; Maharana, K.C.; Singh, S. Mitochondrial dysfunction in the pathogenesis of endothelial dysfunction. Mol. Cell. Biochem. 2024, 479, 1999–2016. [Google Scholar] [CrossRef]
- Li, Y.J.; Jin, X.; Li, D.; Lu, J.; Zhang, X.N.; Yang, S.J.; Zhao, Y.X.; Wu, M. New insights into vascular aging: Emerging role of mitochondria function. Biomed. Pharmacother. 2022, 156, 113954. [Google Scholar] [CrossRef]
- Kozanoglu, I.; Pepedil-Tanrikulu, F. Functions of the endothelium and its role in hematopoietic cell transplantation. Transfus. Apher. Sci. 2022, 61, 103368. [Google Scholar] [CrossRef]
- de la Bastida-Casero, L.; García-León, B.; Tura-Ceide, O.; Oliver, E. The Relevance of the Endothelium in Cardiopulmonary Disorders. Int. J. Mol. Sci. 2024, 25, 9260. [Google Scholar] [CrossRef]
- Lenasi, H. Endothelium at a Glance. In Endothelial Dysfunction: Old Concepts and New Challenges; BoD–Books on Demand: Hamburg, Germany, 2018. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, L.; Serrador, J.M. Subcellular Localization Guides eNOS Function. Int. J. Mol. Sci. 2024, 25, 13402. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Lin, S.; Fagan, K.A.; Li, K.X.; Shaul, P.W.; Cooper, D.M.; Rodman, D.M. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 2000, 275, 17979–17985. [Google Scholar] [CrossRef] [PubMed]
- Devika, N.T.; Jaffar Ali, B.M. Analysing calcium dependent and independent regulation of eNOS in endothelium triggered by extracellular signalling events. Mol. Biosyst. 2013, 9, 2653–2664. [Google Scholar] [CrossRef]
- Durán, W.N.; Breslin, J.W.; Sánchez, F.A. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc. Res. 2010, 87, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Bowman, L.; D’Arsigny, C.L.; Archer, S.L. Soluble guanylate cyclase: A new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clin. Pharmacol. Ther. 2015, 97, 88–102. [Google Scholar] [CrossRef]
- Katakam, P.V.; Wappler, E.A.; Katz, P.S.; Rutkai, I.; Institoris, A.; Domoki, F.; Gáspár, T.; Grovenburg, S.M.; Snipes, J.A.; Busija, D.W. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 752–759. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Bubolz, A.H.; Zhang, D.X.; Gutterman, D.D. Endothelial cytoskeletal elements are critical for flow-mediated dilation in human coronary arterioles. Med. Biol. Eng. Comput. 2008, 46, 469–478. [Google Scholar] [CrossRef]
- Kulovic-Sissawo, A.; Tocantins, C.; Diniz, M.S.; Weiss, E.; Steiner, A.; Tokic, S.; Madreiter-Sokolowski, C.T.; Pereira, S.P.; Hiden, U. Mitochondrial Dysfunction in Endothelial Progenitor Cells: Unraveling Insights from Vascular Endothelial Cells. Biology 2024, 13, 70. [Google Scholar] [CrossRef]
- Zhan, Y.; Cao, J.; Ji, L.; Zhang, M.; Shen, Q.; Xu, P.; Zhuang, X.; Qin, S.; Hua, F.; Sun, L.; et al. Impaired mitochondria of Tregs decreases OXPHOS-derived ATP in primary immune thrombocytopenia with positive plasma pathogens detected by metagenomic sequencing. Exp. Hematol. Oncol. 2022, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, A.; Melo, L.; Clemente-Sanchez, A.; Chao, X.; Ahmadi, A.R.; Peiffer, B.; Sun, Z.; Sesaki, H.; Li, T.; et al. Loss of hepatic DRP1 exacerbates alcoholic hepatitis by inducing megamitochondria and mitochondrial maladaptation. Hepatology 2023, 77, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Feinberg, M.W. Regulation of endothelial cell metabolism: Just go with the flow. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 13–15. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef]
- Wang, J.; Toan, S.; Zhou, H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 2020, 23, 299–314. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Y.; Kim, I.M.; Weintraub, N.L.; Tang, Y. The Impaired Bioenergetics of Diabetic Cardiac Microvascular Endothelial Cells. Front. Endocrinol. 2021, 12, 642857. [Google Scholar] [CrossRef]
- Wang, T.; Wang, X.; Fu, T.; Ma, Y.; Wang, Q.; Zhang, S.; Zhang, X.; Zhou, H.; Chang, X.; Tong, Y. Roles of mitochondrial dynamics and mitophagy in diabetic myocardial microvascular injury. Cell Stress Chaperones 2023, 28, 675–688. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, H.; Chang, X. Involvement of mitochondrial dynamics and mitophagy in diabetic endothelial dysfunction and cardiac microvascular injury. Arch. Toxicol. 2023, 97, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, R.; Xu, K.; Guo, H.; Li, C.; Sun, X. Protective effect of Xinmai’an tablets via mediation of the AMPK/SIRT1/PGC-1α signaling pathway on myocardial ischemia-reperfusion injury in rats. Phytomedicine 2023, 120, 155034. [Google Scholar] [CrossRef]
- He, H.; Qiao, Y.; Zhou, Q.; Wang, Z.; Chen, X.; Liu, D.; Yin, D.; He, M. Iron Overload Damages the Endothelial Mitochondria via the ROS/ADMA/DDAHII/eNOS/NO Pathway. Oxidative Med. Cell Longev. 2019, 2019, 2340392. [Google Scholar] [CrossRef]
- Liao, H.; Qi, Y.; Ye, Y.; Yue, P.; Zhang, D.; Li, Y. Mechanotranduction Pathways in the Regulation of Mitochondrial Homeostasis in Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 625089. [Google Scholar] [CrossRef]
- Sun, D.; Wang, J.; Toan, S.; Muid, D.; Li, R.; Chang, X.; Zhou, H. Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: Focus on mitochondrial quality surveillance. Angiogenesis 2022, 25, 307–329. [Google Scholar] [CrossRef]
- Shen, G.X. Mitochondrial dysfunction, oxidative stress and diabetic cardiovascular disorders. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 106–112. [Google Scholar] [CrossRef]
- Roy Chowdhury, S.K.; Sangle, G.V.; Xie, X.; Stelmack, G.L.; Halayko, A.J.; Shen, G.X. Effects of extensively oxidized low-density lipoprotein on mitochondrial function and reactive oxygen species in porcine aortic endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Sangle, G.V.; Chowdhury, S.K.; Xie, X.; Stelmack, G.L.; Halayko, A.J.; Shen, G.X. Impairment of mitochondrial respiratory chain activity in aortic endothelial cells induced by glycated low-density lipoprotein. Free Radic. Biol. Med. 2010, 48, 781–790. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef]
- Ho, H.J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Ozawa, S.; Mori, K.; Asanuma, K.; Yanagita, M.; Uchida, S.; Nakagawa, T. ENOS deficiency causes podocyte injury with mitochondrial abnormality. Free Radic. Biol. Med. 2015, 87, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Na, K.R.; Shin, J.A.; Suh, K.S.; Kim, J.J.; Lee, K.W.; Choi, D.E. Collecting Duct-Specific CR6-Interacting Factor-1-Deletion Aggravates Renal Inflammation and Fibrosis Induced by Unilateral Ureteral Obstruction. Int. J. Mol. Sci. 2021, 22, 11699. [Google Scholar] [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253. [Google Scholar] [CrossRef]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Busija, D.W.; Rutkai, I.; Dutta, S.; Katakam, P.V. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr. Physiol. 2016, 6, 1529–1548. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Receptor-mediated drug delivery of bispecific therapeutic antibodies through the blood-brain barrier. Front. Drug Deliv. 2023, 3, 1227816. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.H.; Feldsien, T.M.; Mezler, M.; Untucht, C.; Venugopalan, R.; Lefebvre, D.R. Novel Developments to Enable Treatment of CNS Diseases with Targeted Drug Delivery. Pharmaceutics 2023, 15, 1100. [Google Scholar] [CrossRef]
- Shimizu, F.; Nakamori, M. Blood-Brain Barrier Disruption in Neuroimmunological Disease. Int. J. Mol. Sci. 2024, 25, 10625. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Wang, J.; He, L.; Lai, H.; Zhang, T.; Wang, X.; Li, W. Mitochondrial oxidative stress in brain microvascular endothelial cells: Triggering blood-brain barrier disruption. Mitochondrion 2023, 69, 71–82. [Google Scholar] [CrossRef]
- Jing, Y.; Yang, D.X.; Wang, W.; Yuan, F.; Chen, H.; Ding, J.; Geng, Z.; Tian, H.L. Aloin Protects Against Blood-Brain Barrier Damage After Traumatic Brain Injury in Mice. Neurosci. Bull. 2020, 36, 625–638. [Google Scholar] [CrossRef]
- Mao, X.W.; Nishiyama, N.C.; Byrum, S.D.; Stanbouly, S.; Jones, T.; Holley, J.; Sridharan, V.; Boerma, M.; Tackett, A.J.; Willey, J.S.; et al. Spaceflight induces oxidative damage to blood-brain barrier integrity in a mouse model. FASEB J. 2020, 34, 15516–15530. [Google Scholar] [CrossRef] [PubMed]
- Parodi-Rullán, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 1019–1039. [Google Scholar] [CrossRef]
- Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, M.I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Palacios, H.H.; Walrafen, B.; Lipsitt, A.E.; Obrenovich, M.E.; Morales, L. Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int. J. Biochem. Cell Biol. 2009, 41, 1989–2004. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Torres-Yaghi, Y.; Altshuler, M. The diagnosis and natural history of Huntington disease. Handb. Clin. Neurol. 2017, 144, 63–67. [Google Scholar] [CrossRef]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef]
- Meng, K.; Jia, H.; Hou, X.; Zhu, Z.; Lu, Y.; Feng, Y.; Feng, J.; Xia, Y.; Tan, R.; Cui, F.; et al. Mitochondrial Dysfunction in Neurodegenerative Diseases: Mechanisms and Corresponding Therapeutic Strategies. Biomedicines 2025, 13, 327. [Google Scholar] [CrossRef]
- Paul, G.; Elabi, O.F. Microvascular Changes in Parkinson’s Disease- Focus on the Neurovascular Unit. Front. Aging Neurosci. 2022, 14, 853372. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Sun, J.; Dong, Q.; Cui, M. Blood-brain barrier endothelial cells in neurodegenerative diseases: Signals from the “barrier”. Front. Neurosci. 2023, 17, 1047778. [Google Scholar] [CrossRef]
- Grossini, E.; De Marchi, F.; Venkatesan, S.; Mele, A.; Ferrante, D.; Mazzini, L. Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit. Antioxidants 2023, 12, 1887. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Saporta, S.; Sanberg, P.R. Implications of blood-brain barrier disruption in ALS. Amyotroph. Lateral Scler. 2008, 9, 375–376. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Ehrhart, J.; Mustafa, H.; Llauget, A.; Boccio, K.J.; Sanberg, P.R.; Appel, S.H.; Borlongan, C.V. Phenotypic characteristics of human bone marrow-derived endothelial progenitor cells in vitro support cell effectiveness for repair of the blood-spinal cord barrier in ALS. Brain Res. 2019, 1724, 146428. [Google Scholar] [CrossRef]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef] [PubMed]
- Grossini, E.; Garhwal, D.; Venkatesan, S.; Ferrante, D.; Mele, A.; Saraceno, M.; Scognamiglio, A.; Mandrioli, J.; Amedei, A.; De Marchi, F.; et al. The Potential Role of Peripheral Oxidative Stress on the Neurovascular Unit in Amyotrophic Lateral Sclerosis Pathogenesis: A Preliminary Report from Human and In Vitro Evaluations. Biomedicines 2022, 10, 691. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Zakhary, N.I.; Aleya, L.; Bungǎu, S.G.; Bohara, R.A.; Siddiqi, N.J. Aging, Metabolic, and Degenerative Disorders: Biomedical Value of Antioxidants. Oxidative Med. Cell Longev. 2018, 2018, 2098123. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Abo-El-Sooud, K.; Aleya, L.; Bungǎu, S.G.; Najda, A.; Saluja, R. Alleviation of Drugs and Chemicals Toxicity: Biomedical Value of Antioxidants. Oxidative Med. Cell Longev. 2018, 2018, 6276438. [Google Scholar] [CrossRef]
- Jalouli, M.; Rahman, M.A.; Biswas, P.; Rahman, H.; Harrath, A.H.; Lee, I.S.; Kang, S.; Choi, J.; Park, M.N.; Kim, B. Targeting natural antioxidant polyphenols to protect neuroinflammation and neurodegenerative diseases: A comprehensive review. Front. Pharmacol. 2025, 16, 1492517. [Google Scholar] [CrossRef]
- Barbalho, S.M.; Leme Boaro, B.; da Silva Camarinha Oliveira, J.; Patocka, J.; Barbalho Lamas, C.; Tanaka, M.; Laurindo, L.F. Molecular Mechanisms Underlying Neuroinflammation Intervention with Medicinal Plants: A Critical and Narrative Review of the Current Literature. Pharmaceuticals 2025, 18, 133. [Google Scholar] [CrossRef]
- Tao, W.; Zhang, H.; Jiang, X.; Chen, N. Resveratrol combats chronic diseases through enhancing mitochondrial quality. Food Sci. Hum. Wellness 2024, 13, 597–610. [Google Scholar] [CrossRef]
- Azzini, E.; Peña-Corona, S.I.; Hernández-Parra, H.; Chandran, D.; Saleena, L.A.K.; Sawikr, Y.; Peluso, I.; Dhumal, S.; Kumar, M.; Leyva-Gómez, G.; et al. Neuroprotective and anti-inflammatory effects of curcumin in Alzheimer’s disease: Targeting neuroinflammation strategies. Phytother. Res. 2024, 38, 3169–3189. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.Y.; Kim, H.B.; Hwang, E.S.; Park, H.S.; Cho, J.M.; Ham, Y.K.; Kim, J.H.; Mun, M.K.; Maeng, S.; Park, J.H. Naringin enhances long-term potentiation and recovers learning and memory deficits of amyloid-beta induced Alzheimer’s disease-like behavioral rat model. Neurotoxicology 2023, 95, 35–45. [Google Scholar] [CrossRef]
- Shi, R.; Gao, D.; Stoika, R.; Liu, K.; Sik, A.; Jin, M. Potential implications of polyphenolic compounds in neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2024, 64, 5491–5514. [Google Scholar] [CrossRef]
- Dogra, S.K.; Kajla, V.; Garg, S.; Singh, H.; Kumar, D.; Puri, G.R.; Kaur, M.; Kumar, A.; Bilandi, A. Genistein as a neuroprotective agent: A comprehensive review of its potential in neurodegenerative diseases. J. Pharm. Insights Res. 2023, 1, 50–60. [Google Scholar]
- Zhong, G.; Wang, X.; Li, J.; Xie, Z.; Wu, Q.; Chen, J.; Wang, Y.; Chen, Z.; Cao, X.; Li, T.; et al. Insights Into the Role of Copper in Neurodegenerative Diseases and the Therapeutic Potential of Natural Compounds. Curr. Neuropharmacol. 2024, 22, 1650–1671. [Google Scholar] [CrossRef]
- Arias-Sánchez, R.A.; Torner, L.; Fenton Navarro, B. Polyphenols and Neurodegenerative Diseases: Potential Effects and Mechanisms of Neuroprotection. Molecules 2023, 28, 5415. [Google Scholar] [CrossRef] [PubMed]
- Chipofya, E.; Docrat, T.F.; Marnewick, J.L. The Neuroprotective Effect of Rooibos Herbal Tea Against Alzheimer’s Disease: A Review. Mol. Nutr. Food Res. 2025, 69, e202400670. [Google Scholar] [CrossRef]
- Chen, P.; Yao, L.; Yuan, M.; Wang, Z.; Zhang, Q.; Jiang, Y.; Li, L. Mitochondrial dysfunction: A promising therapeutic target for liver diseases. Genes Dis. 2024, 11, 101115. [Google Scholar] [CrossRef]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, Y.; Lu, B.; Xu, J.; Wang, H.; Wang, F.; Lin, N. The role of Drp1—Pink1—Parkin—Mediated mitophagy in perfluorobutane sulfonate- induced hepatocyte damage. Ecotoxicol. Environ. Saf. 2024, 285, 117066. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Kuang, L.; Xiang, X.; Zhang, J.; Zhu, Y.; Wu, Y.; Yan, Q.; Liu, L.; Li, T. Correction: Drp1 regulates mitochondrial dysfunction and dysregulated metabolism in ischemic injury via Clec16a-, BAX-, and GSH- pathways. Cell Death Dis. 2025, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Riva, A.; Moreno, C.; Odena, G.; Mudan, S.; Manyakin, N.; Miquel, R.; Degré, D.; Trepo, E.; Sancho-Bru, P.; et al. Perturbations in Mitochondrial Dynamics Are Closely Involved in the Progression of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2020, 44, 856–865. [Google Scholar] [CrossRef]
- Quan, Y.; Shou, D.; Yang, S.; Cheng, J.; Li, Y.; Huang, C.; Chen, H.; Zhou, Y. Mdivi1 ameliorates mitochondrial dysfunction in non-alcoholic steatohepatitis by inhibiting JNK/MFF signaling. J. Gastroenterol. Hepatol. 2023, 38, 2215–2227. [Google Scholar] [CrossRef]
- Lai, S.; Tang, D.; Feng, J. Mitochondrial targeted therapies in MAFLD. Biochem. Biophys. Res. Commun. 2025, 753, 151498. [Google Scholar] [CrossRef]
- Simões, I.C.M.; Amorim, R.; Teixeira, J.; Karkucinska-Wieckowska, A.; Carvalho, A.; Pereira, S.P.; Simões, R.F.; Szymanska, S.; Dąbrowski, M.; Janikiewicz, J.; et al. The Alterations of Mitochondrial Function during NAFLD Progression-An Independent Effect of Mitochondrial ROS Production. Int. J. Mol. Sci. 2021, 22, 6848. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, S.; Wu, J.; Wang, Y. Mitochondrial metabolic dysfunction and non-alcoholic fatty liver disease: New insights from pathogenic mechanisms to clinically targeted therapy. J. Transl. Med. 2023, 21, 510. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, Y.; Wang, D.; Huang, Z.; Xiao, X.; Zheng, Q.; Li, S.; Long, D.; Feng, L. Mitochondrial Dysfunction in Metabolic Dysfunction Fatty Liver Disease (MAFLD). Int. J. Mol. Sci. 2023, 24, 17514. [Google Scholar] [CrossRef]
- Ordóñez-Vázquez, A.L.; Beltrán-Gall, S.M.; Pal, S.C.; Méndez-Sánchez, N. Editorial: Treatment with Dual Incretin Receptor Agonists to Maintain Normal Glucose Levels May Also Maintain Normal Weight and Control Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD). Med. Sci. Monit. 2022, 28, e938365. [Google Scholar] [CrossRef] [PubMed]
- Vesa, C.M.; Bungau, S.G. Novel Molecules in Diabetes Mellitus, Dyslipidemia and Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 4029. [Google Scholar] [CrossRef] [PubMed]
- Avula, S.; Parikh, S.; Demarest, S.; Kurz, J.; Gropman, A. Treatment of mitochondrial disorders. Curr. Treat. Options Neurol. 2014, 16, 292. [Google Scholar] [CrossRef] [PubMed]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12, 1083. [Google Scholar] [CrossRef]
- Grossini, E.; Venkatesan, S.; Alkabes, M.; Toma, C.; de Cillà, S. Membrane Blue Dual Protects Retinal Pigment Epithelium Cells/Ganglion Cells-Like through Modulation of Mitochondria Function. Biomedicines 2022, 10, 2854. [Google Scholar] [CrossRef]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef]
- Hernandez-Resendiz, S.; Prunier, F.; Girao, H.; Dorn, G.; Hausenloy, D.J. Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 2020, 24, 6571–6585. [Google Scholar] [CrossRef]
- Hong, W.L.; Huang, H.; Zeng, X.; Duan, C.Y. Targeting mitochondrial quality control: New therapeutic strategies for major diseases. Mil. Med. Res. 2024, 11, 59. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- García-González, N.; Gonçalves-Sánchez, J.; Gómez-Nieto, R.; Gonçalves-Estella, J.M.; López, D.E. Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 12485. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Chu, Y.; Yun, Z.; Yan, Y.; Jin, J. Mitochondrial transplantation: Opportunities and challenges in the treatment of obesity, diabetes, and nonalcoholic fatty liver disease. J. Transl. Med. 2022, 20, 483. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Eastwood, J.D.; Alba, D.E.; Velmurugan, S.; Sun, N.; Porciatti, V.; Lee, R.K.; Hauswirth, W.W.; Guy, J.; Yu, H. Gene therapy restores mitochondrial function and protects retinal ganglion cells in optic neuropathy induced by a mito-targeted mutant ND1 gene. Gene Ther. 2022, 29, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Ramón, J.; Vila-Julià, F.; Molina-Granada, D.; Molina-Berenguer, M.; Melià, M.J.; García-Arumí, E.; Torres-Torronteras, J.; Cámara, Y.; Martí, R. Therapy Prospects for Mitochondrial DNA Maintenance Disorders. Int. J. Mol. Sci. 2021, 22, 6447. [Google Scholar] [CrossRef]
- Chen, R.; Chen, J. Mitochondrial transfer—A novel promising approach for the treatment of metabolic diseases. Front. Endocrinol. 2023, 14, 1346441. [Google Scholar] [CrossRef]
- Millington-Ward, S.; Palfi, A.; Shortall, C.; Finnegan, L.K.; Bargroff, E.; Post, I.J.; Maguire, J.; Irnaten, M.; O′ Brien, C.; Kenna, P.F.; et al. AAV-NDI1 Therapy Provides Significant Benefit to Murine and Cellular Models of Glaucoma. Int. J. Mol. Sci. 2024, 25, 8876. [Google Scholar] [CrossRef]
- Kubat, G.B.; Ulger, O.; Akin, S. Requirements for successful mitochondrial transplantation. J. Biochem. Mol. Toxicol. 2021, 35, e22898. [Google Scholar] [CrossRef]
- McCully, J.D.; Del Nido, P.J.; Emani, S.M. Mitochondrial transplantation for organ rescue. Mitochondrion 2022, 64, 27–33. [Google Scholar] [CrossRef]
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.J.; Lee, E.W.; Lee, S.C.; Bae, K.H.; Han, B.S.; Kim, W.K. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Zhang, L.; Hu, G.; Tao, L.; Zhang, F. Mitochondrial transplantation for the treatment of cardiac and noncardiac diseases: Mechanisms, prospective, and challenges. Life Med. 2024, 3, lnae017. [Google Scholar] [PubMed]
- Koene, S.; Smeitink, J. Mitochondrial medicine: Entering the era of treatment. J. Intern. Med. 2009, 265, 193–209. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role | Mechanisms | Action/Involvement | References |
|---|---|---|---|
| Endothelium function/dysfunction | Barrier, nutrient transportation, and waste products elimination; mitochondrial dysfunction, swelling, and apoptosis; enhancement of Aβ production; morphological changes in mitochondria; ATP reduction and ROS increase | Maintaining vascular tone and function; cardio-metabolic disease; AD; liver diseases; aging; renal diseases | [1,43,45,62] |
| Mitochondria in endothelial cells | Oxygen sensors and ROS production; inflammation, redox signaling, AMPK, and PGC pathways; TRPV4 expression and calcium influx; morphology alteration; reduction in mitochondrial function | Aging; cardio-metabolic diseases; neurodegenerative diseases; AD; renal diseases; ALS | [6,7,65,84] |
| ROS release | NO bioavailability reduction, impaired endothelial dependent vasodilation; Ca2+ and eNOS activity regulation; elevated ROS levels | Arterial stiffness; maintaining vascular tone and function; aging | [19,41,46] |
| Mitochondrial structure/function | Mitochondrial dynamics’ regulation; ETC | Maintaining endothelial cell’s function; ATP production | [26] |
| Mitochondrial function targeted drugs | Removal of damaged mitochondria; regulation of AKT, nuclear factor erythroid 2-related factor 2, Janus kinase/signal transducer pathways and the activator of transcription, nucleotide-binding and oligomerization domain-like receptor familypyrin domain-containing 3 inflammasome, and MAPK; anti-inflammation and anti-amyloid | Regulation of health and pathological conditions; alleviation of neurodegenerative disorders and cognitive function; mitigating AD, PD, HD, and ALS progression | [10,91,92,93,94,95,96,97] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossini, E.; Venkatesan, S.; Ola Pour, M.M. Mitochondrial Dysfunction in Endothelial Cells: A Key Driver of Organ Disorders and Aging. Antioxidants 2025, 14, 372. https://doi.org/10.3390/antiox14040372
Grossini E, Venkatesan S, Ola Pour MM. Mitochondrial Dysfunction in Endothelial Cells: A Key Driver of Organ Disorders and Aging. Antioxidants. 2025; 14(4):372. https://doi.org/10.3390/antiox14040372
Chicago/Turabian StyleGrossini, Elena, Sakthipriyan Venkatesan, and Mohammad Mostafa Ola Pour. 2025. "Mitochondrial Dysfunction in Endothelial Cells: A Key Driver of Organ Disorders and Aging" Antioxidants 14, no. 4: 372. https://doi.org/10.3390/antiox14040372
APA StyleGrossini, E., Venkatesan, S., & Ola Pour, M. M. (2025). Mitochondrial Dysfunction in Endothelial Cells: A Key Driver of Organ Disorders and Aging. Antioxidants, 14(4), 372. https://doi.org/10.3390/antiox14040372