Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology

 , and
, and 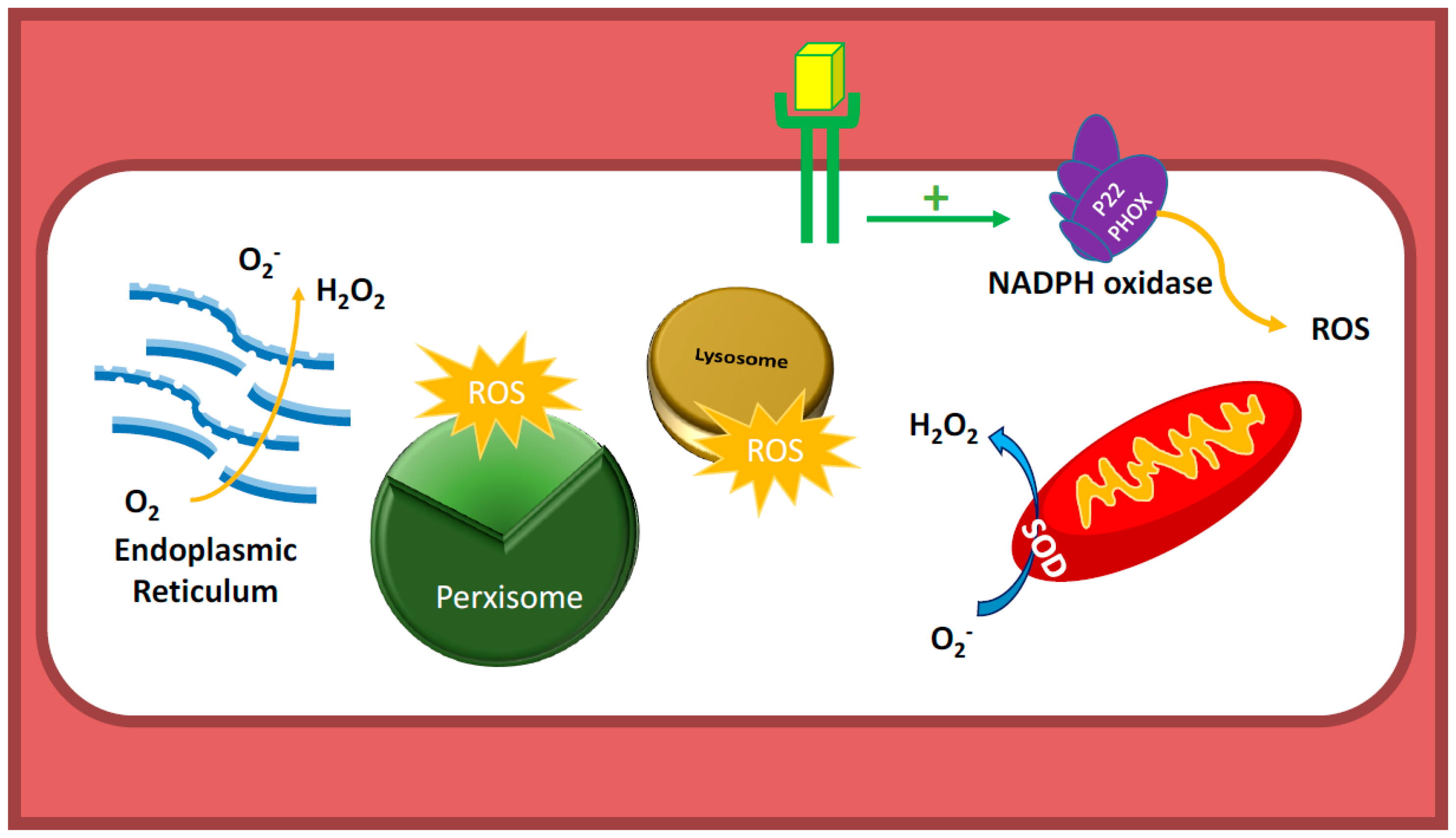{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. ROS Paradox
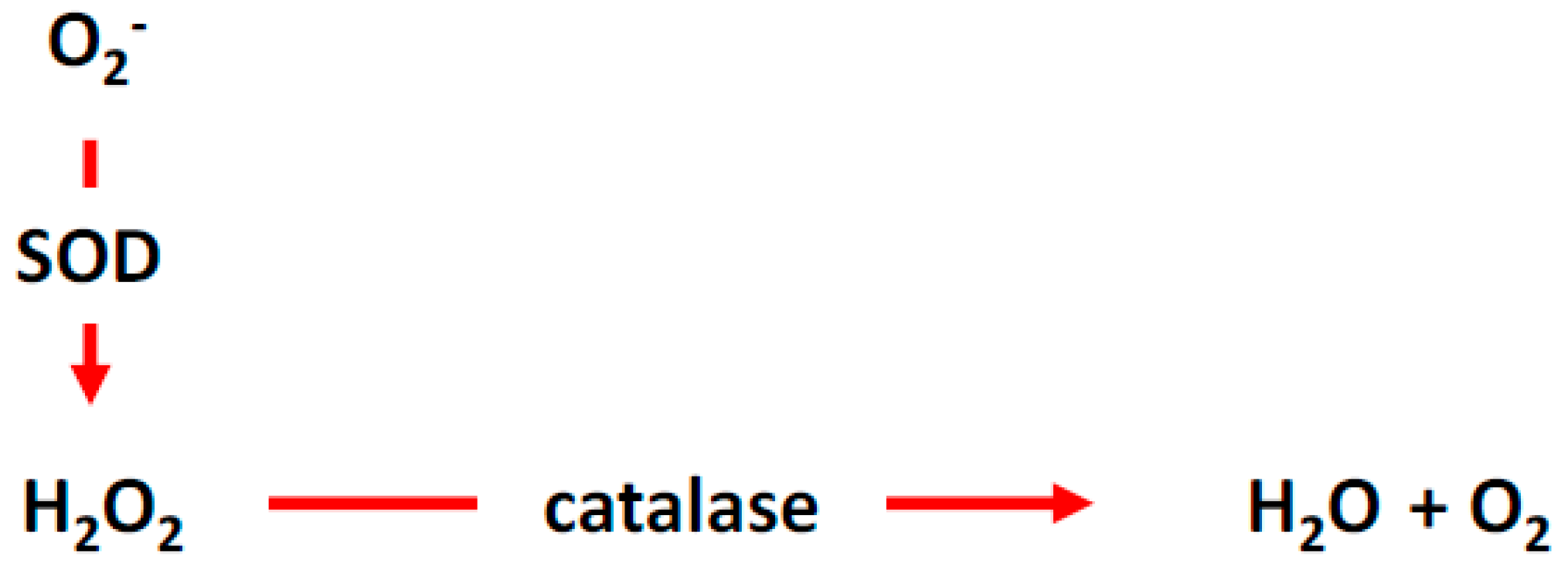
2. Reactive Oxygen Species (ROS)
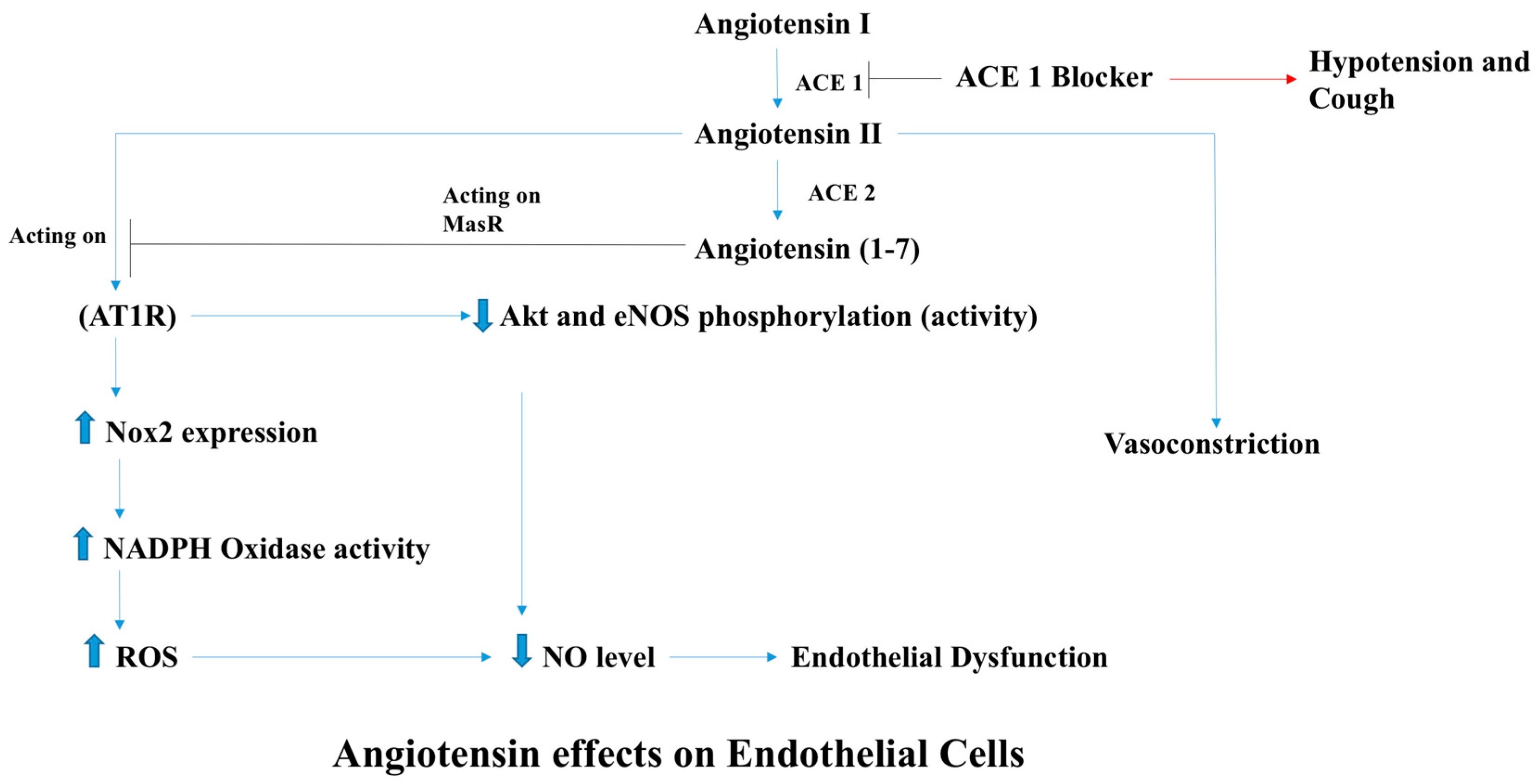
2.1. Endothelial Dysfunction
2.2. Mitochondrial Dysfunction
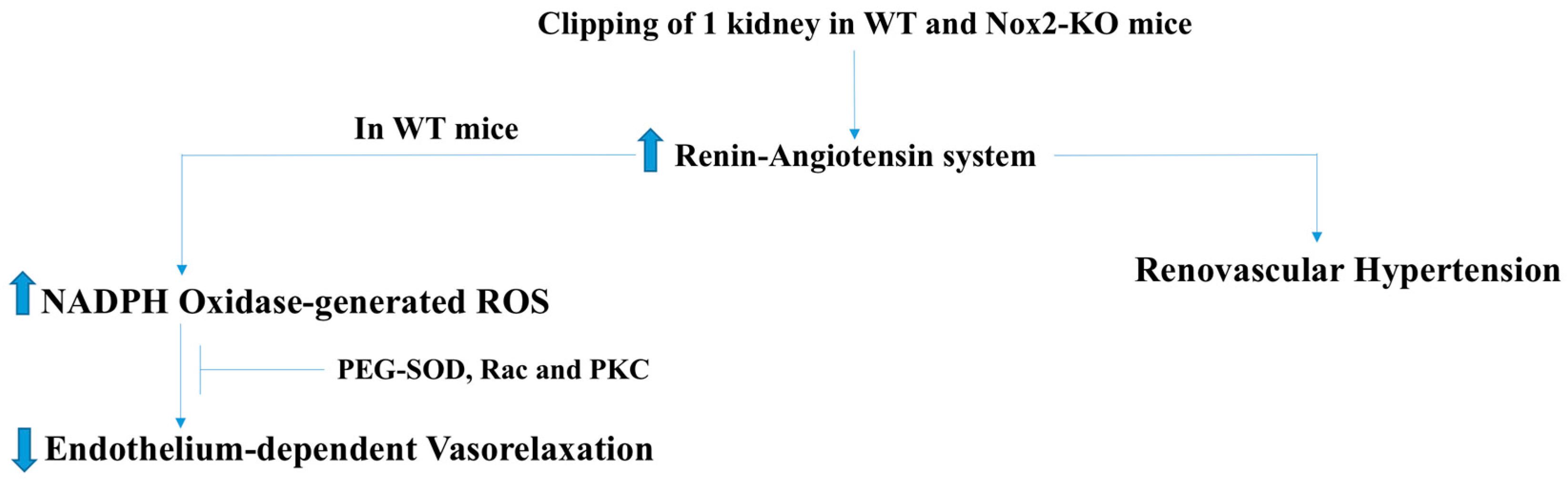
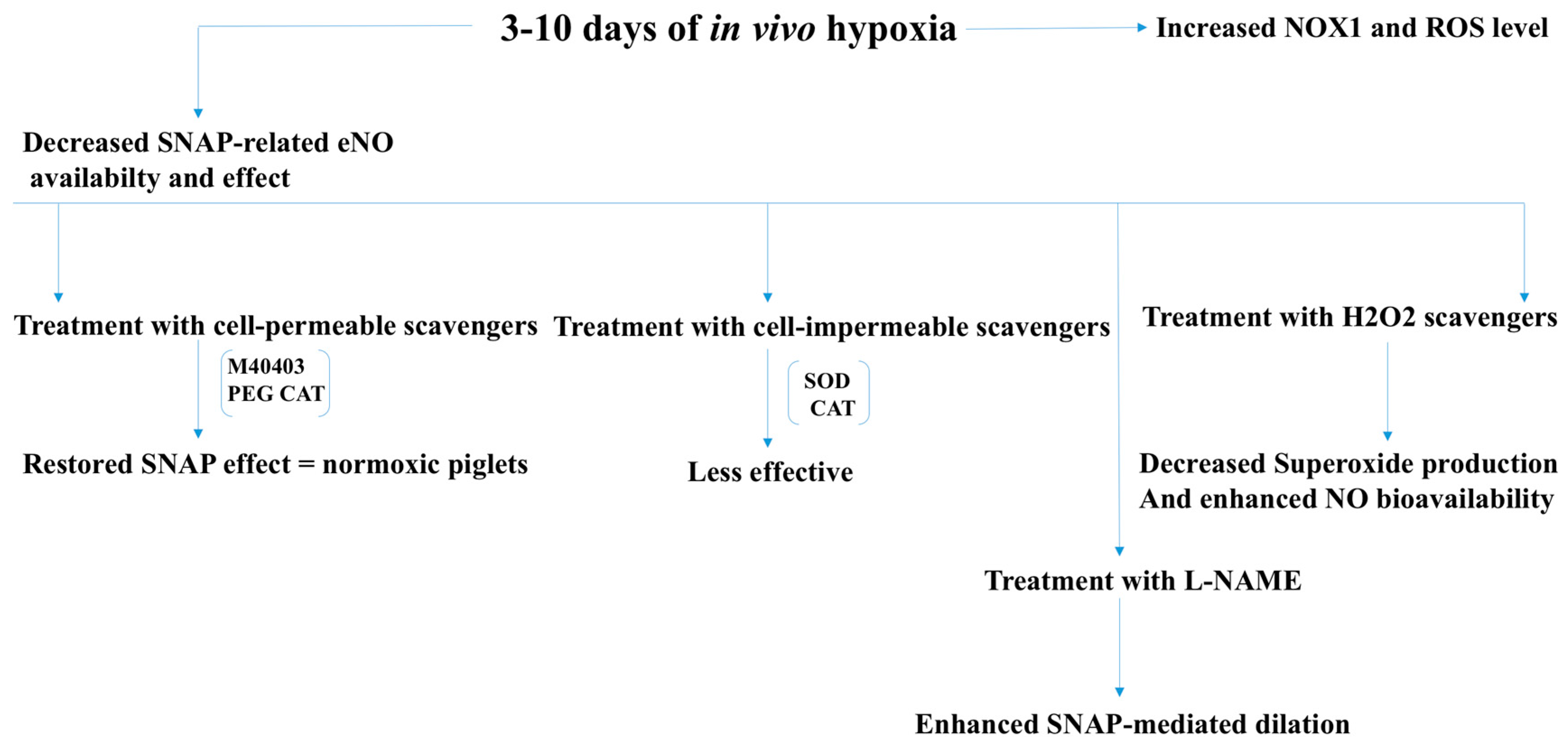
2.3. Pulmonary and Renovascular Hypertension
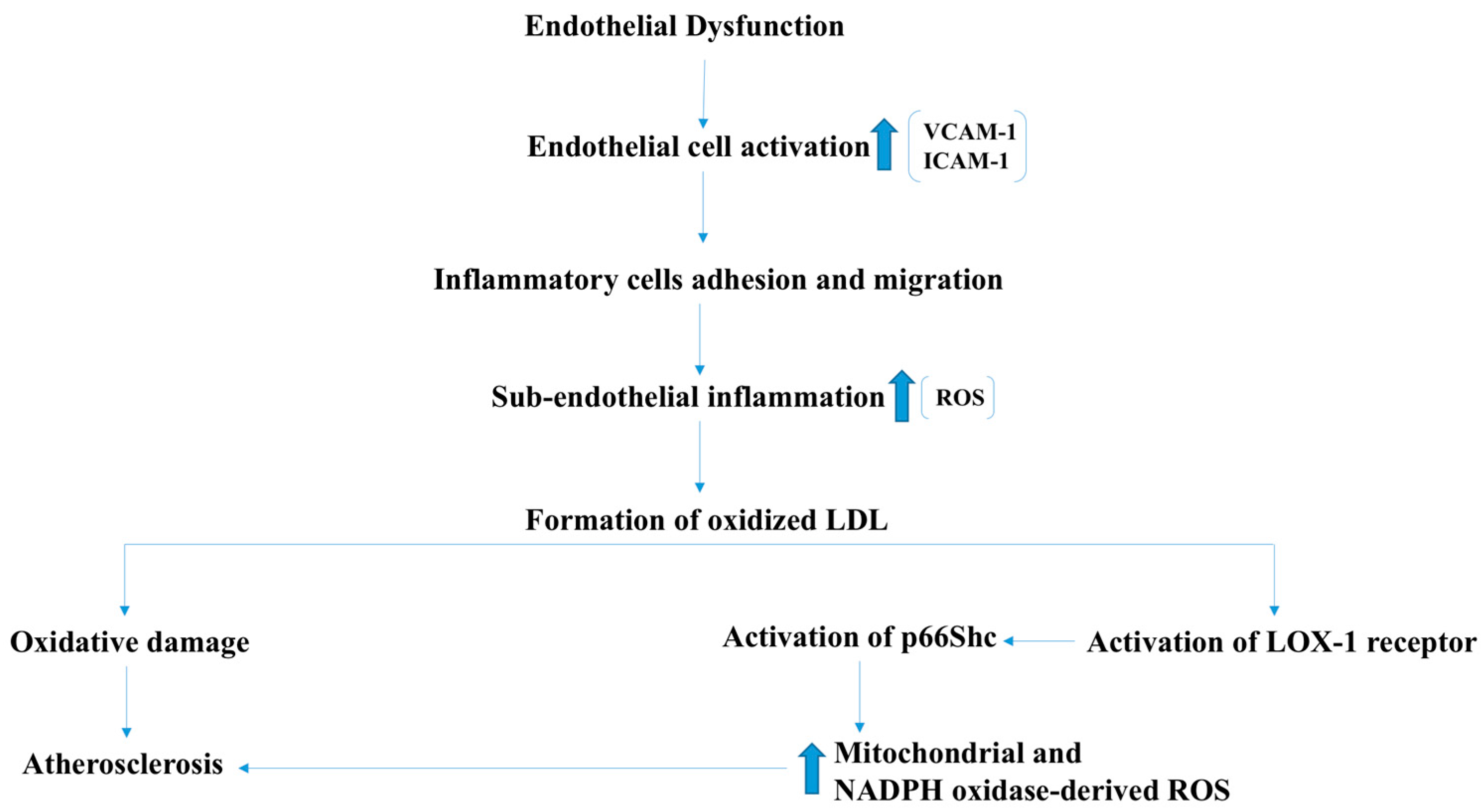
2.4. Atherosclerosis and Ischemic Heart Disease
2.5. Ischemic-Reperfusion Injury
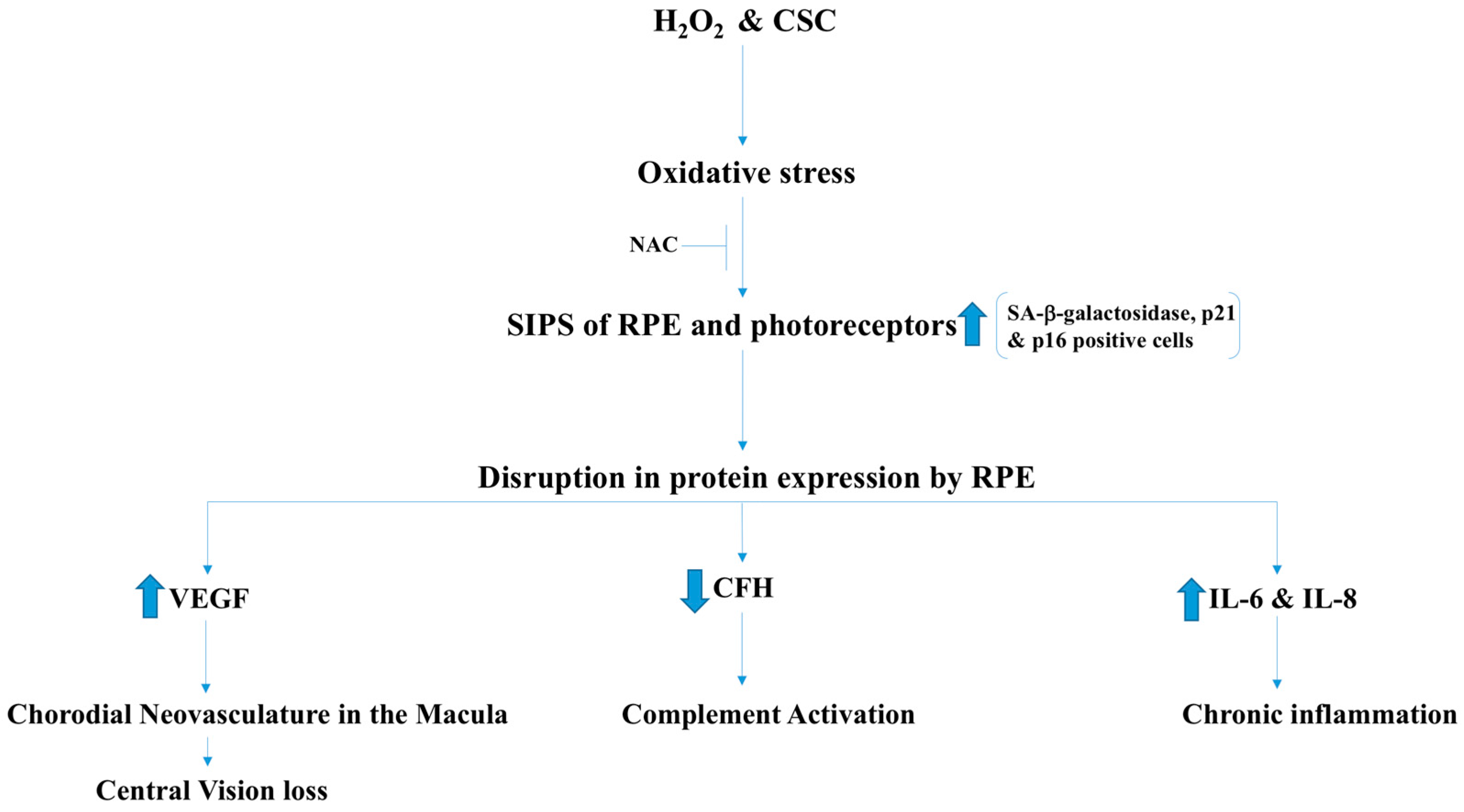
2.6. Age-Related Macular Degeneration (AMD)
3. Beneficial Effects of Sub-Cellular ROS
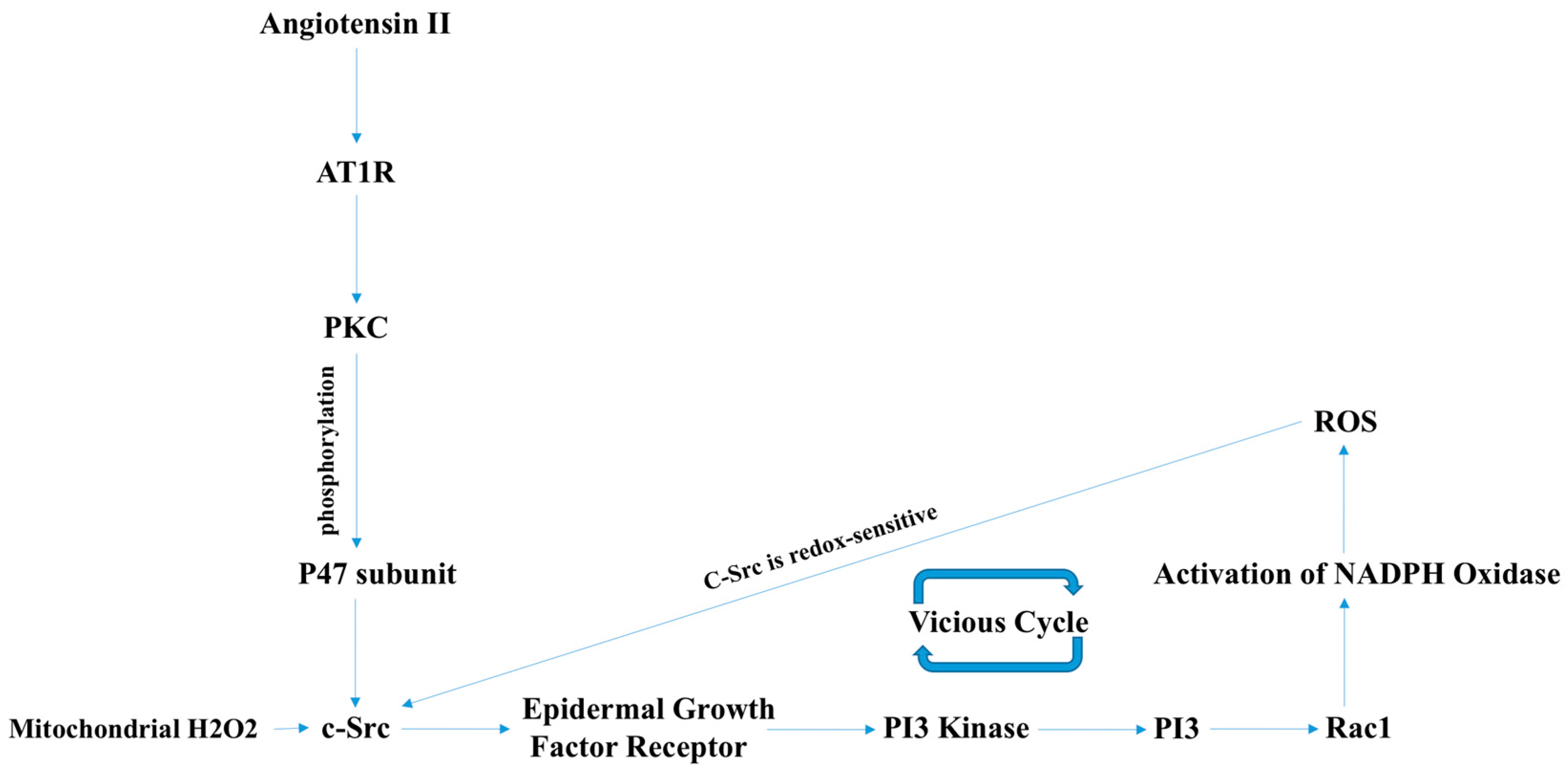
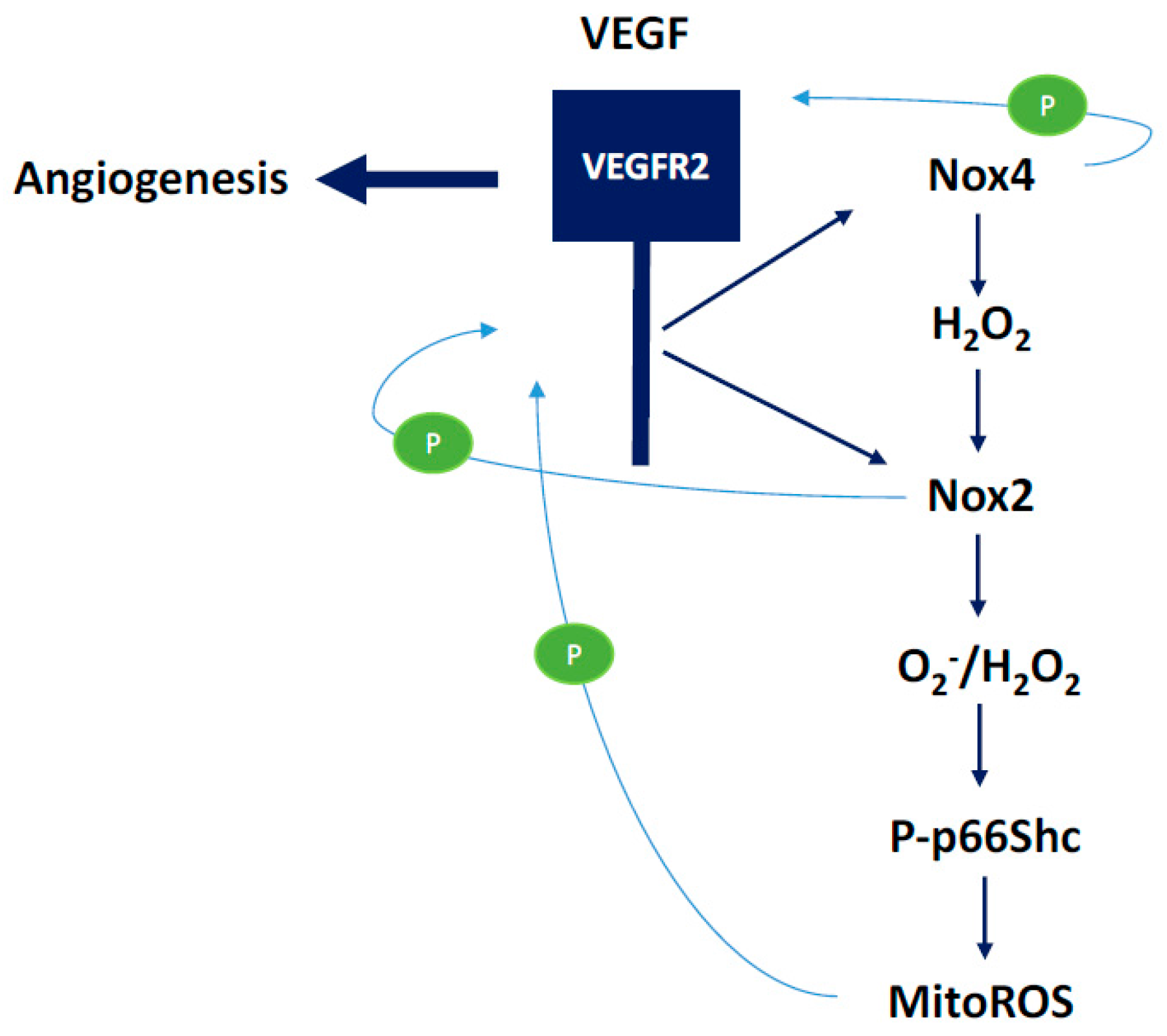
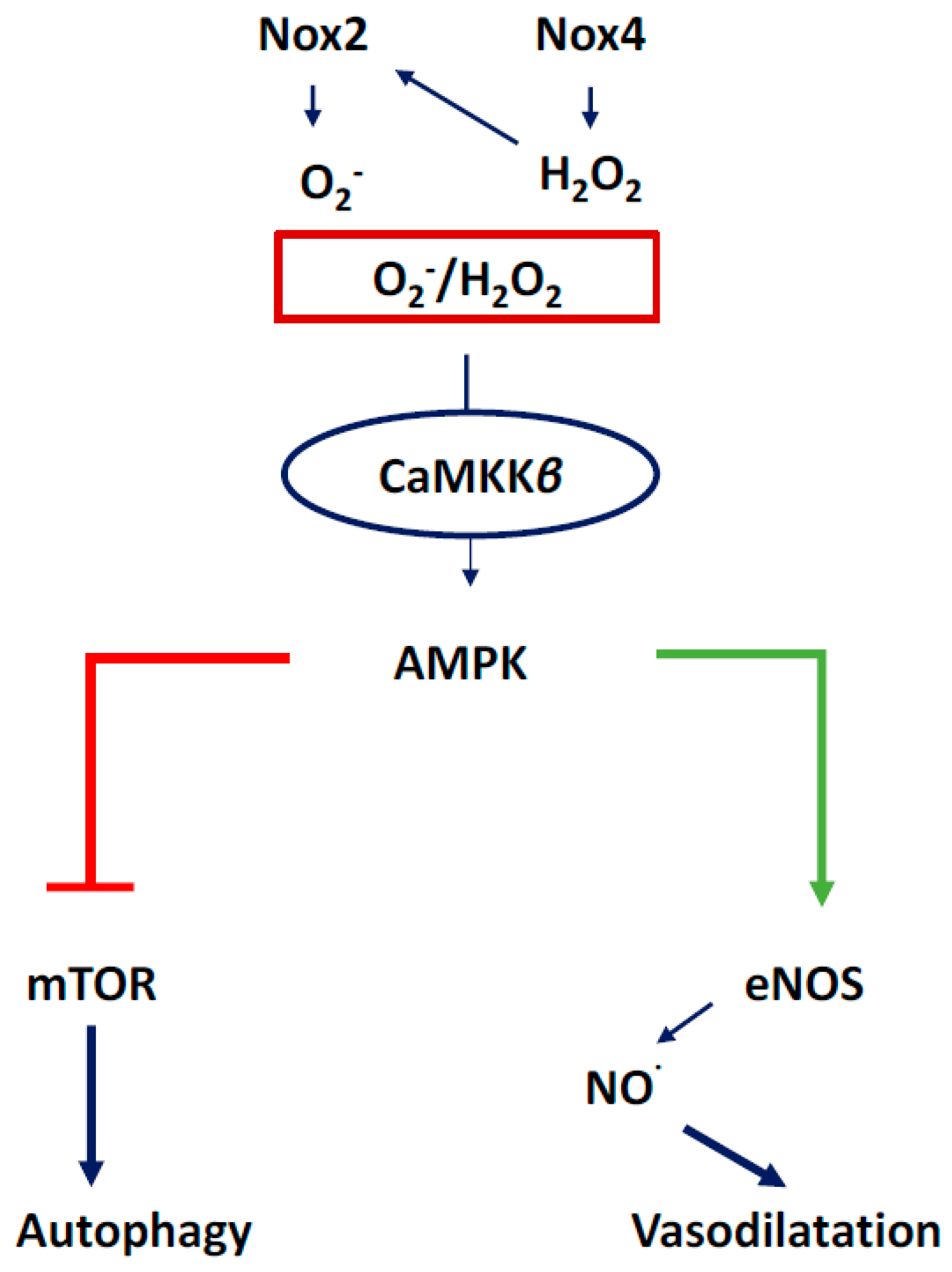
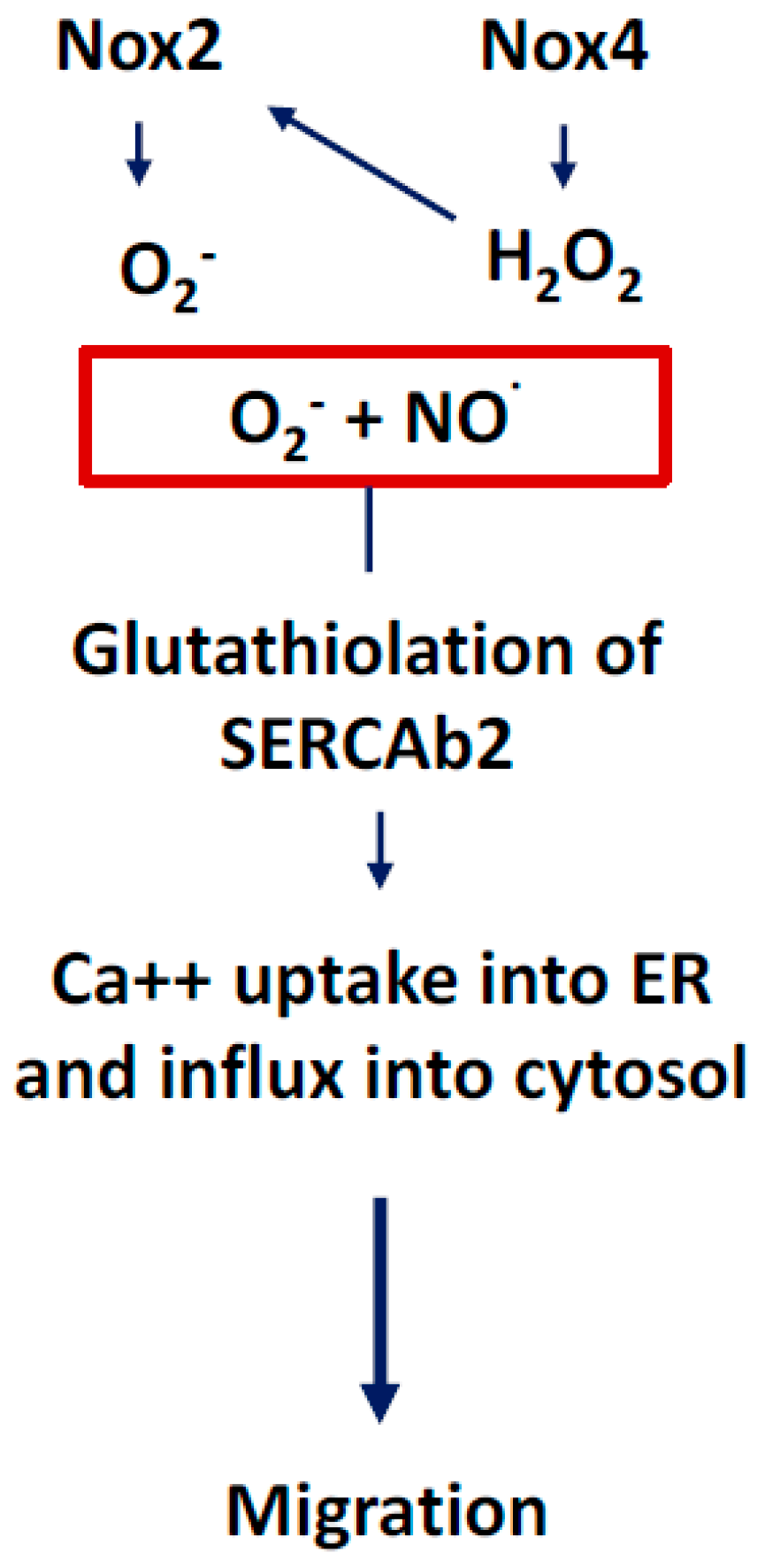
3.1. NOX2-Containing NADPH Oxidase
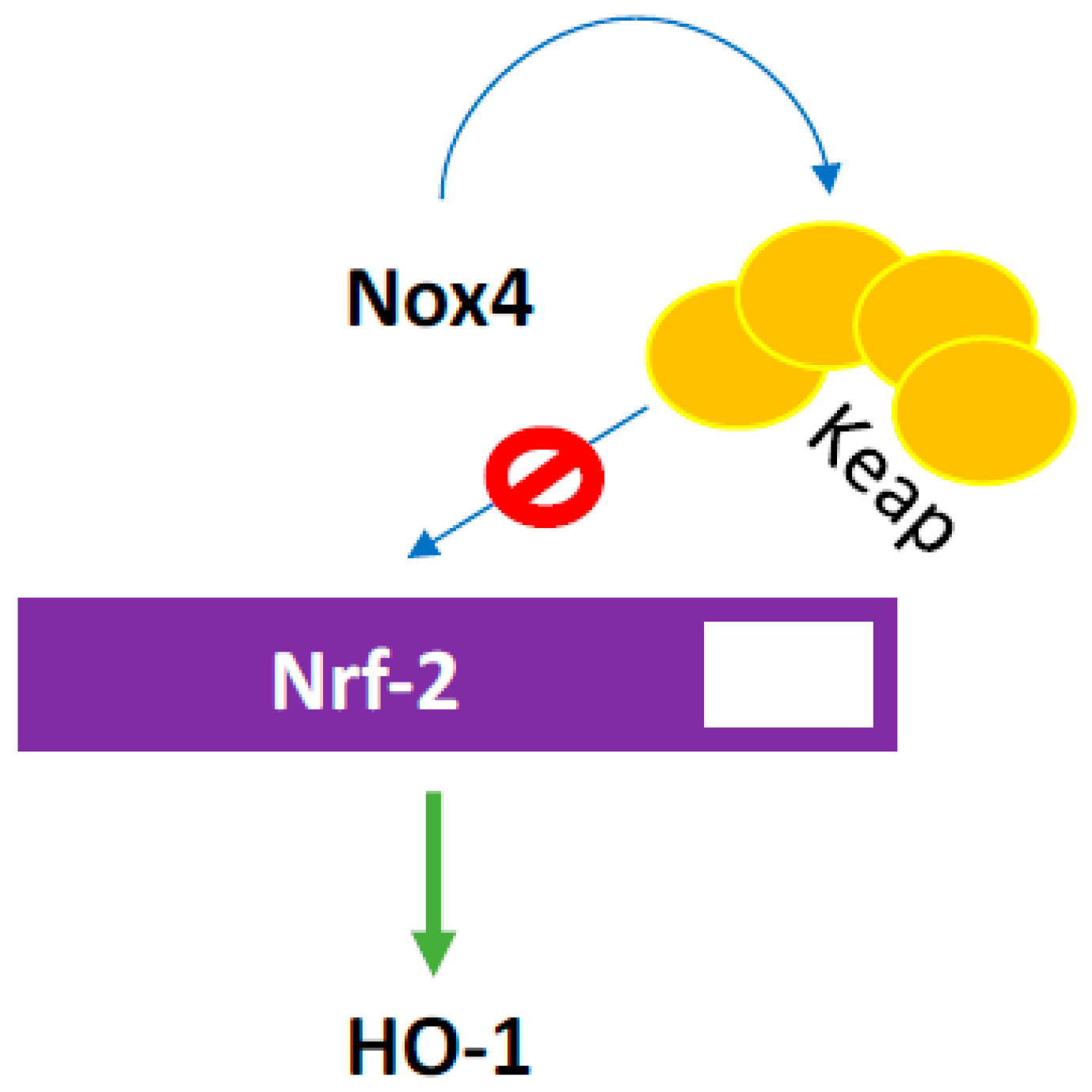
3.2. NOX4
3.3. Mitochondrial ROS
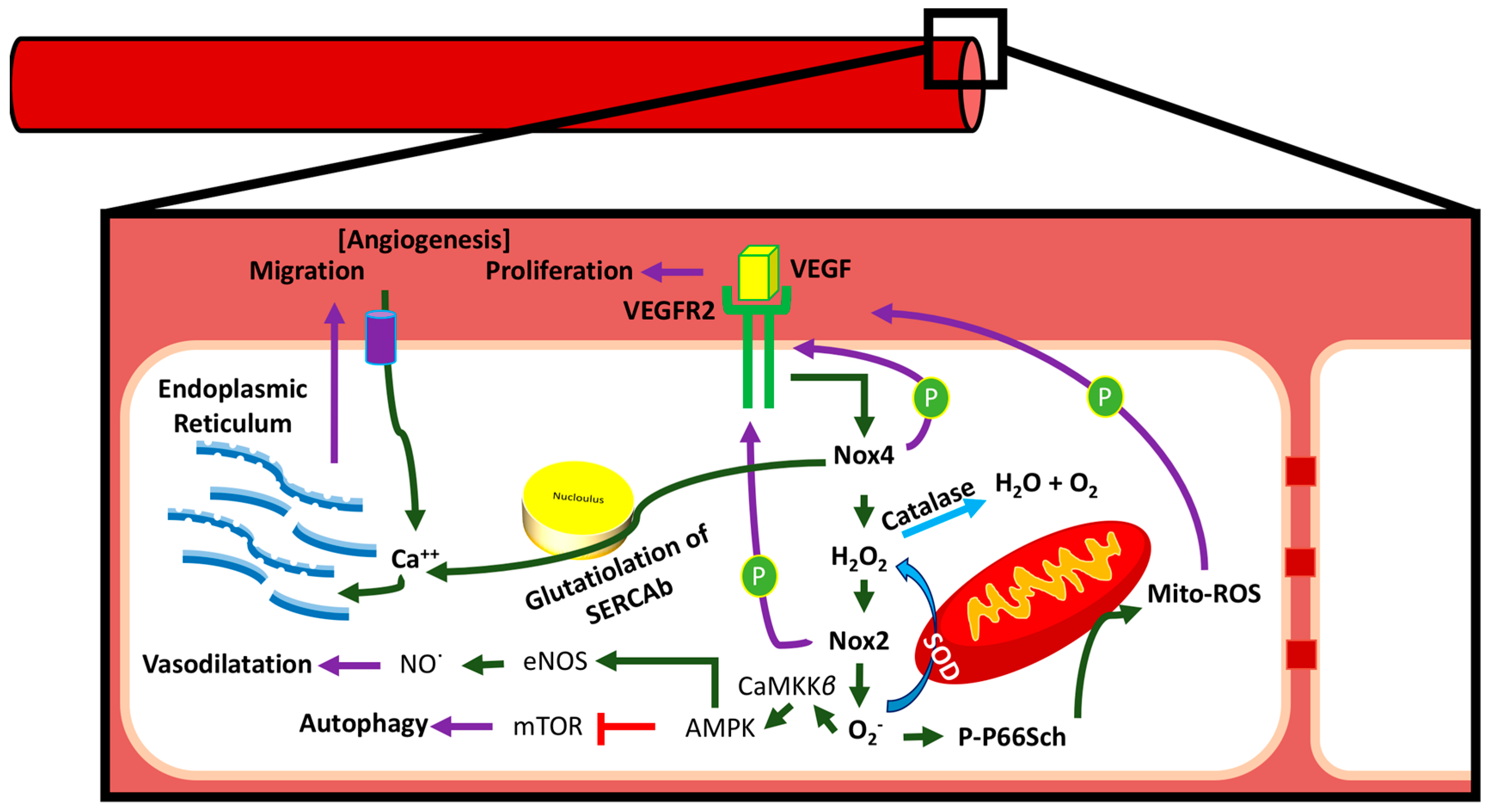
3.4. Communication between Sub-Cellular ROS
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sleight, P. The HOPE Study (Heart Outcomes Prevention Evaluation). J. Renin-Angiotensin-Aldost. Syst. 2000, 1, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Kachra, Z.; Spokes, K.C.; Aird, W.C. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000, 486, 252–256. [Google Scholar] [CrossRef]
- Wang, Y.; Zang, Q.S.; Liu, Z.; Wu, Q.; Maass, D.; Dulan, G.; Shaul, P.; Melito, L.; Frantz, D.; Kilgore, J.; et al. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am. J. Physiol. 2011, 301, C695–C704. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, C749–C764. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Ruth Michaelis, U.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Shafique, E.; Choy, W.C.; Liu, Y.; Feng, J.; Cordeiro, B.; Lyra, A.; Arafah, M.; Yassin-Kassab, A.; Zanetti, A.; Clements, R.; et al. Oxidative stress improves coronary endothelial function through activation of the pro-survival kinase AMPK. Aging 2013, 5, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.; Kai, C.; Pei, Y.; Chunying, L.; Xiaoyun, H.; Christine, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J., Jr. NADPH Oxidase 4 Promotes Endothelial Angiogenesis Through eNOS Activation. Circulation 2011, 124. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Damrauer, S.M.; Lee, M.; Sellke, F.W.; Ferran, C.; Abid, M.R. Endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J.R. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Shafique, E.; Torina, A.; Reichert, K.; Colantuono, B.; Nur, N.; Zeeshan, K.; Ravichandran, V.; Liu, Y.; Feng, J.; Benjamin, L.; et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium. Cardiovasc. Res. 2017, 113, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Rinze, R.; Adlam, D.; Tatham, A.L.; De Bono, J.; Channon, K.M. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: Studies in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007, 100, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hou, X.; Xiao, J.; Kuroda, J.; Ago, T.; Sadoshima, J.; Cohen, R.; Tong, X.Y. Both hydrogen peroxide and transforming growth factor beta 1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.; Walker, S.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free Radic. Biol. Med. 2013, 61, 473–501. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-Induced Production of Mitochondrial Superoxide in Angiotensin II-Mediated Endothelial Oxidative Stress and Hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Kirilyuk, I.A.; Dikalov, S.I. Antihypertensive effect of mitochondria-targeted proxyl nitroxides. Redox Biol. 2015, 4, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zhang, C.; Ma, X.; Miao, H.; Wang, J.; Liu, L.; Chen, S.; Zeng, R.; Chen, Y.; Bihl, J.C. Angiotensin-(1-7) counteracts angiotensin II-induced dysfunction in cerebral endothelial cells via modulating Nox2/ROS and PI3K/NO pathways. Exp. Cell Res. 2015, 336, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Schreiber, J.G.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. gp91phox-Containing NADPH Oxidase Mediates Endothelial Dysfunction in Renovascular Hypertension. Circulation 2004, 109, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Fike, C.D.; Dikalova, A.; Slaughter, J.C.; Kaplowitz, M.R.; Zhang, Y.; Aschner, J.L. Reactive oxygen species-reducing strategies improve pulmonary arterial responses to nitric oxide in piglets with chronic hypoxia-induced pulmonary hypertension. Antioxid. Redox Signal. 2013, 18, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial Dysfunction, Oxidative Stress, and Risk of Cardiovascular Events in Patients with Coronary Artery Disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Lüscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Martin-Padura, I.; de Nigris, F.; Migliaccio, E.; Mansueto, G.; Minardi, S.; Rienzo, M.; Lerman, L.O.; Stendardo, M.; Giorgio, M.; De Rosa, G.; et al. p66Shc Deletion Confers Vascular Protection in Advanced Atherosclerosis in Hypercholesterolemic Apolipoprotein E Knockout Mice. Endothelium 2008, 15, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Bosutti, A.; Grassi, G.; Zanetti, M.; Aleksova, A.; Zecchin, M.; Sinagra, G.; Biolo, G.; Guarnieri, G. Relation between the plasma levels of LDL-cholesterol and the expression of the early marker of inflammation long pentraxin PTX3 and the stress response gene p66(ShcA) in pacemaker-implanted patients. Clin. Exp. Med. 2007, 7, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Azumi, H.; Inoue, N.; Ohashi, Y.; Terashima, M.; Mori, T.; Fujita, H.; Awano, K.; Kobayashi, S.; Maeda, K.; Hata, K.; et al. Superoxide generation in directional coronary atherectomy specimens of patients with angina pectoris: Important role of NAD(P)H oxidase. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Barry-Lane, P.A.; Patterson, C.; Merwe, M.V.; Hu, Z.; Holland, S.M.; Yeh, E.T.; Runge, M.S. P47phox is required for atherosclerotic lesion progression in ApoE–/– mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Abbas, A.K.; Aster, J.C.; Perkins, J.A. Robbins Basic Pathology; Elsevier: Philadelphia, PA, USA, 2018. [Google Scholar]
- Thu, V.T.; Kim, H.K.; Ha, S.H.; Yoo, J.; Park, W.S.; Kim, N.; Oh, G.T.; Han, J. Glutathione peroxidase 1 protects mitochondria against hypoxia/reoxygenation damage in mouse hearts. Pflügers Arch. Eur. J. Physiol. 2010, 460, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Marazita, M.C.; Dugour, A.; Marquioni-Ramella, M.D.; Figueroa, J.M.; Suburo, A.M. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: Implications for Age-related Macular Degeneration. Redox Biol. 2016, 7, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Biol. Med. 2012, 53. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Spokes, K.C.; Shih, S.; Aird, W.C. NADPH Oxidase Activity Selectively Modulates Vascular Endothelial Growth Factor Signaling Pathways. J. Biol. Chem. 2007, 282, 35373–35385. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belarddinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial ROS generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.M.; Chan, E.C.; Liu, G.S.; Jiang, F.; Dusting, G.J. Transforming growth factor-β1 requires NADPH oxidase 4 for angiogenesis in vitro and in vivo. J. Cell. Mol. Med. 2014, 18, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Sudhahar, V.; Kim, S.J.; Chen, G.F.; McKinney, R.D.; Kojda, G.; Fukai, T.; Ushio-Fukai, M. Critical Role of Endothelial Hydrogen Peroxide in Post-Ischemic Neovascularization. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Salloum, F.N.; Fisher, B.J.; Kukreja, R.C.; Fowler, A.A. Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ. Res. 2006, 98, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Sellke, F.W. Antioxidant Therapy: Is it your Gateway to Improved Cardiovascular Health? Pharm. Anal. Acta 2015, 6, 1–10. [Google Scholar] [CrossRef]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14. https://doi.org/10.3390/antiox7010014
Aldosari S, Awad M, Harrington EO, Sellke FW, Abid MR. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants. 2018; 7(1):14. https://doi.org/10.3390/antiox7010014
Chicago/Turabian StyleAldosari, Sarah, Maan Awad, Elizabeth O. Harrington, Frank W. Sellke, and M. Ruhul Abid. 2018. "Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology" Antioxidants 7, no. 1: 14. https://doi.org/10.3390/antiox7010014
APA StyleAldosari, S., Awad, M., Harrington, E. O., Sellke, F. W., & Abid, M. R. (2018). Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants, 7(1), 14. https://doi.org/10.3390/antiox7010014