
Xanthine Oxidoreductase: A Double-Edged Sword in Neurological Diseases





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
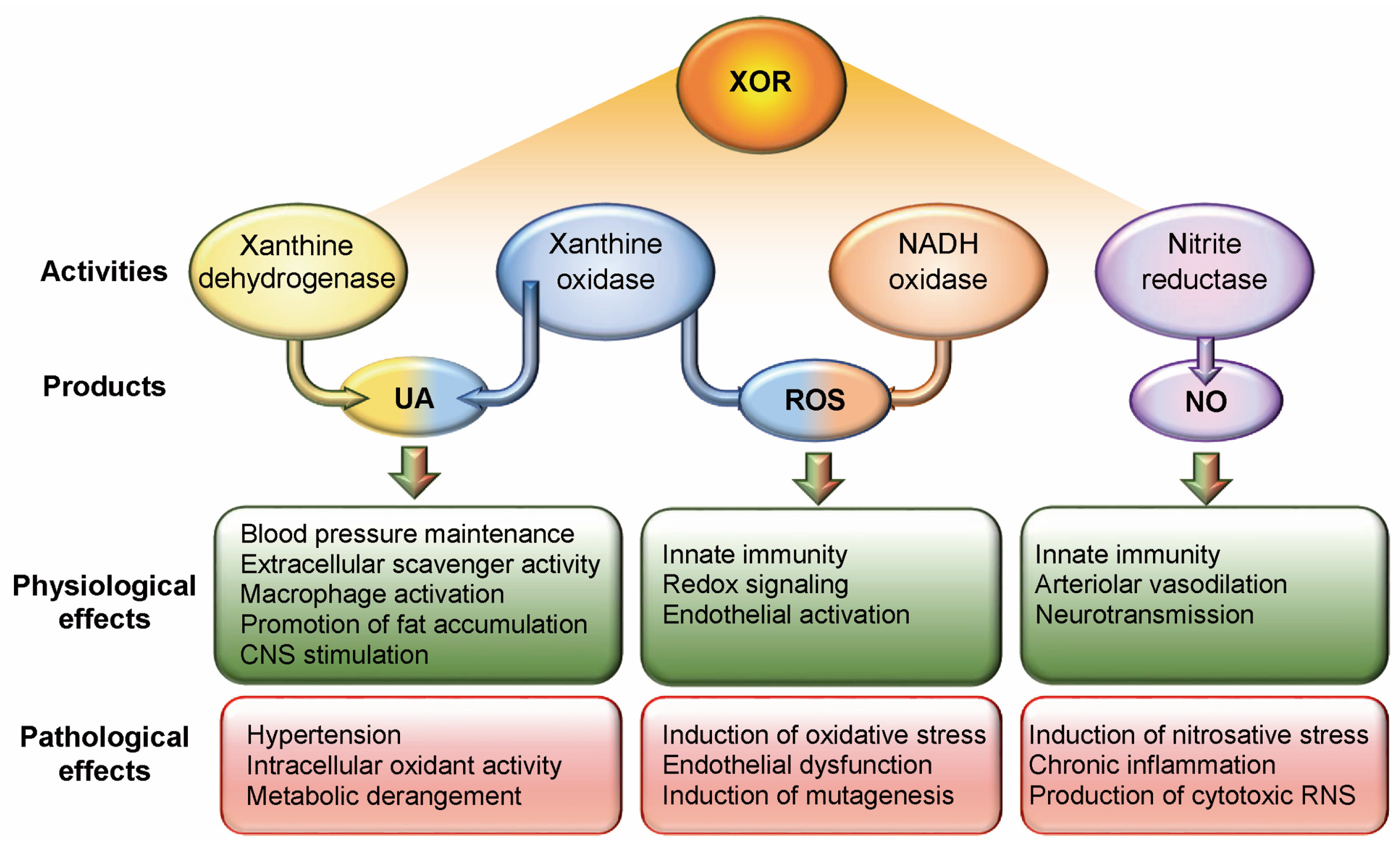
2. Xanthine Oxidoreductase
3. Multiple Sclerosis
4. Alzheimer’s Disease
5. Amyotrophic Lateral Sclerosis
6. Huntington’s Disease
7. Parkinson’s Disease
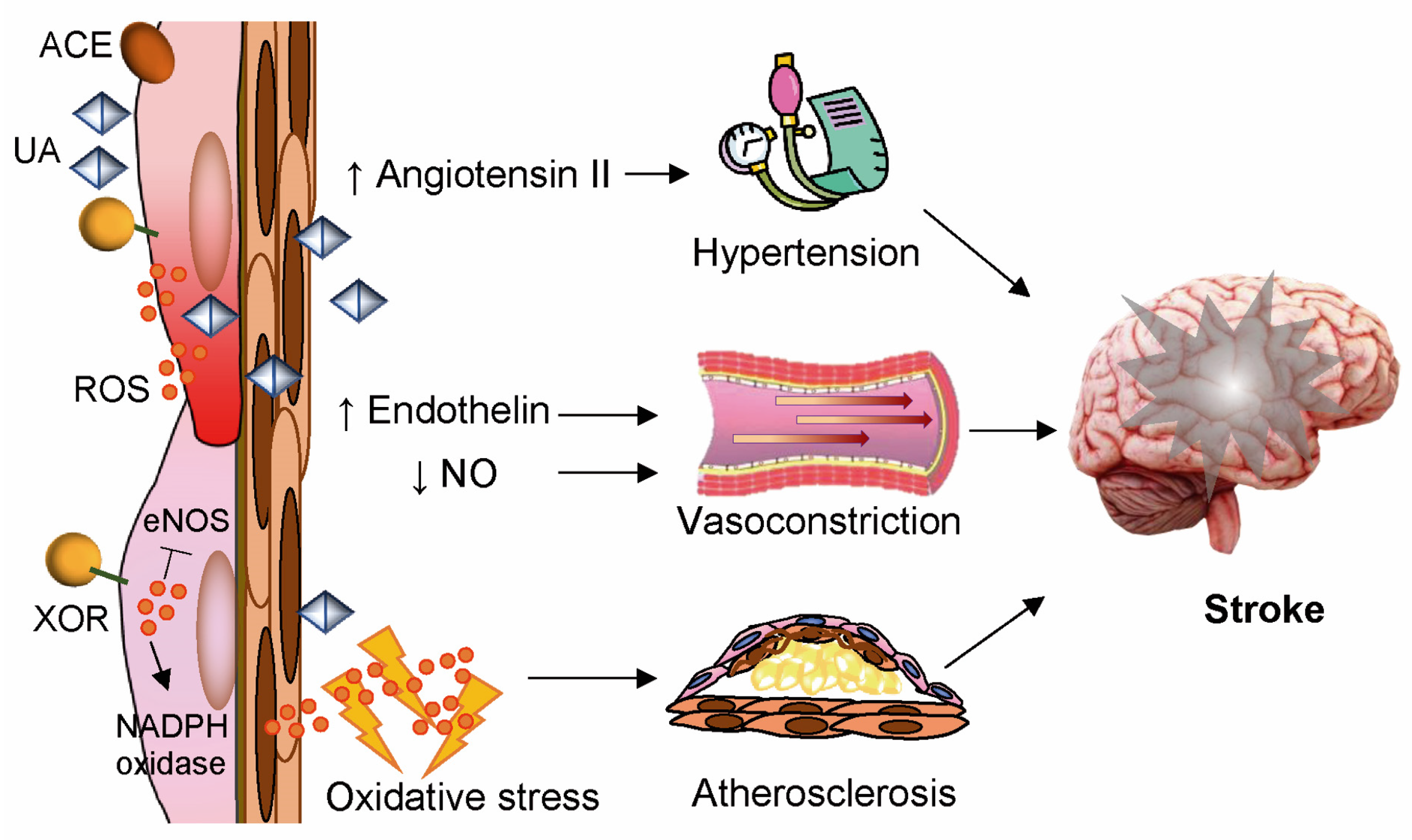
8. Stroke
9. Inosine Treatments
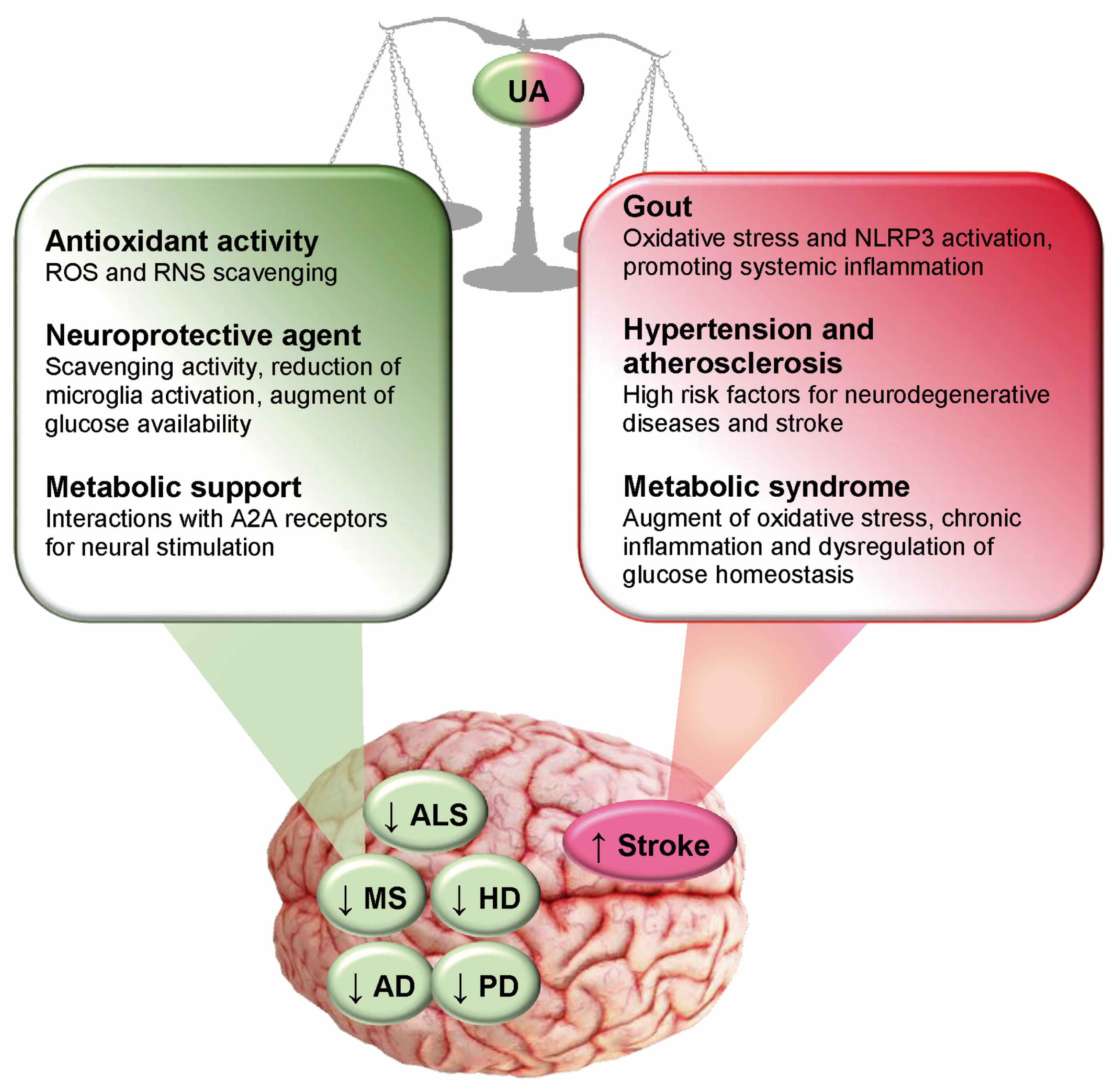
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β |
| ACE | Angiotensin-Converting Enzyme |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| CNS | Central Nervous System |
| COX2 | Cyclooxygenase-2 |
| EAE | Experimental Autoimmune Encephalomyelitis |
| EDSS | Expanded Disability Status Scale |
| ENOS | Endothelial Nitric Oxide Synthase |
| GBD | Global Burden of Diseases, Injuries, and Risk Factors Study |
| GSH | Reduced Glutathione |
| HD | Huntington’s Disease |
| MAP | Mitogen-Activated Protein |
| MS | Multiple Sclerosis |
| NAD | Nicotinamide Adenine Dinucleotide |
| NADH | Nicotinamide Adenine Dinucleotide Reduced |
| NADP | Nicotinamide Adenine Dinucleotide Phosphate |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate Reduced |
| NO | Nitric Oxide |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| PD | Parkinson’s Disease |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| UA | Uric Acid |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| XDH | Xanthine Dehydrogenase |
| XO | Xanthine Oxidase |
| XOR | Xanthine Oxidoreductase |
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting urate to reduce oxidative stress in Parkinson disease. Exp. Neurol. 2017, 298, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.M.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxid. Med. Cell. Longev. 2020, 2020, 7191080. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V.E. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, the Bad, and the Ugly. J. Huntingtons Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef]
- Percário, S.; da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; de Nazaré Araújo Moreira, T.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxid. Med. Cell. Longev. 2020, 2020, 2360872. [Google Scholar] [CrossRef]
- Li, Z.; Bi, R.; Sun, S.; Chen, S.; Chen, J.; Hu, B.; Jin, H. The Role of Oxidative Stress in Acute Ischemic Stroke-Related Thrombosis. Oxid. Med. Cell. Longev. 2022, 2022, 8418820. [Google Scholar] [CrossRef]
- Xu, L.; Li, C.; Wan, T.; Sun, X.; Lin, X.; Yan, D.; Li, J.; Wei, P. Targeting uric acid: A promising intervention against oxidative stress and neuroinflammation in neurodegenerative diseases. Cell Commun. Signal. 2025, 23, 4. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. The C-terminal peptide plays a role in the formation of an intermediate form during the transition between xanthine dehydrogenase and xanthine oxidase. FEBS J. 2015, 282, 3075–3090. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Lanaspa, M.A.; Gaucher, E.A. Uric acid: A danger signal from the RNA world that may have a role in the epidemic of obesity, metabolic syndrome, and cardiorenal disease: Evolutionary considerations. Semin. Nephrol. 2011, 31, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Camandola, S.; Malott, K.F.; Edelhauser, M.A.; Mattson, M.P. The Role of Uric Acid and Methyl Derivatives in the Prevention of Age-Related Neurodegenerative Disorders. Curr. Top. Med. Chem. 2015, 15, 2233–2238. [Google Scholar] [CrossRef]
- Stamp, L.K.; Dalbeth, N. Moving urate-lowering therapy in gout beyond guideline recommendations. Semin. Arthritis Rheum. 2024, 65, 152358. [Google Scholar] [CrossRef]
- Gherghina, M.E.; Peride, I.; Tiglis, M.; Neagu, T.P.; Niculae, A.; Checherita, I.A. Uric Acid and Oxidative Stress-Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int. J. Mol. Sci. 2022, 23, 3188. [Google Scholar] [CrossRef]
- Yu, W.; Cheng, J.D. Uric acid and cardiovascular disease: An update from molecular mechanism to clinical perspective. Front. Pharmacol. 2020, 11, 582680. [Google Scholar] [CrossRef]
- Nakayama, A.; Kurajoh, M.; Toyoda, Y.; Takada, T.; Ichida, K.; Matsuo, H. Dysuricemia. Biomedicines 2023, 11, 3169. [Google Scholar] [CrossRef]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- Wang, F.; Yuan, Q.; Chen, F.; Pang, J.; Pan, C.; Xu, F.; Chen, Y. Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front. Cell Dev. Biol. 2021, 9, 742483. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bolognesi, A.; Polito, L. Pathophysiology of circulating xanthine oxidoreductase: New emerging roles for a multi-tasking enzyme. Biochim. Biophys. Acta 2014, 1842, 1502–1517. [Google Scholar] [CrossRef]
- van Golen, R.F.; Reiniers, M.J.; Olthof, P.B.; van Gulik, T.M.; Heger, M. Sterile inflammation in hepatic ischemia/reperfusion injury: Present concepts and potential therapeutics. J. Gastroenterol. Hepatol. 2013, 28, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Liddell, J.R.; Hilton, J.B.W.; Kysenius, K.; Billings, J.L.; Nikseresht, S.; McInnes, L.E.; Hare, D.J.; Paul, B.; Mercer, S.W.; Belaidi, A.A.; et al. Microglial ferroptotic stress causes non-cell autonomous neuronal death. Mol. Neurodegener. 2024, 19, 14. [Google Scholar] [CrossRef]
- Battelli, M.G.; Bortolotti, M.; Bolognesi, A.; Polito, L. Pro-Aging Effects of Xanthine Oxidoreductase Products. Antioxidants 2020, 9, 839. [Google Scholar] [CrossRef]
- Wang, L.; Li, B.; Quan, M.Y.; Li, L.; Chen, Y.; Tan, G.J.; Zhang, J.; Liu, X.P.; Guo, L. Mechanism of oxidative stress p38MAPK-SGK1 signaling axis in experimental autoimmune encephalomyelitis (EAE). Oncotarget 2017, 8, 42808–42816. [Google Scholar] [CrossRef]
- Dąbrowska-Bouta, B.; Strużyńska, L.; Sidoryk-Węgrzynowicz, M.; Sulkowski, G. Memantine Modulates Oxidative Stress in the Rat Brain following Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2021, 22, 11330. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijević, M.; Kotur-Stevuljević, J.; Stojić-Vukanić, Z.; Vujnović, I.; Pilipović, I.; Nacka-Aleksić, M.; Leposavić, G. Sex Difference in Oxidative Stress Parameters in Spinal Cord of Rats with Experimental Autoimmune Encephalomyelitis: Relation to Neurological Deficit. Neurochem. Res. 2017, 42, 481–492. [Google Scholar] [CrossRef]
- Trifunovic, S.; Stevanovic, I.; Milosevic, A.; Ristic, N.; Janjic, M.; Bjelobaba, I.; Savic, D.; Bozic, I.; Jakovljevic, M.; Tesovic, K.; et al. The Function of the Hypothalamic-Pituitary-Adrenal Axis During Experimental Autoimmune Encephalomyelitis: Involvement of Oxidative Stress Mediators. Front. Neurosci. 2021, 15, 649485. [Google Scholar] [CrossRef]
- Khalatbari Mohseni, G.; Hosseini, S.A.; Majdinasab, N.; Cheraghian, B. Effects of N-acetylcysteine on oxidative stress biomarkers, depression, and anxiety symptoms in patients with multiple sclerosis. Neuropsychopharmacol. Rep. 2023, 43, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.L.; Gutiérrez, F.; Iglesias, F.; Martín-Polo, J.; Merino, S.; Martín-Serradilla, J.I.; Laherrán, E.; Tejero, M.A. Serum uric acid levels in multiple sclerosis patients inversely correlate with disability. Neurol. Sci. 2011, 32, 347–350. [Google Scholar] [CrossRef]
- Liu, B.; Shen, Y.; Xiao, K.; Tang, Y.; Cen, L.; Wei, J. Serum uric acid levels in patients with multiple sclerosis: A meta-analysis. Neurol. Res. 2012, 34, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Lanzillo, R.; Costabile, T.; Russo, C.; Carotenuto, A.; Sasso, G.; Postiglione, E.; De Luca Picione, C.; Vastola, M.; Maniscalco, G.T.; et al. Uric acid in relapsing-remitting multiple sclerosis: A 2-year longitudinal study. J. Neurol. 2015, 262, 961–967. [Google Scholar] [CrossRef]
- GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Oksanen, M.; Hyötyläinen, I.; Trontti, K.; Rolova, T.; Wojciechowski, S.; Koskuvi, M.; Viitanen, M.; Levonen, A.L.; Hovatta, I.; Roybon, L.; et al. NF-E2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin-1 mutated Alzheimer’s disease astrocytes. Glia 2020, 68, 589–599. [Google Scholar] [CrossRef]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxidative Med. Cell. Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, J.; Tian, Q.; Tian, N.; Tian, Q.; He, X.; Wang, Y.; Dong, Z. Uric Acid Mitigates Cognitive Deficits via TFEB-Mediated Microglial Autophagy in Mice Models of Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 3678–3696. [Google Scholar] [CrossRef]
- Johnson, R.J.; Tolan, D.R.; Bredesen, D.; Nagel, M.; Sánchez-Lozada, L.G.; Fini, M.; Burtis, S.; Lanaspa, M.A.; Perlmutter, D. Could Alzheimer’s disease be a maladaptation of an evolutionary survival pathway mediated by intracerebral fructose and uric acid metabolism? Am. J. Clin. Nutr. 2023, 117, 455–466. [Google Scholar] [CrossRef]
- Lu, N.; Dubreuil, M.; Zhang, Y.; Neogi, T.; Rai, S.K.; Ascherio, A.; Hernán, M.A.; Choi, H.K. Gout and the risk of Alzheimer’s disease: A population-based, BMI-matched cohort study. Ann. Rheum. Dis. 2016, 75, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. Oxidative damage in neurodegeneration: Roles in the pathogenesis and progression of Alzheimer disease. Physiol. Rev. 2024, 104, 103–197. [Google Scholar] [CrossRef]
- Bhole, R.P.; Chikhale, R.V.; Rathi, K.M. Current biomarkers and treatment strategies in Alzheimer disease: An overview and future perspectives. IBRO Neurosci. Rep. 2023, 16, 8–42. [Google Scholar] [CrossRef]
- Paganoni, S.; Zhang, M.; Quiroz Zárate, A.; Jaffa, M.; Yu, H.; Cudkowicz, M.E.; Wills, A.M. Uric acid levels predict survival in men with amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kiriyama, T.; Kobayashi, Y.; Horikawa, H.; Ueno, S. Clinical outcomes and serum uric acid levels in elderly patients with amyotrophic lateral sclerosis aged ≥70 years. Am. J. Neurodegener. Dis. 2013, 2, 140–144. [Google Scholar]
- Xu, L.Q.; Hu, W.; Guo, Q.F.; Xu, G.R.; Wang, N.; Zhang, Q.J. Serum Uric Acid Levels Predict Mortality Risk in Male Amyotrophic Lateral Sclerosis Patients. Front. Neurol. 2021, 12, 602663. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, É.J.; Liu, D.; Johns, D.R.; Cudkowicz, M.E.; Paganoni, S.; Schwarzschild, M.A.; Leitner, M.; Ascherio, A. Serum urate at trial entry and ALS progression in EMPOWER. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 120–125. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Q.; Ke, Y.; Hao, J.; Lu, L.; Lu, N.; Chen, X. Serum uric acid levels in patients with amyotrophic lateral sclerosis: A meta-analysis. Sci. Rep. 2018, 8, 1100. [Google Scholar] [CrossRef]
- Iazzolino, B.; Grassano, M.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Cabras, S.; Callegaro, S.; Matteoni, E.; Di Pede, F.; et al. High serum uric acid levels are protective against cognitive impairment in amyotrophic lateral sclerosis. J. Neurol. 2024, 271, 955–961. [Google Scholar] [CrossRef]
- Auinger, P.; Kieburtz, K.; McDermott, M.P. The relationship between uric acid levels and Huntington’s disease progression. Mov. Disord. 2010, 25, 224–228. [Google Scholar] [CrossRef]
- Graham, S.F.; Kumar, P.; Bahado-Singh, R.O.; Robinson, A.; Mann, D.; Green, B.D. Novel Metabolite Biomarkers of Huntington’s Disease as Detected by High-Resolution Mass Spectrometry. J. Proteome Res. 2016, 15, 1592–1601. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Haque, A.; Aboufadel, S.; Snell, C.; Fischer, R.S.; Granger, S.W.; Granger, D.A.; Thomas, E.A. Uric Acid as a Potential Peripheral Biomarker for Disease Features in Huntington’s Patients. Front. Neurosci. 2020, 14, 73. [Google Scholar] [CrossRef]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953, Erratum in: Lancet Neurol. 2021, 20, e7. [Google Scholar] [CrossRef]
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Parkinson’s Progression Markers Initiative. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: A cross-sectional study. Lancet Neurol. 2023, 22, 407–417. [Google Scholar] [CrossRef]
- Dong-Chen, X.; Yong, C.; Yang, X.; Chen-Yu, S.; Li-Hua, P. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal. Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, Y.; Liu, P.; Xu, X. Xanthine oxidoreductase mediates genotoxic drug-induced autophagy and apoptosis resistance by uric acid accumulation and TGF-β-activated kinase 1 (TAK1) activation. FASEB J. 2023, 37, e22723. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.L.; Chen, X.; Hou, X.O.; Yuan, X.; Yuan, B.S.; Yuan, Y.Q.; Zhang, Q.L.; Cao, X.; Liu, C.F.; Luo, W.F.; et al. Urate promotes SNCA/α-synuclein clearance via regulating mTOR-dependent macroautophagy. Exp. Neurol. 2017, 297, 138–147. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, Y.J.; Lee, J.E.; Kim, Y.S.; Lee, J.W.; Kim, H.; Shin, J.Y.; Lee, P.H. Uric acid regulates α-synuclein transmission in Parkinsonian models. Front. Aging Neurosci. 2023, 15, 1117491. [Google Scholar] [CrossRef]
- Liang, L.P.; Kavanagh, T.J.; Patel, M. Glutathione deficiency in Gclm null mice results in complex I inhibition and dopamine depletion following paraquat administration. Toxicol. Sci. 2013, 134, 366–373. [Google Scholar] [CrossRef]
- Chang, K.H.; Chen, C.M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Bakshi, R.; Macklin, E.A.; Hung, A.Y.; Hayes, M.T.; Hyman, B.T.; Wills, A.M.; Gomperts, S.N.; Growdon, J.H.; Ascherio, A.; Scherzer, C.R.; et al. Associations of Lower Caffeine Intake and Plasma Urate Levels with Idiopathic Parkinson’s Disease in the Harvard Biomarkers Study. J. Park. Dis. 2020, 10, 505–510. [Google Scholar] [CrossRef]
- Koros, C.; Simitsi, A.M.; Papagiannakis, N.; Bougea, A.; Prentakis, A.; Papadimitriou, D.; Pachi, I.; Antonelou, R.; Angelopoulou, E.; Beratis, I.; et al. Serum uric acid level as a putative biomarker in Parkinson’s disease patients carrying GBA1 mutations: 2-Year data from the PPMI study. Park. Relat. Disord. 2021, 84, 1–4. [Google Scholar] [CrossRef]
- Hasimoglu, Y.G.; Chen, X.; Bakshi, R.; Schwarzschild, M.A.; Macklin, E.A. Does Serum Urate Change as Parkinson’s Disease Progresses? J. Park. Dis. 2020, 10, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zheng, J.; Ma, J.; Wang, Z.; Sun, W.; Li, M.; Huang, S.; Hu, S. Low serum uric acid levels are associated with the nonmotor symptoms and brain gray matter volume in Parkinson’s disease. Neurol. Sci. 2022, 43, 1747–1754. [Google Scholar] [CrossRef]
- Grażyńska, A.; Adamczewska, K.; Antoniuk, S.; Bień, M.; Toś, M.; Kufel, J.; Urbaś, W.; Siuda, J. The Influence of Serum Uric Acid Level on Non-Motor Symptoms Occurrence and Severity in Patients with Idiopathic Parkinson’s Disease and Atypical Parkinsonisms-A Systematic Review. Medicina 2021, 57, 972. [Google Scholar] [CrossRef] [PubMed]
- Koros, C.; Simitsi, A.M.; Papagiannakis, N.; Bougea, A.; Prentakis, A.; Papadimitriou, D.; Pachi, I.; Beratis, I.; Stanitsa, E.; Angelopoulou, E.; et al. Serum Uric Acid as a Putative Biomarker in Prodromal Parkinson’s Disease: Longitudinal Data from the PPMI Study. J. Parkinsons Dis. 2023, 13, 811–818, Erratum in: J. Park. Dis. 2024, 14, 1075. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Kuo, G.; Kumbhar, R.; Blair, W.; Dawson, V.L.; Dawson, T.M.; Mao, X. Emerging targets of α-synuclein spreading in α-synucleinopathies: A review of mechanistic pathways and interventions. Mol. Neurodegener. 2025, 20, 10. [Google Scholar] [CrossRef]
- Starr, L.A.; McKay, L.E.; Peter, K.N.; Seyfarth, L.M.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. Attenuation of Dopaminergic Neurodegeneration in a C. elegans Parkinson’s Model through Regulation of Xanthine Dehydrogenase (XDH-1) Expression by the RNA Editase, ADR-2. J. Dev. Biol. 2023, 11, 20. [Google Scholar] [CrossRef]
- Thies, J.L.; Willicott, K.; Craig, M.L.; Greene, M.R.; DuGay, C.N.; Caldwell, G.A.; Caldwell, K.A. Xanthine Dehydrogenase Is a Modulator of Dopaminergic Neurodegeneration in Response to Bacterial Metabolite Exposure in C. elegans. Cells 2023, 12, 1170. [Google Scholar] [CrossRef]
- Haryuni, R.D.; Nukui, T.; Piao, J.L.; Shirakura, T.; Matsui, C.; Sugimoto, T.; Baba, K.; Nakane, S.; Nakatsuji, Y. Elevated Serum Xanthine Oxidase and Its Correlation with Antioxidant Status in Patients with Parkinson’s Disease. Biomolecules 2024, 14, 490. [Google Scholar] [CrossRef]
- Song, Y.; Racette, B.A.; Camacho-Soto, A.; Searles Nielsen, S. Biologic targets of prescription medications and risk of neurodegenerative disease in United States Medicare beneficiaries. PLoS ONE 2023, 18, e0285011. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. The role of xanthine oxidoreductase and uric acid in metabolic syndrome. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: Not only oxidative stress. Atherosclerosis 2014, 237, 562–567. [Google Scholar] [CrossRef]
- Chen, J.; Liu, J.; Gu, Z.; Fan, J.; Lei, S.; Zhang, Q.; Pan, K.; Wang, Y. Adherence to oxidative balance score is inversely associated with the prevalence of stroke: Results from National Health and Nutrition Examination Survey 1999–2018. Front. Neurol. 2024, 15, 1348011. [Google Scholar] [CrossRef]
- Yu, H.; Chen, X.; Guo, X.; Chen, D.; Jiang, L.; Qi, Y.; Shao, J.; Tao, L.; Hang, J.; Lu, G.; et al. The clinical value of serum xanthine oxidase levels in patients with acute ischemic stroke. Redox Biol. 2023, 60, 102623. [Google Scholar] [CrossRef]
- Chen, X.; Zeng, Q.; Tao, L.; Yuan, J.; Hang, J.; Lu, G.; Shao, J.; Li, Y.; Yu, H. Machine Learning-Based Clinical Prediction Models for Acute Ischemic Stroke Based on Serum Xanthine Oxidase Levels. World Neurosurg. 2024, 184, e695–e707. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.Y.; Lee, J.; Yoon, S.; Kim, E.G.; Lee, E.; Kim, N.; Lee, S.; Gym, H.; Park, S.I. Effects of uric acid on ischemic diseases, stratified by lipid levels: A drug-target, nonlinear Mendelian randomization study. Sci. Rep. 2024, 14, 1338. [Google Scholar] [CrossRef]
- Otani, N.; Hoshiyama, E.; Ouchi, M.; Takekawa, H.; Suzuki, K. Uric acid and neurological disease: A narrative review. Front. Neurol. 2023, 14, 1164756. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Yu, S. Allopurinol and the risk of stroke in older adults receiving medicare. BMC Neurol. 2016, 16, 164. [Google Scholar] [CrossRef]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Bolognesi, A. Chronic kidney disease: Which role for xanthine oxidoreductase activity and products? Pharmacol. Res. 2022, 184, 106407. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Carta, A.R.; Kachroo, A.; Schwarzschild, M.A. Pathophysiological roles for purines: Adenosine, caffeine and urate. Prog. Brain Res. 2010, 183, 183–208. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, N.; Aoyama, K. Glutathione-Mediated Neuroprotective Effect of Purine Derivatives. Int. J. Mol. Sci. 2023, 24, 13067. [Google Scholar] [CrossRef]
- Paganoni, S.; Schwarzschild, M.A. Urate as a Marker of Risk and Progression of Neurodegenerative Disease. Neurotherapeutics 2017, 14, 148–153. [Google Scholar] [CrossRef]
- Parkinson Study Group SURE-PD Investigators. Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: A randomized clinical trial. JAMA Neurol. 2014, 71, 141–150. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Macklin, E.A.; Bakshi, R.; Battacharyya, S.; Logan, R.; Espay, A.J.; Hung, A.Y.; Bwala, G.; Goetz, C.G.; Russell, D.S.; et al. Parkinson Study Group SURE-PD Investigators, Sex differences by design and outcome in the Safety of Urate Elevation in PD (SURE-PD) trial. Neurology 2019, 93, e1328–e1338. [Google Scholar] [CrossRef]
- Basile, M.S.; Bramanti, P.; Mazzon, E. Inosine in Neurodegenerative Diseases: From the Bench to the Bedside. Molecules 2022, 27, 4644. [Google Scholar] [CrossRef]
- Nicholson, K.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Bakshi, R.; Grasso, D.L.; Wills, A.M.; Jahandideh, S.; Taylor, A.A.; et al. Pilot trial of inosine to elevate urate levels in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1522–1533. [Google Scholar] [CrossRef]
- Walk, D.; Nicholson, K.; Locatelli, E.; Chan, J.; Macklin, E.A.; Ferment, V.; Manousakis, G.; Chase, M.; Connolly, M.; Dagostino, D.; et al. Randomized trial of inosine for urate elevation in amyotrophic lateral sclerosis. Muscle Nerve 2023, 67, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Spitsin, S.; Hooper, D.C.; Leist, T.; Streletz, L.J.; Mikheeva, T.; Koprowskil, H. Inactivation of peroxynitrite in multiple sclerosis patients after oral administration of inosine may suggest possible approaches to therapy of the disease. Mult. Scler. 2001, 7, 313–319. [Google Scholar] [CrossRef]
- Markowitz, C.E.; Spitsin, S.; Zimmerman, V.; Jacobs, D.; Udupa, J.K.; Hooper, D.C.; Koprowski, H. The treatment of multiple sclerosis with inosine. J. Altern. Complement. Med. 2009, 15, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Llull, L.; Laredo, C.; Renú, A.; Pérez, B.; Vila, E.; Obach, V.; Urra, X.; Planas, A.; Amaro, S.; Chamorro, Á. Uric Acid Therapy Improves Clinical Outcome in Women with Acute Ischemic Stroke. Stroke 2015, 46, 2162–2167. [Google Scholar] [CrossRef]
- Seifar, F.; Dinasarapu, A.R.; Jinnah, H.A. Uric Acid in Parkinson’s Disease: What Is the Connection? Mov. Disord. 2022, 37, 2173–2183. [Google Scholar] [CrossRef]
- Johnson, T.A.; Jinnah, H.A.; Kamatani, N. Shortage of Cellular ATP as a Cause of Diseases and Strategies to Enhance ATP. Front. Pharmacol. 2019, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Sekine, M.; Fujiwara, M.; Okamoto, K.; Ichida, K.; Nagata, K.; Hille, R.; Nishino, T. Significance and amplification methods of the purine salvage pathway in human brain cells. J. Biol. Chem. 2024, 300, 107524. [Google Scholar] [CrossRef]
- Watanabe, H.; Hattori, T.; Kume, A.; Misu, K.; Ito, T.; Koike, Y.; Johnson, T.A.; Kamitsuji, S.; Kamatani, N.; Sobue, G. Improved Parkinsons disease motor score in a single-arm open-label trial of febuxostat and inosine. Medicine 2020, 99, e21576. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase in Drug Metabolism: Beyond a Role as a Detoxifying Enzyme. Curr. Med. Chem. 2016, 23, 4027–4036. [Google Scholar] [CrossRef]
- Aliena-Valero, A.; Rius-Pérez, S.; Baixauli-Martín, J.; Torregrosa, G.; Chamorro, Á.; Pérez, S.; Salom, J.B. Uric Acid Neuroprotection Associated to IL-6/STAT3 Signaling Pathway Activation in Rat Ischemic Stroke. Mol. Neurobiol. 2021, 58, 408–423. [Google Scholar] [CrossRef]
- Bolognesi, A.; Bortolotti, M.; Battelli, M.G.; Polito, L. Gender Influence on XOR Activities and Related Pathologies: A Narrative Review. Antioxidants 2024, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Mijailovic, N.R.; Vesic, K.; Borovcanin, M.M. The Influence of Serum Uric Acid on the Brain and Cognitive Dysfunction. Front. Psychiatry 2022, 13, 828476. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Kamiya, A.; Karnik, S.S.; Rohilla, S.; Dubey, S.K.; Taliyan, R. Novel Gene Therapy Approaches for Targeting Neurodegenerative Disorders: Focusing on Delivering Neurotrophic Genes. Mol. Neurobiol. 2025, 62, 386–411. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine Oxidoreductase: A Double-Edged Sword in Neurological Diseases. Antioxidants 2025, 14, 483. https://doi.org/10.3390/antiox14040483
Bortolotti M, Polito L, Battelli MG, Bolognesi A. Xanthine Oxidoreductase: A Double-Edged Sword in Neurological Diseases. Antioxidants. 2025; 14(4):483. https://doi.org/10.3390/antiox14040483
Chicago/Turabian StyleBortolotti, Massimo, Letizia Polito, Maria Giulia Battelli, and Andrea Bolognesi. 2025. "Xanthine Oxidoreductase: A Double-Edged Sword in Neurological Diseases" Antioxidants 14, no. 4: 483. https://doi.org/10.3390/antiox14040483
APA StyleBortolotti, M., Polito, L., Battelli, M. G., & Bolognesi, A. (2025). Xanthine Oxidoreductase: A Double-Edged Sword in Neurological Diseases. Antioxidants, 14(4), 483. https://doi.org/10.3390/antiox14040483