Regulation of Mitochondrial Dynamics by Proteolytic Processing and Protein Turnover


Abstract
:1. Introduction
2. Links between Protein Turnover and Mitochondrial Function
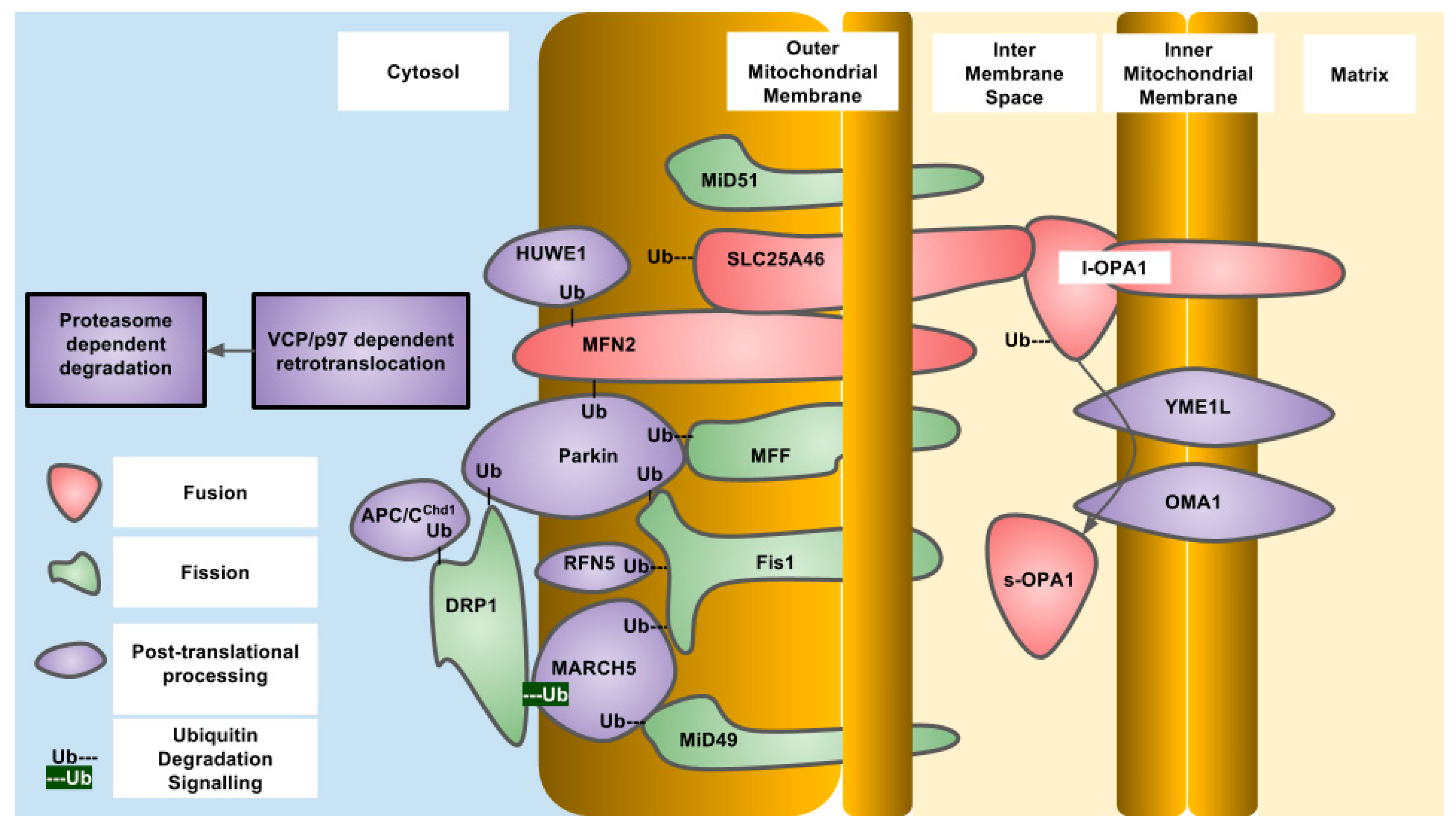
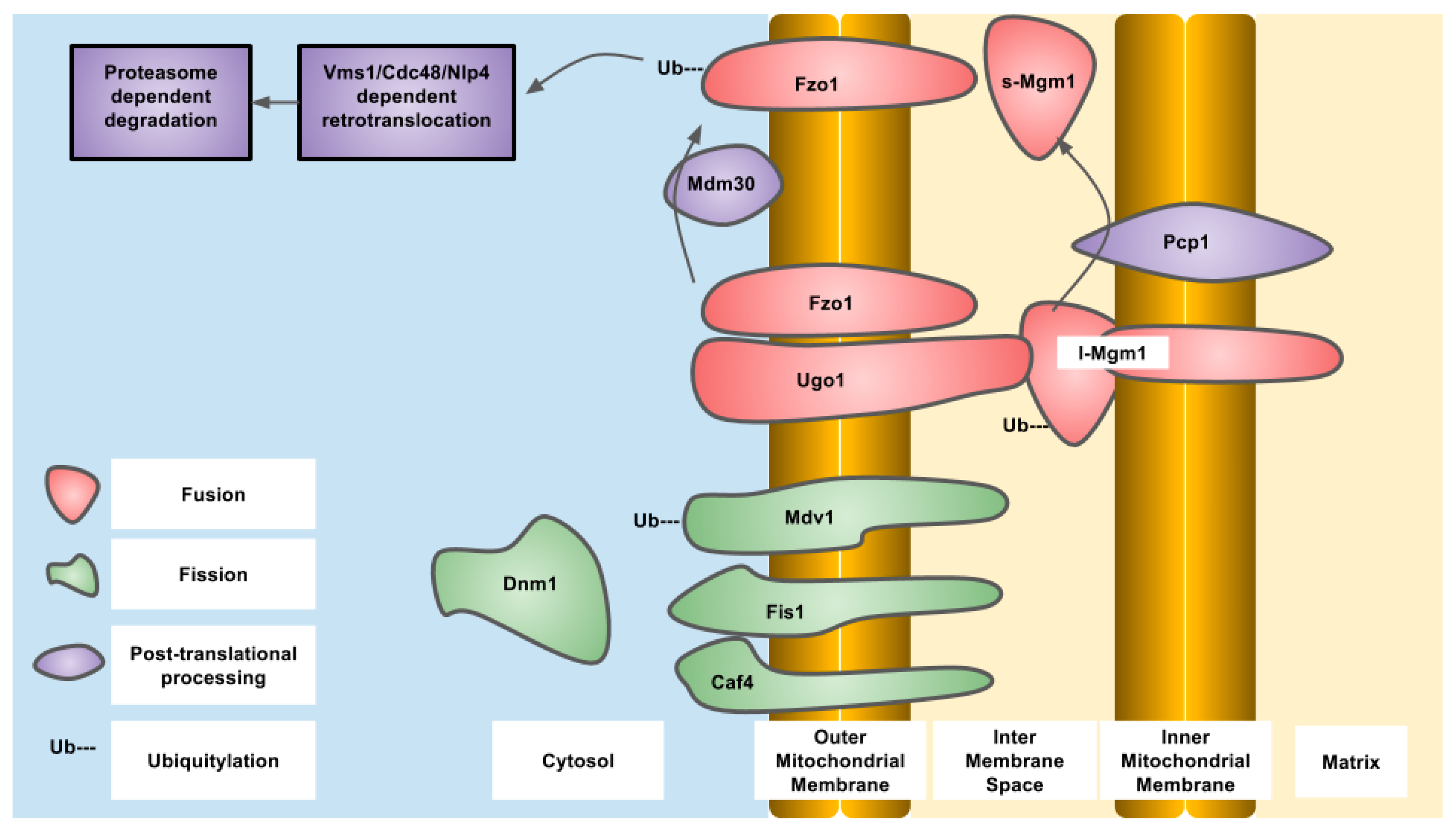
3. Inner Mitochondrial Membrane Fission and Fusion
4. Outer Mitochondrial Membrane Fission and Fusion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Shaw, J.M. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005, 39, 503–536. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 2009, 20, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, Z.; Zhang, L.; Zhou, Z.; Zheng, X.; Liu, G.; Bu, G.; Fraser, P.E.; Xu, H.; Zhang, Y.-W. Appoptosin interacts with mitochondrial outer-membrane fusion proteins and regulates mitochondrial morphology. J. Cell Sci. 2016, 129, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chan, D.C. New insights into mitochondrial fusion. FEBS Lett. 2007, 581, 2168–2173. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Klionsky, D.J. Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 2008, 283, 32386–32393. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetics in human evolution and disease: Implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.G.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Corbett, M.; Vandewalle, J.; Jarvela, I.; Lawrence, O.; Meldrum, C.; Bauters, M.; Govaerts, K.; Vandeleur, L.; Van Esch, H.; et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 2008, 82, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Sheu, S.-S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Xu, L.; Yu, Y.; Zhu, W.; Andrews, D.W.; Yoon, Y.; Kuo, T.H. Regulation of Ca2+-induced permeability transition by Bcl-2 is antagonized by Drp1 and hFis1. Mol. Cell Biochem. 2005, 272, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hatakeyama, S.; Oyamada, K.; Oda, Y.; Nishimura, T.; Nakayama, K.I. Large-scale analysis of the human ubiquitin-related proteome. Proteomics 2005, 5, 4145–4151. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.; Westermann, B. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Geva-Zatorsky, N.; Issaeva, I.; Cohen, A.; Dekel, E.; Danon, T.; Cohen, L.; Mayo, A.; Alon, U. Proteome half-life dynamics in living human cells. Science 2011, 331, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Belle, A.; Tanay, A.; Bitincka, L.; Shamir, R.; O’Shea, E.K. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 13004–13009. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Christiano, R.; Nagaraj, N.; Fröhlich, F.; Walther, T.C. Global proteome turnover analyses of the Yeasts S. cerevisiae and S. pombe. Cell Rep. 2014, 9, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Donaldson, G.; Christianson, S.; Zahayko, N.; Khalimonchuk, O. Stress-triggered activation of the metalloprotease Oma1 involves its C-terminal region and is important for mitochondrial stress protection in yeast. J. Biol. Chem. 2014, 289, 13259–13272. [Google Scholar] [CrossRef] [PubMed]
- Herlan, M.; Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J. Biol. Chem. 2003, 278, 27781–27788. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.L.; McQuibban, G.A. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1). J. Biol. Chem. 2012, 287, 40131–40139. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Czauderna, F.; Paulick, K.; Schwarzer, R.; Kaufmann, J.; Santel, A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci. 2005, 118, 3049–3059. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Santel, A.; Schwarzer, R.; Dames, S.; Giese, K.; Klippel, A.; Kaufmann, J. Knockdown of MTP18, a novel phosphatidylinositol 3-kinase-dependent protein, affects mitochondrial morphology and induces apoptosis. J. Biol. Chem. 2004, 279, 31544–31555. [Google Scholar] [CrossRef] [PubMed]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef] [PubMed]
- Anton, F.; Dittmar, G.; Langer, T.; Escobar-Henriques, M. Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol. Cell 2013, 49, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Hirose, S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol. Biol. Cell 2008, 19, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.-R.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbé, S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M.J.; Leboucher, G.P.; Livnat-Levanon, N.; Glickman, M.H.; Weissman, A.M. Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol. Biol. Cell 2008, 19, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.; Weinbach, N.; Westermann, B. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol. Biol. Cell 2003, 14, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Westermann, B.; Langer, T. Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J. Cell Biol. 2006, 173, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Sesaki, H.; Jensen, R.E. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J. Cell Biol. 2001, 152, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Sesaki, H.; Jensen, R.E. Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J. Biol. Chem. 2004, 279, 28298–28303. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Horner, J.; Song, C.; McCaffery, J.M.; Nunnari, J. Mitochondrial outer and inner membrane fusion requires a modified carrier protein. J. Cell Biol. 2009, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.-Y.; Gingras, A.-C.; et al. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Vashisht, A.A.; Wan, J.; Jen, J.C.; Claypool, S.M.; Wohlschlegel, J.A.; Koehler, C.M. Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2-mediated hyperfusion of mitochondria. Mol. Biol. Cell 2017, 28, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.R.; Thomenius, M.J.; Johnson, E.S.; Freel, C.D.; Wu, J.Q.; Coloff, J.L.; Yang, C.-S.; Tang, W.; An, J.; Ilkayeva, O.R.; et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol. Biol. Cell 2011, 22, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, J.; Wu, R.; Ma, J.; Du, G.; Jiao, R.; Tian, Y.; Zheng, Z.; Yuan, Z. DJ-1 promotes the proteasomal degradation of Fis1: Implications of DJ-1 in neuronal protection. Biochem. J. 2012, 447, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Onoue, K.; Jofuku, A.; Ban-Ishihara, R.; Ishihara, T.; Maeda, M.; Koshiba, T.; Itoh, T.; Fukuda, M.; Otera, H.; Oka, T.; et al. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J. Cell Sci. 2013, 126, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Chen, C.; Punj, V.; Machida, K. The TBC1D15 oncoprotein controls stem cell self-renewal through destabilization of the Numb-p53 complex. PLoS ONE 2013, 8, e57312. [Google Scholar] [CrossRef] [PubMed]
- Tieu, Q.; Okreglak, V.; Naylor, K.; Nunnari, J. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J. Cell Biol. 2002, 158, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Fekkes, P.; Shepard, K.A.; Yaffe, M.P. Gag3p, an outer membrane protein required for fission of mitochondrial tubules. J. Cell Biol. 2000, 151, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Cerveny, K.L.; McCaffery, J.M.; Jensen, R.E. Division of mitochondria requires a novel DMN1-interacting protein, Net2p. Mol. Biol. Cell 2001, 12, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chan, D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Cherok, E.; Das, S.; Li, S.; Roelofs, B.A.; Ge, S.X.; Polster, B.M.; Boyman, L.; Lederer, W.J.; Wang, C.; et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol. Biol. Cell 2016, 27, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Clinton, R.W.; Francy, C.A.; Ramachandran, R.; Qi, X.; Mears, J.A. Dynamin-related Protein 1 Oligomerization in Solution Impairs Functional Interactions with Membrane-anchored Mitochondrial Fission Factor. J. Biol. Chem. 2016, 291, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Qin, S.; Jiang, C. Parkin-induced ubiquitination of Mff promotes its association with p62/SQSTM1 during mitochondrial depolarization. Acta Biochim. Biophys. Sin. 2015, 47, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Balicza, P.; Weaver, D.; Naghdi, S.; Joseph, S.K.; Várnai, P.; Gyuris, T.; Horváth, A.; Nagy, L.; Seifert, E.L.; et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Okano, Y. Human Misato regulates mitochondrial distribution and morphology. Exp. Cell Res. 2007, 313, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.M.; Rüegg, M.; Niemann, A.; Suter, U. Targeting and function of the mitochondrial fission factor GDAP1 are dependent on its tail-anchor. PLoS ONE 2009, 4, e5160. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Half-Life (h) [49] | Half-Life (Mins) [47] | Ubiquitylation Site [48] | E3 Ligase | Deubiquitinase | Function |
|---|---|---|---|---|---|---|
| CAF4 | 7.2 | 34 | Not reported | |||
| DNM1 | 10.6 | 31 | Not reported | |||
| FIS1 | 9.1 | Not reported | ||||
| FZO1 | 3 | 24 | 79 370 398 464 | MDM30 MDM30 | UBP12 UBP2 | Mitochondrial fusion Degradation |
| MDV1 | 7.3 | 25 | 126 | |||
| MGM1 | 8.4 | Not reported | 549 | |||
| UGO1 | Not reported | Not reported | Not reported |
| Protein | Half-Life (h) | Ubiquitylation Site [60] | E3 Ligase | Deubiquitinase | Function |
|---|---|---|---|---|---|
| DRP1 | Not reported | MARCH5 Parkin APC/CCdh1 | Mitochondrial translocation—non degradative Degradation Degradation after mitosis | ||
| Fis1 | Not reported | MARCH5 Parkin RFN5 | Degradation Degradation Degradation | ||
| GDAP1 | 172 173 188 | ||||
| MFF | 28 251 | Parkin | |||
| MFN1 | 89 91 161 178 181 393 423 467 475 681 746 759 | ||||
| MFN2 | 3.9 | 79 154 158 171 307 316 406 416 420 460 719 720 730 737 | Parkin HUWE1 | USP30 | |
| MiD49 | Not reported | MARCH5 | Degradation | ||
| MiD51 | Not reported | ||||
| MSTO1 | 80 89 105 203 206 | ||||
| MTP18 | Not reported | ||||
| OPA1 | 228 568 | ||||
| SLC25A46 | 76 | ||||
| TBC1D15 | 90 103 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, S.; McStay, G.P. Regulation of Mitochondrial Dynamics by Proteolytic Processing and Protein Turnover. Antioxidants 2018, 7, 15. https://doi.org/10.3390/antiox7010015
Ali S, McStay GP. Regulation of Mitochondrial Dynamics by Proteolytic Processing and Protein Turnover. Antioxidants. 2018; 7(1):15. https://doi.org/10.3390/antiox7010015
Chicago/Turabian StyleAli, Sumaira, and Gavin P. McStay. 2018. "Regulation of Mitochondrial Dynamics by Proteolytic Processing and Protein Turnover" Antioxidants 7, no. 1: 15. https://doi.org/10.3390/antiox7010015
APA StyleAli, S., & McStay, G. P. (2018). Regulation of Mitochondrial Dynamics by Proteolytic Processing and Protein Turnover. Antioxidants, 7(1), 15. https://doi.org/10.3390/antiox7010015