
Competitive Ligand-Induced Recruitment of Coactivators to Specific PPARα/δ/γ Ligand-Binding Domains Revealed by Dual-Emission FRET and X-Ray Diffraction of Cocrystals
 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Recombinant PPARα/δ/γ-LBD Expression and Purification
2.2. Single- and Dual-FRET Assays
2.3. Cocrystallization of PPARα/γ-LBD with Coregulator Peptides
2.4. X-Ray Diffraction: Data Collection and Model Refinement
3. Results
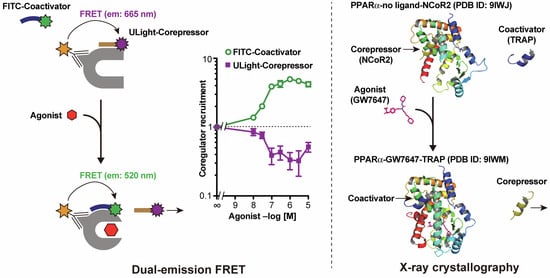
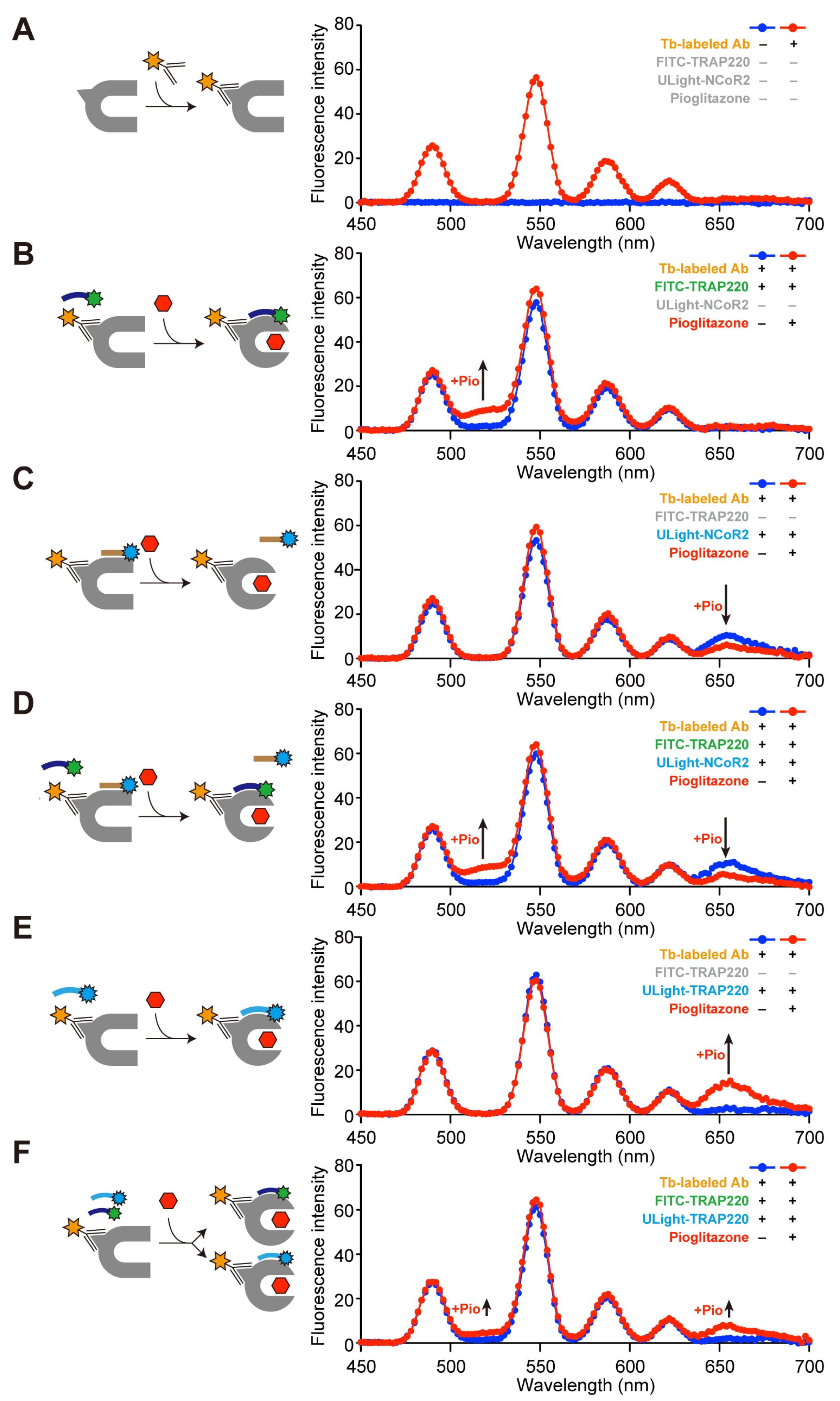
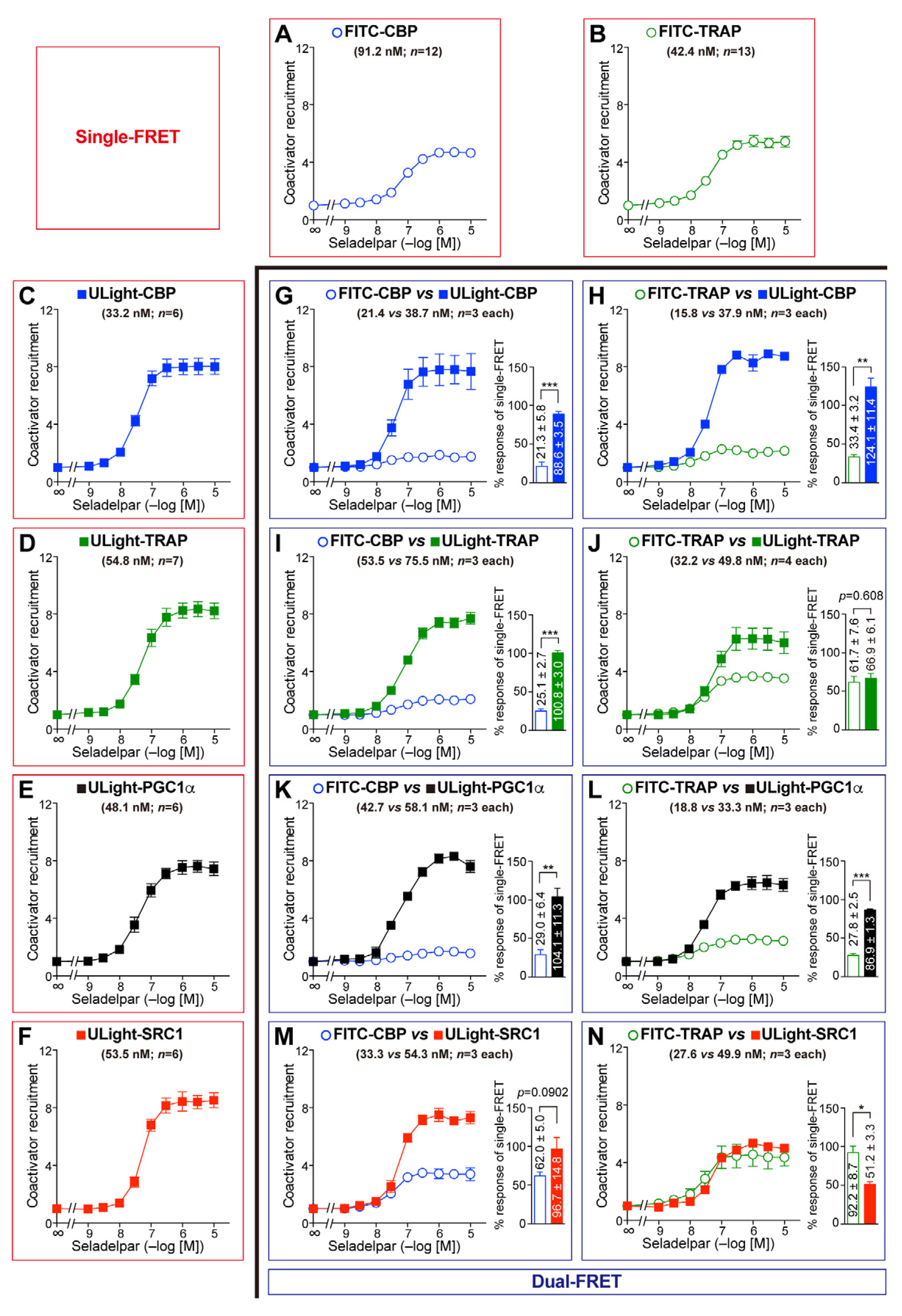
3.1. Dual-FRET Assay to Monitor Coregulator Attachment
3.2. Simultaneous Corepressor Dissociation and Coactivator Recruitment Revealed by the Dual-FRET Assay
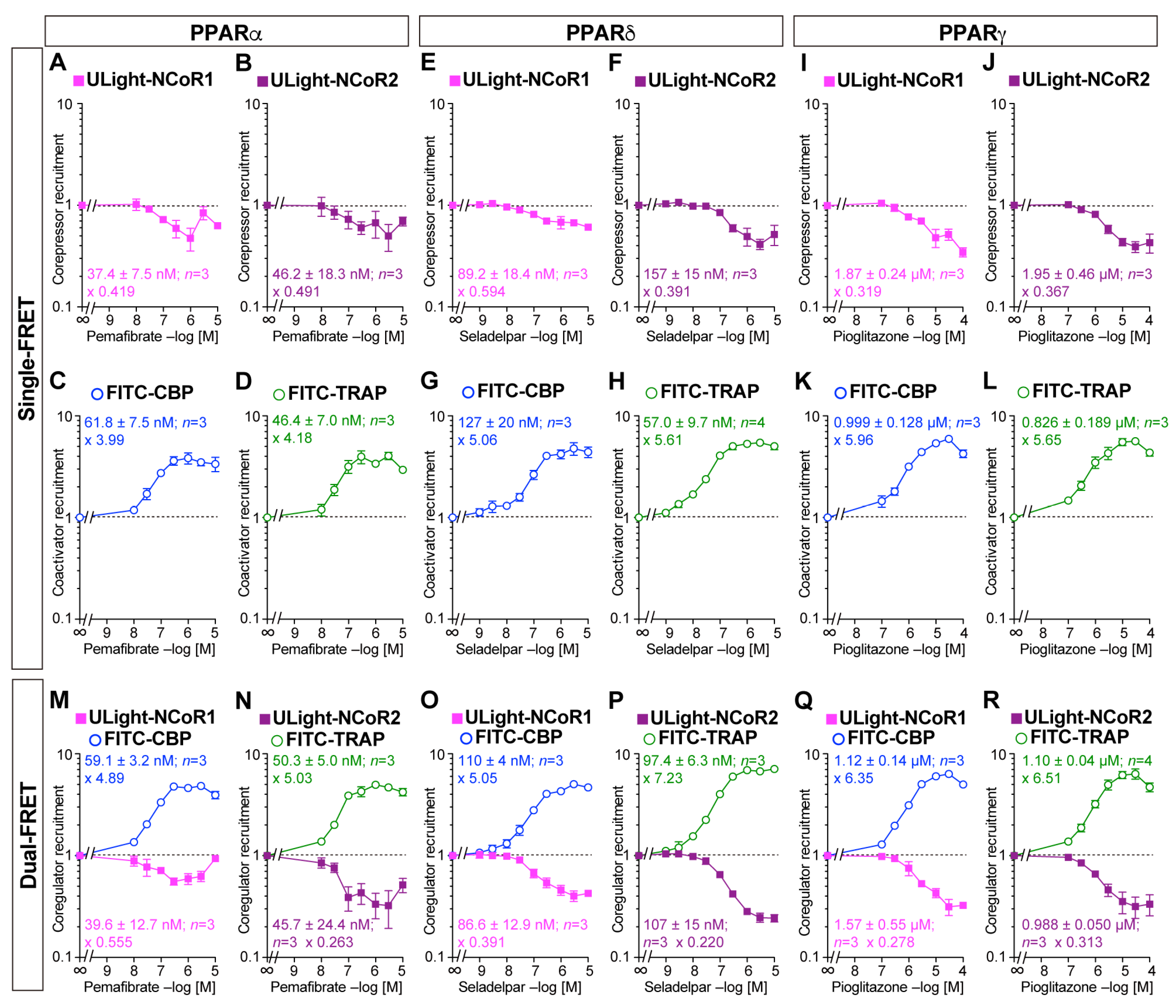
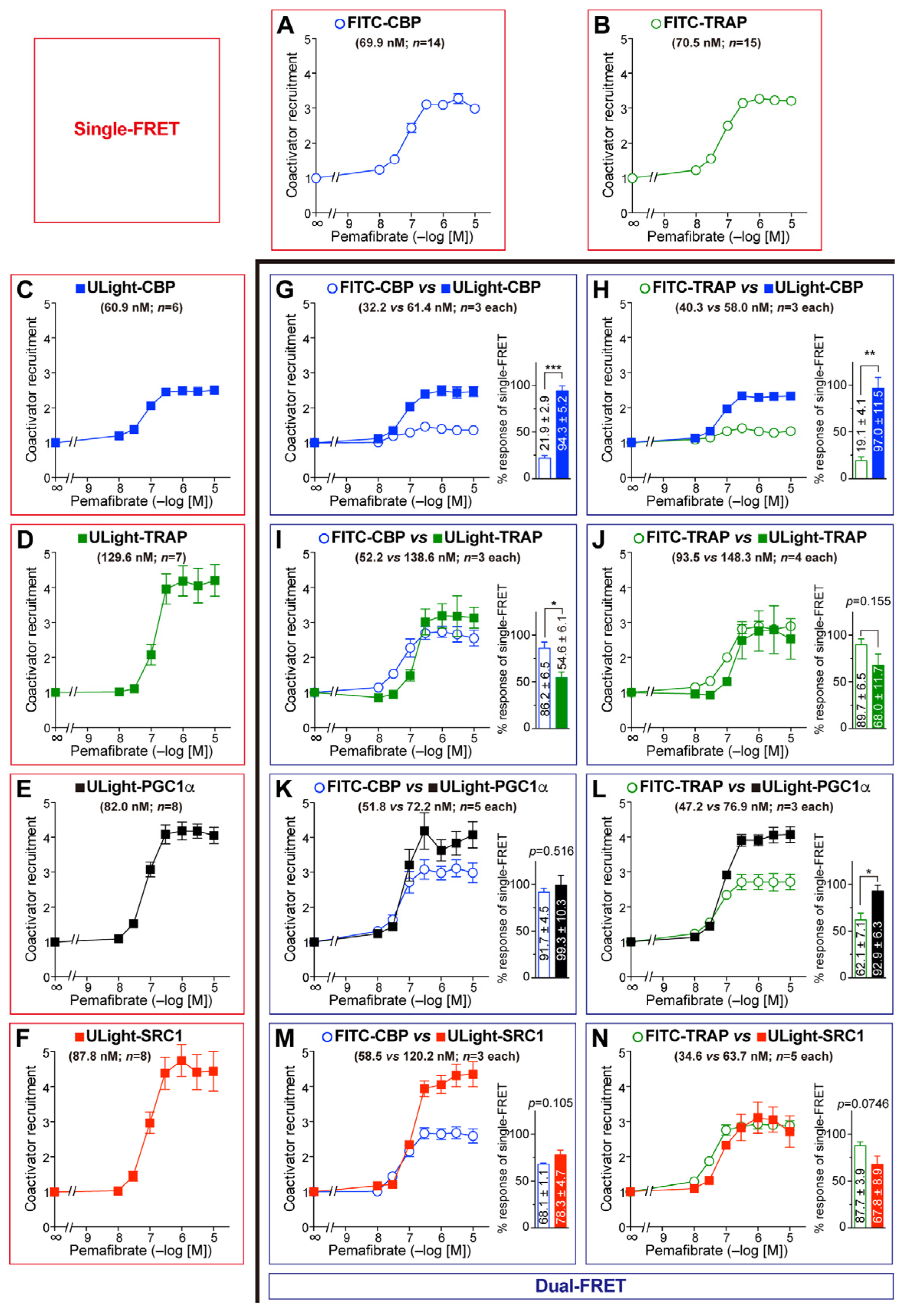
3.3. Competitive Coactivator Recruitment Revealed by Dual-FRET Assay
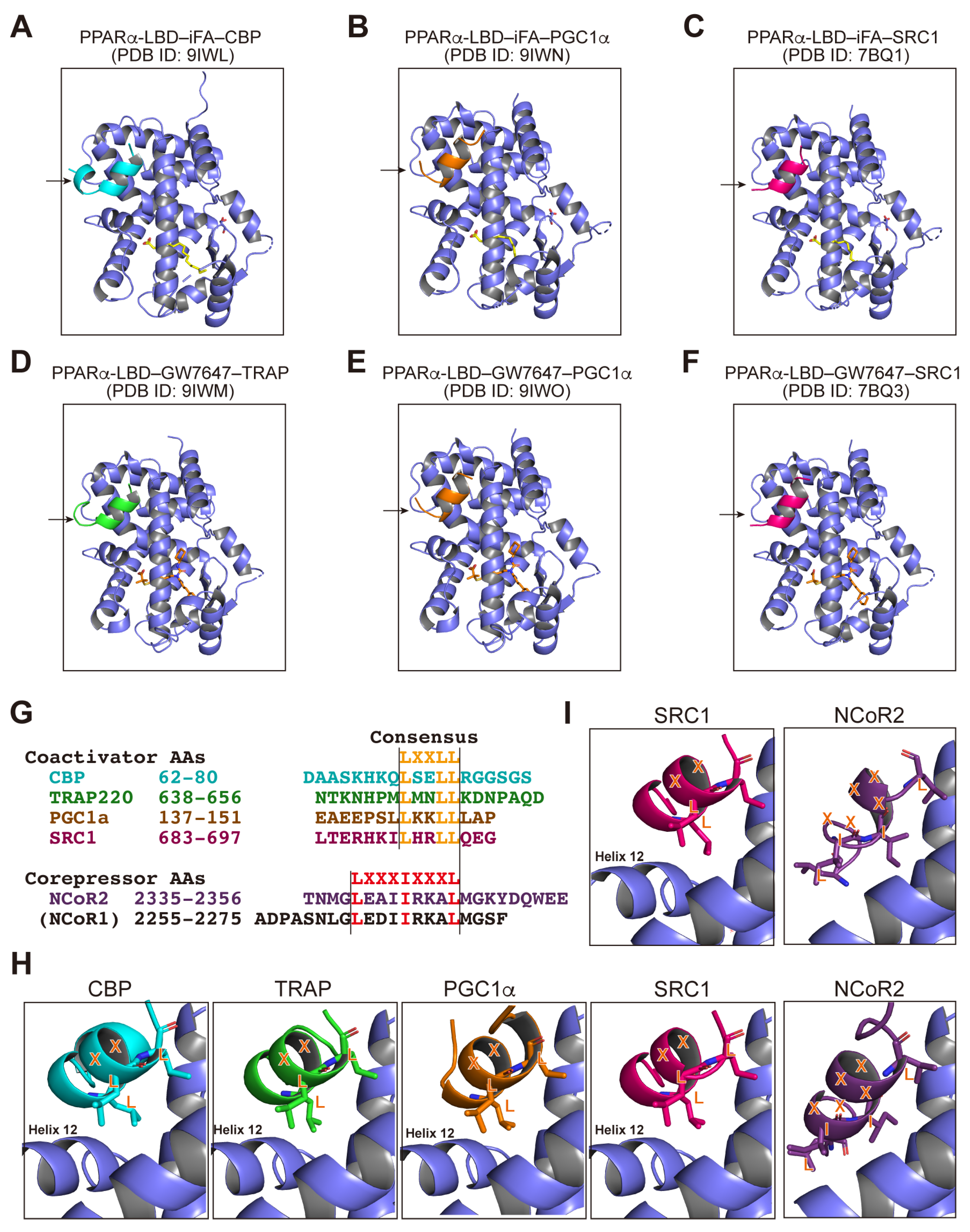
3.4. PPARα/γ-LBD–Corepressor Cocrystal Structures
3.5. PPARα-LBD–Coactivator Cocrystal Structures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kamata, S.; Yamamoto, J.; Ohtani, H.; Tosaka, Y.; Yoshikawa, S.; Akahoshi, N.; Ishii, I. 2D DIGE proteomic analysis reveals fasting-induced protein remodeling through organ-specific transcription factor(s) in mice. FEBS Open Bio. 2018, 8, 1524–1543. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- ClinCalc DrugStats Database. Available online: https://clincalc.com/DrugStats/ (accessed on 18 April 2025).
- Hoy, S.M. Seladelpar: First Approval. Drugs 2024, 84, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and development of PPAR modulators in health and disease: An update of clinical evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishii, I. Current clinical trial status and future prospects of PPAR-targeted drugs for treating nonalcoholic fatty liver disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef]
- Tzeng, J.; Byun, J.; Park, J.Y.; Yamamoto, T.; Schesing, K.; Tian, B.; Sadoshima, J.; Oka, S. An ideal PPAR response element bound to and activated by PPARα. PLoS ONE 2015, 10, e0134996. [Google Scholar] [CrossRef]
- Qi, C.; Zhu, Y.; Reddy, J.K. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem. Biophys. 2000, 32, 187–204. [Google Scholar] [CrossRef]
- Feige, J.N.; Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell. Biol. 2007, 17, 292–301. [Google Scholar] [CrossRef]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-regulated gene expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Schwabe, J.W. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell. Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef]
- Xu, H.E.; Stanley, T.B.; Montana, V.G.; Lambert, M.H.; Shearer, B.G.; Cobb, J.E.; McKee, D.D.; Galardi, C.M.; Plunket, K.D.; Nolte, R.T.; et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 2002, 415, 813–817. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications–a review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Eichner, L.J.; Shaw, R.J.; Auwerx, J. Transcriptional coregulators: Fine-tuning metabolism. Cell Metab. 2014, 20, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Kao, H.Y.; Love, J.D.; Li, C.; Banayo, E.; Gooch, J.T.; Krishna, V.; Chatterjee, K.; Evans, R.M.; Schwabe, J.W. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999, 13, 3209–3216. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Alexander, M.Y.; Tutar, Y.; Wilkinson, F.L.; Venditti, A. Oxidative stress in metabolic disorders and drug-induced injury: The potential role of Nrf2 and PPARs activators. Oxid. Med. Cell. Longev. 2017, 2017, 2508909. [Google Scholar] [CrossRef]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome proliferator-activated receptors (PPARs) and oxidative stress in physiological conditions and in cancer. Antioxidants 2021, 10, 1734. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Kashiwagi, N.; Shimamura, A.; Yashiro, S.; Komori, Y.; Hosoda, A.; Akahoshi, N.; Ishii, I. Different coactivator recruitment to human PPARα/δ/γ ligand-binding domains by eight PPAR agonists to treat nonalcoholic fatty liver disease. Biomedicines 2024, 12, 624. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα ligand-binding domain structures with endogenous fatty acids and fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Honda, A.; Kamata, S.; Akahane, M.; Machida, Y.; Uchii, K.; Shiiyama, Y.; Habu, Y.; Miyawaki, S.; Kaneko, C.; Oyama, T.; et al. Functional and structural insights into human PPARα/δ/γ subtype selectivity of bezafibrate, fenofibric acid, and pemafibrate. Int. J. Mol. Sci. 2022, 23, 4726. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishikawa, R.; Akahane, M.; Fujita, A.; Kaneko, C.; Miyawaki, S.; Habu, Y.; Shiiyama, Y.; Uchii, K.; et al. Functional and structural insights into the human PPARα/δ/γ targeting preferences of anti-NASH investigational drugs, lanifibranor, seladelpar, and elafibranor. Antioxidants 2023, 12, 1523. [Google Scholar] [CrossRef] [PubMed]
- Kamata, S.; Oyama, T.; Ishii, I. Preparation of co-crystals of human PPARα-LBD and ligand for high-resolution X-ray crystallography. STAR Protoc. 2021, 2, 100364. [Google Scholar] [CrossRef]
- Honda, A.; Kamata, S.; Satta, C.; Machida, Y.; Uchii, K.; Terasawa, K.; Nemoto, A.; Oyama, T.; Ishii, I. Structural basis for anti-non-alcoholic fatty liver disease and diabetic dyslipidemia drug saroglitazar as a PPAR α/γ dual agonist. Biol. Pharm. Bull. 2021, 44, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lazar, M.A. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 1999, 402, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Perissi, V.; Staszewski, L.M.; McInerney, E.M.; Kurokawa, R.; Krones, A.; Rose, D.W.; Lambert, M.H.; Milburn, M.V.; Glass, C.K.; Rosenfeld, M.G. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999, 13, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.A.; Zhang, J.H.; Harrison, R.K.; McGeehan, G.M. Development of a homogeneous, fluorescence resonance energy transfer-based in vitro recruitment assay for peroxisome proliferator-activated receptor delta via selection of active LXXLL coactivator peptides. Anal. Biochem. 2002, 304, 63–69. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Qiu, X.; Xu, J.; Cardoso Dos Santos, M.; Hildebrandt, N. Multiplexed biosensing and bioimaging using lanthanide-based time-gated Förster resonance energy transfer. Acc. Chem. Res. 2022, 55, 551–564. [Google Scholar] [CrossRef]
- Rajapakse, H.E.; Reddy, D.R.; Mohandessi, S.; Butlin, N.G.; Miller, L.W. Luminescent terbium protein labels for time-resolved microscopy and screening. Angew. Chem. Int. Ed. Engl. 2009, 48, 4990–4992. [Google Scholar] [CrossRef] [PubMed]
- Kupcho, K.R.; Stafslien, D.K.; DeRosier, T.; Hallis, T.M.; Ozers, M.S.; Vogel, K.W. Simultaneous monitoring of discrete binding events using dual-acceptor terbium-based LRET. J. Am. Chem. Soc. 2007, 129, 13372–13373. [Google Scholar] [CrossRef] [PubMed]
- Cottet, M.; Faklaris, O.; Maurel, D.; Scholler, P.; Doumazane, E.; Trinquet, E.; Pin, J.-P.; Durroux, T. BRET and time-resolved FRET strategy to study GPCR oligomerization: From cell lines toward native tissues. Front. Endocrinol. 2012, 3, 92. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Hermans, M.P.; Fruchart-Najib, J.; Kodama, T. Selective peroxisome proliferator-activated receptor alpha modulators (SPPARMα) in the metabolic syndrome: Is pemafibrate light at the end of the tunnel? Curr. Atheroscler. Rep. 2021, 23, 3. [Google Scholar] [CrossRef]
- Ashraf, T.; Abunada, O.; Seerani, N.; Ali, K.; Muhammad, A.; Mir, S.L.; Shah, S.A.M.; Hassaan, M.; Kumar, V.; Abbas, W.; et al. The role of Seladelpar in primary biliary cholangitis: A systematic review and meta-analysis. BMC Gastroenterol. 2025, 25, 224. [Google Scholar] [CrossRef]
- Azhari, H.F.; Dawson, J. Clinical implications of fracture severity risk with pioglitazone: A systematic review and meta-analysis of clinical randomized trials. Front. Pharmacol. 2025, 16, 1357309. [Google Scholar] [CrossRef]
- Al Subait, A.; Alghamdi, R.H.; Ali, R.; Alsharidah, A.; Huwaizi, S.; Alkhodier, R.A.; Almogren, A.S.; Alzomia, B.A.; Alaskar, A.; Boudjelal, M. Discovery of PPAR alpha lipid pathway modulators that do not bind directly to the receptor as potential anti-cancer compounds. Int. J. Mol. Sci. 2025, 26, 736. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Mosure, S.A.; Zheng, J.; Brust, R.; Bass, J.; Nichols, A.; Solt, L.A.; Griffin, P.R.; Kojetin, D.J. A molecular switch regulating transcriptional repression and activation of PPARγ. Nat. Commun. 2020, 11, 956. [Google Scholar] [CrossRef]
- Stallcup, M.R.; Poulard, C. Gene-specific actions of transcriptional coregulators facilitate physiological plasticity: Evidence for a physiological coregulator code. Trends Biochem. Sci. 2020, 45, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Reddy, J.K. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta 2007, 1771, 936–951. [Google Scholar] [CrossRef]
- Stashi, E.; York, B.; O’Malley, B.W. Steroid receptor coactivators: Servants and masters for control of systems metabolism. Trends Endocrinol. Metab. 2014, 25, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Brown, M. Molecular determinants for the tissue specificity of SERMs. Science 2002, 295, 2465–2468. [Google Scholar] [CrossRef]
- Frkic, R.L.; Marshall, A.C.; Blayo, A.L.; Pukala, T.L.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. PPARγ in complex with an antagonist and inverse agonist: A tumble and trap mechanism of the activation helix. iScience 2018, 5, 69–79. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Katzenellenbogen, J.A. A dual-acceptor time-resolved Föster resonance energy transfer assay for simultaneous determination of thyroid hormone regulation of corepressor and coactivator binding to the thyroid hormone receptor: Mimicking the cellular context of thyroid hormone action. Anal. Biochem. 2009, 386, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Hilal, T.; Puetter, V.; Otto, C.; Parczyk, K.; Bader, B. A dual estrogen receptor TR-FRET assay for simultaneous measurement of steroid site binding and coactivator recruitment. J. Biomol. Screen. 2010, 15, 268–278. [Google Scholar] [CrossRef]
- Geißler, D.; Stufler, S.; Löhmannsröben, H.G.; Hildebrandt, N. Six-color time-resolved Förster resonance energy transfer for ultrasensitive multiplexed biosensing. J. Am. Chem. Soc. 2013, 135, 1102–1109. [Google Scholar] [CrossRef]
- Bunt, G.; Wouters, F.S. FRET from single to multiplexed signaling events. Biophys. Rev. 2017, 9, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, M.J.; Busby, S.A.; Pascal, B.D.; Southern, M.R.; Griffin, P.R. A two-stage differential hydrogen deuterium exchange method for the rapid characterization of protein/ligand interactions. J. Biomol. Tech. 2007, 18, 194–204. [Google Scholar] [PubMed]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamata, S.; Honda, A.; Yashiro, S.; Kaneko, C.; Komori, Y.; Shimamura, A.; Masuda, R.; Oyama, T.; Ishii, I. Competitive Ligand-Induced Recruitment of Coactivators to Specific PPARα/δ/γ Ligand-Binding Domains Revealed by Dual-Emission FRET and X-Ray Diffraction of Cocrystals. Antioxidants 2025, 14, 494. https://doi.org/10.3390/antiox14040494
Kamata S, Honda A, Yashiro S, Kaneko C, Komori Y, Shimamura A, Masuda R, Oyama T, Ishii I. Competitive Ligand-Induced Recruitment of Coactivators to Specific PPARα/δ/γ Ligand-Binding Domains Revealed by Dual-Emission FRET and X-Ray Diffraction of Cocrystals. Antioxidants. 2025; 14(4):494. https://doi.org/10.3390/antiox14040494
Chicago/Turabian StyleKamata, Shotaro, Akihiro Honda, Sayaka Yashiro, Chihiro Kaneko, Yuna Komori, Ayumi Shimamura, Risa Masuda, Takuji Oyama, and Isao Ishii. 2025. "Competitive Ligand-Induced Recruitment of Coactivators to Specific PPARα/δ/γ Ligand-Binding Domains Revealed by Dual-Emission FRET and X-Ray Diffraction of Cocrystals" Antioxidants 14, no. 4: 494. https://doi.org/10.3390/antiox14040494
APA StyleKamata, S., Honda, A., Yashiro, S., Kaneko, C., Komori, Y., Shimamura, A., Masuda, R., Oyama, T., & Ishii, I. (2025). Competitive Ligand-Induced Recruitment of Coactivators to Specific PPARα/δ/γ Ligand-Binding Domains Revealed by Dual-Emission FRET and X-Ray Diffraction of Cocrystals. Antioxidants, 14(4), 494. https://doi.org/10.3390/antiox14040494