Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology

 ,
,  ,
, 
Abstract
:1. Introduction
- (1)
- Which cardiac signaling pathways regulate mitochondrial morphology and function under pathophysiological conditions?
- (2)
- How do the cardiac signaling pathways modulate the activities of mitochondrial fission and fusion proteins?
- (3)
- Do abnormal morphologies of cardiac mitochondria contribute to the development of cardiac pathology or are they just an adaptation/maladaptation to pathological changes in the heart?
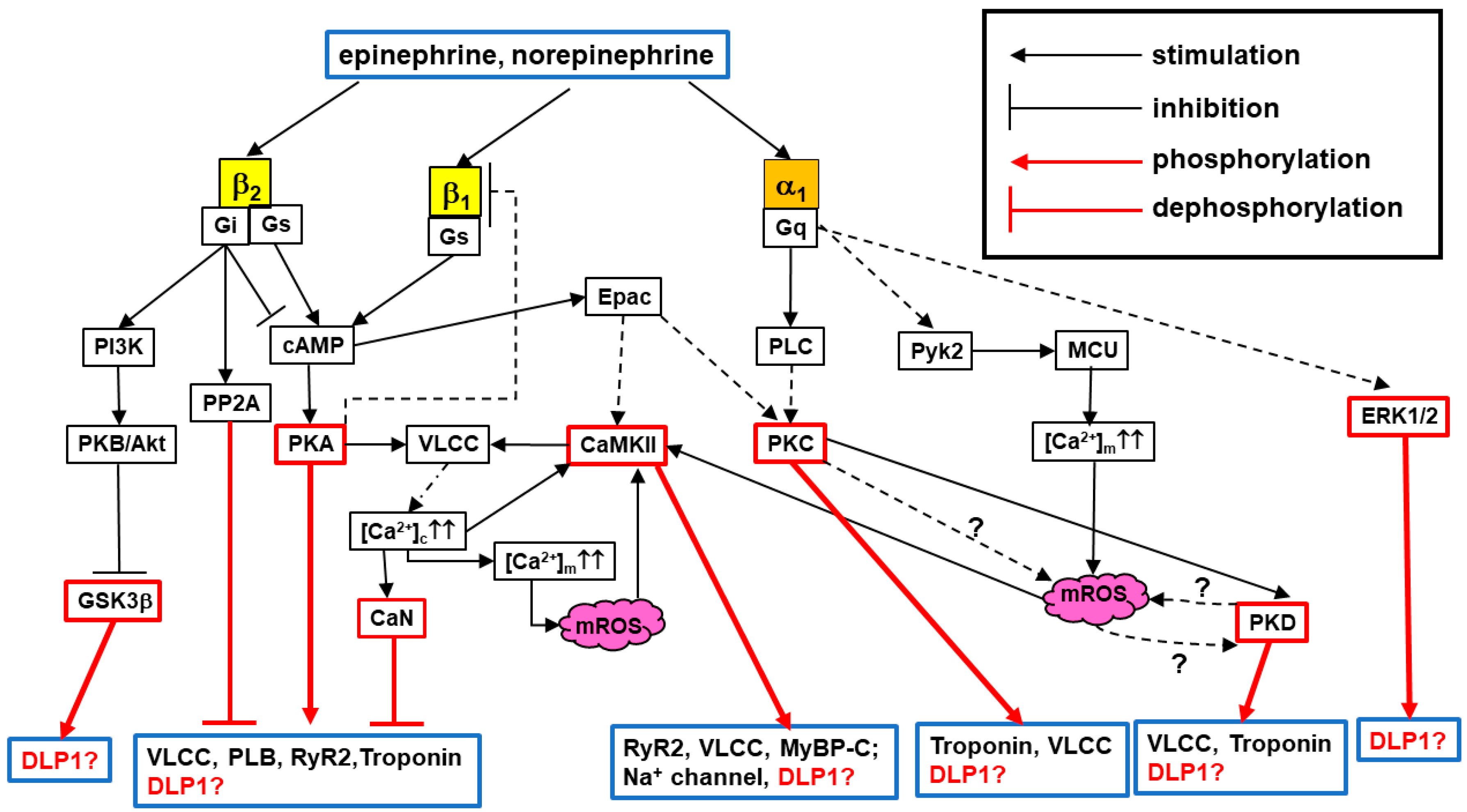
2. Overview: Adrenergic Regulation of Mitochondrial Functions in Cardiomyocytes
- (1)
- β-AR signaling dramatically increases CaT, promoting the acceleration of Ca2+ uptake from the cytosol to the mitochondrial matrix via mitochondrial Ca2+ uniporter (MCU), thus increasing mitochondrial matrix Ca2+ ([Ca2+]m) and consequently activating the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) to enhance ATP production [71,72].
- (2)
- (1)
- Does DLP1 translocation occur under physiological [Ca2+]c elevation, such as increased CaT under acute β-AR stimulation?
- (2)
- Does DLP1 translocation occur via increased [Ca2+]m and/or [Ca2+]c?
- (3)
- Are mitochondrial dynamics, especially fission, involved in the process for energy matching and/or mROS production under AR stimulation (see also Section 4)?
3. DLP1-Mediated Mitochondrial Fission in Non-Cardiac and Cardiac Cells
- (1)
- B-insert structure of DLP1 normally blocks the Mff-binding sites in DLP1.
- (2)
- B-insert binds to cardiolipin at the OMM, which allows the exposure of the Mff-binding site of DLP1 for DLP1-Mff interaction.
- (3)
- Binding of DLP1 dimers to Mff allows DLP1 assemblies to form functionally active DLP1 oligomers on the OMM in the form of a helical ring. GTP-induced constriction of the DLP1 helical ring plays a crucial role in mitochondrial membrane fission.
- (1)
- Different cell types used for the overexpression of DLP1 mutants which possess distinct mitochondrial shapes/networks in resting conditions (i.e., highly elongated and interconnected, or mixture of small and tubular mitochondria).
- (2)
- Involvement of different upstream molecules (i.e., kinases and phosphatases) listed above.
- (3)
- Co-existence of additional PTMs within DLP1 (or in other fission/fusion proteins) caused by the different upstream molecules in addition to S637 phosphorylation.
4. Molecular Mechanisms Underlying the Modulation of DLP1 Function by Adrenergic Signaling in Cardiomyocytes
4.1. Suitable Models for Investigating Post-Translational Modifications of DLP1 and Their Roles in Cardiomyocytes
- (1)
- (2)
- (3)
4.2. Overview of DLP1 Phosphorylation by Adrenergic Signaling in Cardiomyocytes
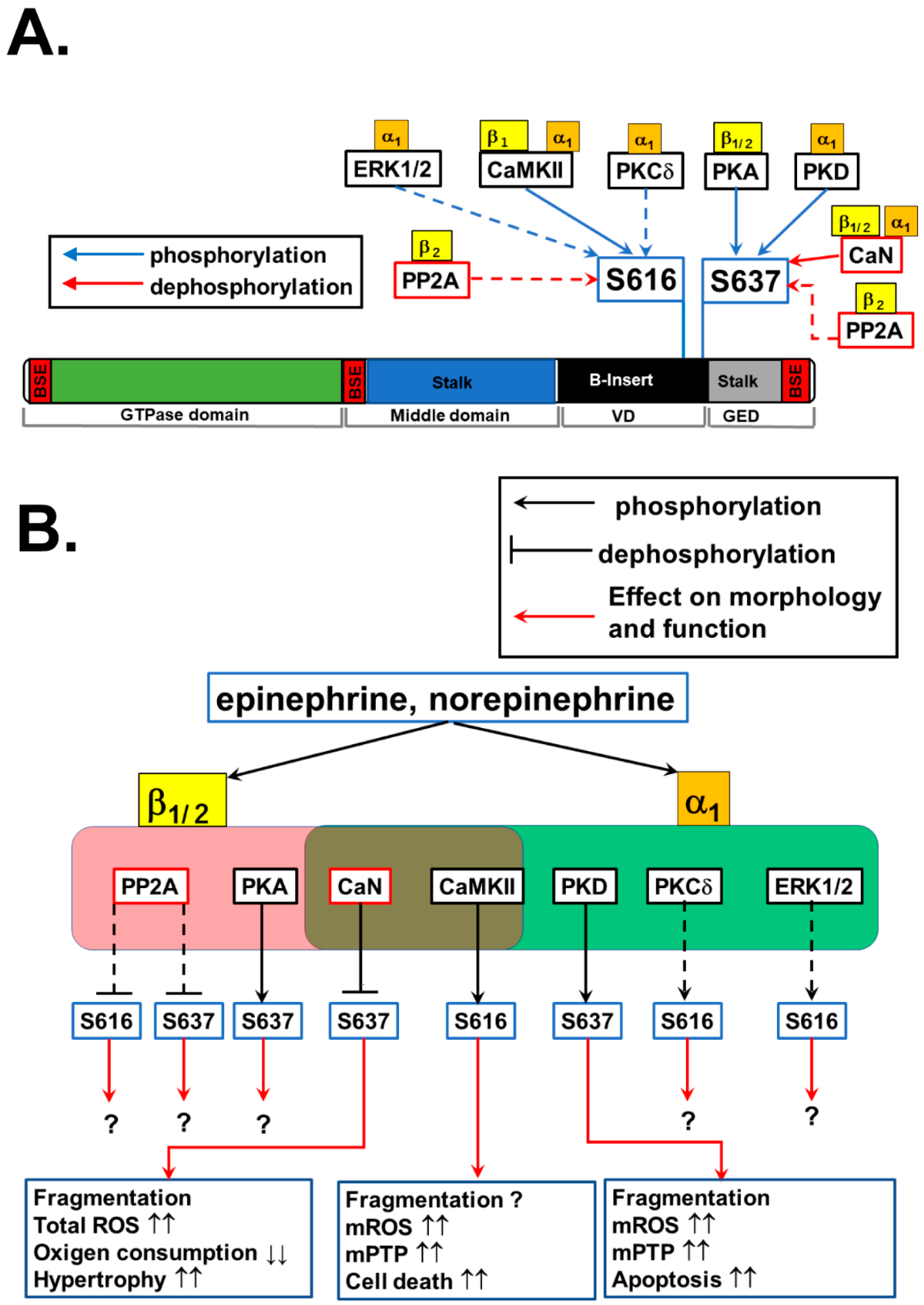
4.3. Phosphorylation of DLP1 at S616 by β-Adrenoceptor Signaling
4.4. S616 Phosphorylation of DLP1 by α1-Adrenoceptor Signaling
4.5. Role of DLP1 Phosphorylation at S616 in Mitochondrial Morphology and Function during Adrenergic Stimulation
4.6. Phosphorylation of DLP1 at S637 by β-Adrenoceptor Signaling
4.7. Phosphorylation of DLP1 at S637 by α1-Adrenoceptor Signaling
4.8. Role of DLP1 Phosphorylation at S637 on Mitochondrial Morphology and Function during Adrenergic Stimulation: Interplay of S637 and S616 Phosphorylation
4.9. Role of Adrenergic Signaling-Mediated DLP1 Phosphorylation in the Development of Heart Failure
- (1)
- (2)
- Accumulation of DLP1 to the OMM and the subsequent enhancement of mitochondrial fission by PTMs of DLP1 increase the efficiency of the electron transport chain (ETC) by affecting ultrastructural and spatial organization of the respiratory chain and ATP synthase, which stimulates forward electron flow through the ETC and ATP production [196].
- (3)
- Fragmented mitochondria by PTMs of DLP1 can sustain higher [Ca2+]m levels without propagation of Ca2+ to the neighboring mitochondria [197], which may also activate the TCA cycle to increase ATP generation.
4.10. Other Post-Translational Modifications of DLP1 by Adrenergic Signaling
5. Summary and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Vafai, S.B.; Mootha, V.K. Mitochondrial Disorders as Windows into an Ancient Organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of Oxidative Phosphorylation, the Mitochondrial Membrane Potential, and their Role in Human Disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Ryan, M.T. Mitochondrial OXPHOS Complex Assembly Lines. Nat. Cell Biol. 2018, 20, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of Mitochondria and Mitochondrial Function within Cells as another Level of Mitochondrial Complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial Dynamics in Mammalian Health and Disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Bioenergetic Role of Mitochondrial Fusion and Fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial Dynamics in Adaptive and Maladaptive Cellular Stress Responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial Dynamics in the Adult Cardiomyocytes: Which Roles for a Highly Specialized Cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial Dynamics in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendelin, M.; Beraud, N.; Guerrero, K.; Andrienko, T.; Kuznetsov, A.V.; Olivares, J.; Kay, L.; Saks, V.A. Mitochondrial Regular Arrangement in Muscle Cells: A “Crystal-Like” Pattern. Am. J. Physiol. Cell. Physiol. 2005, 288, C757–C767. [Google Scholar] [CrossRef] [PubMed]
- Beraud, N.; Pelloux, S.; Usson, Y.; Kuznetsov, A.V.; Ronot, X.; Tourneur, Y.; Saks, V. Mitochondrial Dynamics in Heart Cells: Very Low Amplitude High Frequency Fluctuations in Adult Cardiomyocytes and Flow Motion in Non Beating Hl-1 Cells. J. Bioenerg. Biomembr. 2009, 41, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Lenaers, G.; Hajnoczky, G. Mitochondrial Fusion is Frequent in Skeletal Muscle and Supports Excitation-Contraction Coupling. J. Cell Biol. 2014, 205, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Cupo, R.R.; Gao, E.; Csordas, G.; Slovinsky, W.S.; Paillard, M.; Cheng, L.; Ibetti, J.; Chen, S.R.; Chuprun, J.K.; et al. Mitochondrial Fusion Dynamics is Robust in the Heart and Depends on Calcium Oscillations and Contractile Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E859–E868. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.H.; McCaffery, J.M.; Chan, D.C. Mouse Lines with Photo-Activatable Mitochondria to Study Mitochondrial Dynamics. Genesis 2012, 50, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.D.; Glancy, B.; Balaban, R.S. The Electrochemical Transmission in I-Band Segments of the Mitochondrial Reticulum. Biochim. Biophys. Acta 2016, 1857, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial Reticulum for Cellular Energy Distribution in Muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Combs, C.A.; Femnou, A.; Sun, J.; Murphy, E.; Subramaniam, S.; Balaban, R.S. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell. Rep. 2017, 19, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, P.; Bisetto, S.; Yoon, Y.; Chen, Q.; Sheu, S.S.; Wang, W. A Novel Fission-Independent Role of Dynamin-Related Protein 1 in Cardiac Mitochondrial Respiration. Cardiovasc. Res. 2017, 113, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.; Lucena, J.R.; et al. Impaired Mitochondrial Biogenesis is a Common Feature to Myocardial Hypertrophy and End-Stage Ischemic Heart Failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Diakos, N.A.; Navankasattusas, S.; Abel, E.D.; Rutter, J.; McCreath, L.; Ferrin, P.; McKellar, S.H.; Miller, D.V.; Park, S.Y.; Richardson, R.S.; et al. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch during Mechanical Unloading of the Failing Human Heart: Implications for Cardiac Reloading and Conditioning. JACC Basic Transl. Sci. 2016, 1, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Bautista-Hernandez, V.; Fynn-Thompson, F.; Del Nido, P.; Tian, R. Impaired Mitochondrial Biogenesis Precedes Heart Failure in Right Ventricular Hypertrophy in Congenital Heart Disease. Circ. Heart Fail. 2011, 4, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the Myocardial Ultrastructure and Changes of the Cytoskeleton in Dilated Cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial Calcium Overload is a Key Determinant in Heart Failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed]
- Wust, R.C.; de Vries, H.J.; Wintjes, L.T.; Rodenburg, R.J.; Niessen, H.W.; Stienen, G.J. Mitochondrial Complex I Dysfunction and Altered NAD(P)H Kinetics in Rat Myocardium in Cardiac Right Ventricular Hypertrophy and Failure. Cardiovasc. Res. 2016, 111, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Schwarzer, M.; Chen, D.; Schrepper, A.; Amorim, P.A.; Schoepe, M.; Nguyen, T.D.; Mohr, F.W.; Khalimonchuk, O.; Weimer, B.C.; et al. Proteomic Remodelling of Mitochondrial Oxidative Pathways in Pressure Overload-Induced Heart Failure. Cardiovasc. Res. 2010, 85, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial Energetics in the Heart in Obesity-Related Diabetes: Direct Evidence for Increased Uncoupled Respiration and Activation of Uncoupling Proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, Apoptosis, and Heart Failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global Proteomics and Pathway Analysis of Pressure-Overload-Induced Heart Failure and its Attenuation by Mitochondrial-Targeted Peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial Permeability Transition Pore Opening as a Promising Therapeutic Target in Cardiac Diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial Ischemia Decreases Oxidative Phosphorylation through Cytochrome Oxidase in Subsarcolemmal Mitochondria. Am. J. Physiol. 1997, 273, H1544–H1554. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Slabe, T.J.; Stoll, M.S.; Minkler, P.E.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Ischemia, rather than Reperfusion, Inhibits Respiration through Cytochrome Oxidase in the Isolated, Perfused Rabbit Heart: Role of Cardiolipin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H258–H267. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell. Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Hom, J.; Yu, T.; Yoon, Y.; Porter, G.; Sheu, S.S. Regulation of Mitochondrial Fission by Intracellular Ca2+ in Rat Ventricular Myocytes. Biochim. Biophys. Acta 2010, 1797, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial Fission and Fusion in Human Diseases. N. Engl. J. Med. 2014, 370, 1074. [Google Scholar] [PubMed]
- Chen, L.; Knowlton, A.A. Mitochondria and Heart Failure: New Insights into an Energetic Problem. Minerva Cardioangiol. 2010, 58, 213–229. [Google Scholar] [PubMed]
- Bernstein, D.; Fajardo, G.; Zhao, M. The role of beta-adrenergic receptors in heart failure: differential regulation of cardiotoxicity and cardioprotection. Prog. Pediatr. Cardiol. 2011, 31, 35–38. [Google Scholar] [CrossRef] [PubMed]
- O-Uchi, J.; Lopes, C.M. Combined Blockade of Beta- and Alpha(1)-Adrenoceptors in Left Ventricular Remodeling Induced by Hypertension: Beneficial or Not? Hypertens. Res. 2010, 33, 984–985. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.Y.; Xiao, R.P. Beta-Adrenergic Receptor Subtype Signaling in Heart: From Bench to Bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.; Chaudhry, M. Effect of Alpha1-Adrenergic Receptors in Cardiac Pathophysiology. Am. Heart J. 2006, 152, 842–850. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac Beta-Adrenergic Signaling during Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The Sympathetic Nervous System in Heart Failure Physiology, Pathophysiology, and Clinical Implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac Excitation-Contraction Coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Guo, T. Calcium Signaling in Cardiac Ventricular Myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Zhu, W.; Rahman, M.S.; Liu, M.; Li, D.; Su, S.; Zhang, N.; Hu, X.; Yu, H.; Gupta, M.P.; et al. Molecular Regulation of Mitochondrial Dynamics in Cardiac Disease. Biochim. Biophys. Acta 2017, 1864, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New Insights into the Function and Regulation of Mitochondrial Fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent Advances into the Understanding of Mitochondrial Fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Dynamic Regulation of Mitochondrial Fission through Modification of the Dynamin-Related Protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Jhun, B.S.; O-Uchi, J.; Adaniya, S.M.; Mancini, T.J.; Cao, J.L.; King, M.E.; Landi, A.K.; Ma, H.; Shin, M.; Yang, D.; et al. Protein Kinase D Activation Induces Mitochondrial Fragmentation and Dysfunction in Cardiomyocytes. J. Physiol. 2018, 596, 827–855. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII Induces Permeability Transition through Drp1 Phosphorylation during Chronic Beta-AR Stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible Phosphorylation of Drp1 by Cyclic AMP-Dependent Protein Kinase and Calcineurin Regulates Mitochondrial Fission and Cell Death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 Regulates Mitochondria Morphology and Apoptotic Cell Death Via Local Modulation of cAMP/PKA Signalling. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Pennanen, C.; Parra, V.; Lopez-Crisosto, C.; Morales, P.E.; Del Campo, A.; Gutierrez, T.; Rivera-Mejias, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial Fission is Required for Cardiomyocyte Hypertrophy Mediated by a Ca2+-Calcineurin Signaling Pathway. J. Cell. Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E.; Michel, M.C. Adrenergic and Muscarinic Receptors in the Human Heart. Pharmacol. Rev. 1999, 51, 651–690. [Google Scholar] [PubMed]
- Woodcock, E.A.; Du, X.J.; Reichelt, M.E.; Graham, R.M. Cardiac Alpha 1-Adrenergic Drive in Pathological Remodelling. Cardiovasc. Res. 2008, 77, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.P. Beta-Adrenergic Signaling in the Heart: Dual Coupling of the Beta2-Adrenergic Receptor to G(s) and G(i) Proteins. Science 2001, 2001, re15. [Google Scholar]
- O’Connell, T.D.; Jensen, B.C.; Baker, A.J.; Simpson, P.C. Cardiac Alpha1-Adrenergic Receptors: Novel Aspects of Expression, Signaling Mechanisms, Physiologic Function, and Clinical Importance. Pharmacol. Rev. 2013, 66, 308–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endoh, M.; Blinks, J.R. Actions of Sympathomimetic Amines on the Ca2+ Transients and Contractions of Rabbit Myocardium: Reciprocal Changes in Myofibrillar Responsiveness to Ca2+ Mediated through Alpha- and Beta-Adrenoceptors. Circ. Res. 1988, 62, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.C.; O’Connell, T.D.; Simpson, P.C. Alpha-1-Adrenergic Receptors: Targets for Agonist Drugs to Treat Heart Failure. J. Mol. Cell. Cardiol. 2011, 51, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Blinks, J.R. The Nature of the Antagonism by Methoxamine of the Chronotropic and Inotropic Effects of Catecholamines. Naunyn Schmiedebergs. Arch. Exp. Pathol. Pharmakol. 1964, 248, 73–84. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Morel, E.; Dominguez-Rodriguez, A.; Llach, A.; Lezoualc’h, F.; Benitah, J.P.; Gomez, A.M. Epac in Cardiac Calcium Signaling. J. Mol. Cell. Cardiol. 2013, 58, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.P.; Zhu, W.; Zheng, M.; Cao, C.; Zhang, Y.; Lakatta, E.G.; Han, Q. Subtype-Specific Alpha1- and Beta-Adrenoceptor Signaling in the Heart. Trends Pharmacol. Sci. 2006, 27, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell. Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell. Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, A.; Song, Z.; Liu, H.; Zhou, A.; Shi, G.; Wang, Q.; Gu, L.; Liu, M.; Xie, L.H.; Qu, Z.; et al. Mitochondrial Ca2+ Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- O-Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic Signaling Regulates Mitochondrial Ca2+ Uptake through Pyk2-Dependent Tyrosine Phosphorylation of the Mitochondrial Ca2+ Uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Lipsius, S.L.; Zima, A.V. Reactive Oxygen Species Contribute to the Development of Arrhythmogenic Ca2+ Waves during Beta-Adrenergic Receptor Stimulation in Rabbit Cardiomyocytes. J. Physiol. 2012, 590, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Osadchii, O.E. Cardiac Hypertrophy Induced by Sustained Beta-Adrenoreceptor Activation: Pathophysiological Aspects. Heart Fail. Rev. 2007, 12, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.C.; O’Connell, T.D.; Simpson, P.C. Alpha-1-Adrenergic Receptors in Heart Failure: The Adaptive Arm of the Cardiac Response to Chronic Catecholamine Stimulation. J. Cardiovasc. Pharmacol. 2014, 63, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.W.; Sakata, Y.; Davis, M.G.; Sah, V.P.; Wang, Y.; Liggett, S.B.; Chien, K.R.; Brown, J.H.; Dorn, G.W., 2nd. Enhanced Galphaq Signaling: A Common Pathway Mediates Cardiac Hypertrophy and Apoptotic Heart Failure. Proc. Natl. Acad. Sci. USA 1998, 95, 10140–10145. [Google Scholar] [CrossRef] [PubMed]
- Mende, U.; Kagen, A.; Cohen, A.; Aramburu, J.; Schoen, F.J.; Neer, E.J. Transient Cardiac Expression of Constitutively Active Galphaq Leads to Hypertrophy and Dilated Cardiomyopathy by Calcineurin-Dependent and Independent Pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 13893–13898. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Li, R.; Feng, X.; James Kang, Y. The Involvement of Cytochrome C Oxidase in Mitochondrial Fusion in Primary Cultures of Neonatal Rat Cardiomyocytes. Cardiovasc. Toxicol. 2018, 18, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Lechuga-Vieco, A.V.; Del Mar Munoz, M.; Nieto-Arellano, R.; Torroja, C.; Sanchez-Cabo, F.; Jimenez, C.; Gonzalez-Guerra, A.; Carrascoso, I.; Beninca, C.; et al. Ablation of the Stress Protease OMA1 Protects Against Heart Failure in Mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.C.; Taam, G.M.; Singal, P.K.; Beamish, R.E.; Dhalla, N.S. Protection Against Adrenochrome-Induced Myocardial Damage by various Pharmacological Interventions. Br. J. Exp. Pathol. 1980, 61, 242–255. [Google Scholar] [PubMed]
- Li, Y.; Fang, J.; Hua, Y.; Wang, C.; Mu, D.; Zhou, K. The Study of Fetal Rat Model of Intra-Amniotic Isoproterenol Injection Induced Heart Dysfunction and Phenotypic Switch of Contractile Proteins. Biomed. Res. Int. 2014, 2014, 360687. [Google Scholar] [CrossRef] [PubMed]
- Mikusova, A.; Kralova, E.; Tylkova, L.; Novotova, M.; Stankovicova, T. Myocardial Remodelling Induced by Repeated Low Doses of Isoproterenol. Can. J. Physiol. Pharmacol. 2009, 87, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Rajapurohitam, V.; Kilic, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of Mitochondrial Fusion-Fission Proteins during Post-Infarction Remodeling: The Effect of NHE-1 Inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-Phosphorylated Mitofusin 2 is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 that Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 to Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Mihara, K. Mitochondrial Dynamics: Functional Link with Apoptosis. Int. J. Cell. Biol. 2012, 2012, 821676. [Google Scholar] [CrossRef] [PubMed]
- Al-Husseini, A.; Wijesinghe, D.S.; Farkas, L.; Kraskauskas, D.; Drake, J.I.; Van Tassel, B.; Abbate, A.; Chalfant, C.E.; Voelkel, N.F. Increased Eicosanoid Levels in the Sugen/Chronic Hypoxia Model of Severe Pulmonary Hypertension. PLoS ONE 2015, 10, e0120157. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188. [Google Scholar] [PubMed]
- Kraus, F.; Ryan, M.T. The Constriction and Scission Machineries Involved in Mitochondrial Fission. J. Cell. Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Kobayashi, S.; Fujiki, Y. Peroxisome Division is Impaired in a CHO Cell Mutant with an Inactivating Point-Mutation in Dynamin-Like Protein 1 Gene. Exp. Cell Res. 2006, 312, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Adachi, Y.; Yamada, T.; Suzuki, T.L.; Otomo, T.; McBride, H.M.; Yoshimori, T.; Iijima, M.; Sesaki, H. A Brain-Enriched Drp1 Isoform Associates with Lysosomes, Late Endosomes, and the Plasma Membrane. J. Biol. Chem. 2018, 293, 11809–11822. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Pitts, K.R.; McNiven, M.A. Mammalian Dynamin-Like Protein DLP1 Tubulates Membranes. Mol. Biol. Cell 2001, 12, 2894–2905. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Pitts, K.R.; Dahan, S.; McNiven, M.A. A Novel Dynamin-Like Protein Associates with Cytoplasmic Vesicles and Tubules of the Endoplasmic Reticulum in Mammalian Cells. J. Cell Biol. 1998, 140, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-Related Protein Drp1 is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Pitts, K.R.; Yoon, Y.; Krueger, E.W.; McNiven, M.A. The Dynamin-Like Protein DLP1 is Essential for Normal Distribution and Morphology of the Endoplasmic Reticulum and Mitochondria in Mammalian Cells. Mol. Biol. Cell 1999, 10, 4403–4417. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Cribbs, J.T. Allosteric Modulation of Drp1 Mechanoenzyme Assembly and Mitochondrial Fission by the Variable Domain. J. Biol. Chem. 2012, 287, 10990–11001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural Insights into Oligomerization and Mitochondrial Remodelling of Dynamin 1-Like Protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Wenger, J.; Klinglmayr, E.; Frohlich, C.; Eibl, C.; Gimeno, A.; Hessenberger, M.; Puehringer, S.; Daumke, O.; Goettig, P. Functional Mapping of Human Dynamin-1-Like GTPase Domain Based on X-Ray Structure Analyses. PLoS ONE 2013, 8, e71835. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-Glucose Stimulation Increases Reactive Oxygen Species Production through the Calcium and Mitogen-Activated Protein Kinase-Mediated Activation of Mitochondrial Fission. Antioxid. Redox Signal. 2011, 14, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Clinton, R.W.; Francy, C.A.; Ramachandran, R.; Qi, X.; Mears, J.A. Dynamin-Related Protein 1 Oligomerization in Solution Impairs Functional Interactions with Membrane-Anchored Mitochondrial Fission Factor. J. Biol. Chem. 2016, 291, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; van der Bliek, A.M. The Novel Tail-Anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an Essential Factor for Mitochondrial Recruitment of Drp1 during Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Itoyama, A.; Michiyuki, S.; Honsho, M.; Yamamoto, T.; Moser, A.; Yoshida, Y.; Fujiki, Y. Mff Functions with Pex11pbeta and DLP1 in Peroxisomal Fission. Biol. Open 2013, 2, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, New Components of the Mitochondrial Fission Machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlen, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nister, M. Human MIEF1 Recruits Drp1 to Mitochondrial Outer Membranes and Promotes Mitochondrial Fusion rather than Fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor Proteins MiD49 and MiD51 can Act Independently of Mff and Fis1 in Drp1 Recruitment and are Specific for Mitochondrial Fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of Mitochondrial Fusion Rescues Mff-Deficient Cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chan, D.C. The Mitochondrial Fission Receptor Mff Selectively Recruits Oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and Independent Roles of the Drp1 Adaptors Mff, MiD49 and MiD51 in Mitochondrial Fission. J. Cell. Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, A.B.; Lee, S.H.; To, M.; Lee, A.; Shin, J.Y.; Bustamante, C. Optimized Two-Color Super Resolution Imaging of Drp1 during Mitochondrial Fission with a Slow-Switching Dronpa Variant. Proc. Natl. Acad. Sci. USA 2014, 111, 13093–13098. [Google Scholar] [CrossRef] [PubMed]
- Colasante, C.; Chen, J.; Ahlemeyer, B.; Baumgart-Vogt, E. Peroxisomes in Cardiomyocytes and the Peroxisome/Peroxisome Proliferator-Activated Receptor-Loop. Thromb. Haemost. 2015, 113, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, J.; Li, Y.; Wu, S.; Luo, J.; Yang, H.; Subbiah, R.; Chatham, J.; Zhelyabovska, O.; Yang, Q. Peroxisome Proliferator-Activated Receptor {Delta} is an Essential Transcriptional Regulator for Mitochondrial Protection and Biogenesis in Adult Heart. Circ. Res. 2010, 106, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, P.; He, L.; Li, Y.; Luo, J.; Cheng, L.; Qin, Q.; Brako, L.A.; Lo, W.K.; Lewis, W.; et al. Cardiomyocyte-Restricted Deletion of PPARbeta/Delta in PPARalpha-Null Mice Causes Impaired Mitochondrial Biogenesis and Defense, but no further Depression of Myocardial Fatty Acid Oxidation. PPAR Res. 2011, 2011, 372854. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Lin, T.K.; Yang, D.I.; Yang, J.L.; Liou, C.W.; Chen, S.D. Peroxisome Proliferator-Activated Receptor-Gamma Dependent Pathway Reduces the Phosphorylation of Dynamin-Related Protein 1 and Ameliorates Hippocampal Injury Induced by Global Ischemia in Rats. J. Biomed. Sci. 2016, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liu, X.H.; Han, M.Z.; Wang, Y.M.; Sun, X.L.; Yu, N.; Li, T.; Su, B.; Chen, Z.Y. Blockage of GSK3beta-Mediated Drp1 Phosphorylation Provides Neuroprotection in Neuronal and Mouse Models of Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Q.; Zhang, P.; Sun, L.; Peng, C.; Yuan, Z.; Cheng, J. C-Abl-Mediated Drp1 Phosphorylation Promotes Oxidative Stress-Induced Mitochondrial Fragmentation and Neuronal Cell Death. Cell. Death Dis. 2017, 8, e3117. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Iniguez-Lluhi, J.A.; Stadler, J.; Chang, C.R.; Arnoult, D.; Keller, P.J.; Hong, Y.; Blackstone, C.; Feldman, E.L. SUMOylation of the Mitochondrial Fission Protein Drp1 Occurs at Multiple Nonconsensus Sites within the B Domain and is Linked to its Activity Cycle. FASEB J. 2009, 23, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, S.; Zunino, R.; McBride, H.M. Bax/Bak Promote Sumoylation of DRP1 and its Stable Association with Mitochondria during Apoptotic Cell Death. J. Cell Biol. 2007, 177, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Zunino, R.; Schauss, A.; Rippstein, P.; Andrade-Navarro, M.; McBride, H.M. The SUMO Protease SENP5 is Required to Maintain Mitochondrial Morphology and Function. J. Cell. Sci. 2007, 120, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Hildick, K.L.; Luo, J.; Dearden, L.; Wilkinson, K.A.; Henley, J.M. SENP3-Mediated deSUMOylation of Dynamin-Related Protein 1 Promotes Cell Death Following Ischaemia. EMBO J. 2013, 32, 1514–1528. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Zhang, Y.; Beketaev, I.; Segura, A.M.; Yu, W.; Xi, Y.; Chang, J.; Wang, J. SENP5, a SUMO Isopeptidase, Induces Apoptosis and Cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 78, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Huang, E.; Irrcher, I.; Rashidian, J.; Ishihara, N.; Lagace, D.C.; Slack, R.S.; Park, D.S. CDK5 Phosphorylates DRP1 and Drives Mitochondrial Defects in NMDA-Induced Neuronal Death. Hum. Mol. Genet. 2015, 24, 4573–4583. [Google Scholar] [CrossRef] [PubMed]
- Gawlowski, T.; Suarez, J.; Scott, B.; Torres-Gonzalez, M.; Wang, H.; Schwappacher, R.; Han, X.; Yates, J.R., 3rd; Hoshijima, M.; Dillmann, W. Modulation of Dynamin-Related Protein 1 (DRP1) Function by Increased O-Linked-Beta-N-Acetylglucosamine Modification (O-GlcNAc) in Cardiac Myocytes. J. Biol. Chem. 2012, 287, 30024–30034. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Qi, X. Inhibition of Excessive Mitochondrial Fission Reduced Aberrant Autophagy and Neuronal Damage Caused by LRRK2 G2019S Mutation. Hum. Mol. Genet. 2013, 22, 4545–4561. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1-Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Zaja, I.; Bai, X.; Liu, Y.; Kikuchi, C.; Dosenovic, S.; Yan, Y.; Canfield, S.G.; Bosnjak, Z.J. Cdk1, PKCdelta and Calcineurin-Mediated Drp1 Pathway Contributes to Mitochondrial Fission-Induced Cardiomyocyte Death. Biochem. Biophys. Res. Commun. 2014, 453, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Strack, S.; Wilson, T.J.; Cribbs, J.T. Cyclin-Dependent Kinases Regulate Splice-Specific Targeting of Dynamin-Related Protein 1 to Microtubules. J. Cell Biol. 2013, 201, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.; Cho, H.M.; Kim, H.J.; Jeong, J.; Park, S.K.; Hwang, E.M.; Park, J.Y.; Kim, W.R.; Kim, H.; Sun, W. CDK5-Dependent Inhibitory Phosphorylation of Drp1 during Neuronal Maturation. Exp. Mol. Med. 2014, 46, e105. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial Division is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.J.; Qi, X. Drp1 Phosphorylation by MAPK1 Causes Mitochondrial Dysfunction in Cell Culture Model of Huntington’s Disease. Biochem. Biophys. Res. Commun. 2018, 496, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Disatnik, M.H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant Mitochondrial Fission in Neurons Induced by Protein Kinase C{Delta} Under Oxidative Stress Conditions in Vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, S.S.; Van Dyke, A.M.; Chandra, R.; LeGates, T.A.; Contreras, M.; Dharmasri, P.A.; Higgs, H.N.; Lobo, M.K.; Thompson, S.M.; Blanpied, T.A. Long-Term Potentiation Requires a Rapid Burst of Dendritic Mitochondrial Fission during Induction. Neuron 2018. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.S.; Tan, V.P.; Brown, J.H.; Miyamoto, S. RhoA Regulates Drp1 Mediated Mitochondrial Fission through ROCK to Protect Cardiomyocytes. Cell. Signal. 2018, 50, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Kim, D.H.; Choi, J.E.; Chang, E.J.; Seung, -Y. Increased Phosphorylation of Dynamin-Related Protein 1 and Mitochondrial Fission in Okadaic Acid-Treated Neurons. Brain Res. 2012, 1454, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An Alternative Mitophagy Pathway Mediated by Rab9 Protects the Heart Against Ischemia. J. Clin. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-Dependent Protein Kinase Phosphorylation of Drp1 Regulates its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z.A. Fine-Tuning of Drp1/Fis1 Availability by AKAP121/Siah2 Regulates Mitochondrial Adaptation to Hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM Kinase I Alpha-Induced Phosphorylation of Drp1 Regulates Mitochondrial Morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell. Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Din, S.; Mason, M.; Volkers, M.; Johnson, B.; Cottage, C.T.; Wang, Z.; Joyo, A.Y.; Quijada, P.; Erhardt, P.; Magnuson, N.S.; et al. Pim-1 Preserves Mitochondrial Morphology by Inhibiting Dynamin-Related Protein 1 Translocation. Proc. Natl. Acad. Sci. USA 2013, 110, 5969–5974. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-Related Protein 1 (Drp1)-Mediated Diastolic Dysfunction in Myocardial Ischemia-Reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Slupe, A.M.; Merrill, R.A.; Flippo, K.H.; Lobas, M.A.; Houtman, J.C.; Strack, S. A Calcineurin Docking Motif (LXVP) in Dynamin-Related Protein 1 Contributes to Mitochondrial Fragmentation and Ischemic Neuronal Injury. J. Biol. Chem. 2013, 288, 12353–12365. [Google Scholar] [CrossRef] [PubMed]
- Dickey, A.S.; Strack, S. PKA/AKAP1 and PP2A/Bbeta2 Regulate Neuronal Morphogenesis Via Drp1 Phosphorylation and Mitochondrial Bioenergetics. J. Neurosci. 2011, 31, 15716–15726. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.A.; Slupe, A.M.; Strack, S. N-Terminal Phosphorylation of Protein Phosphatase 2A/Bbeta2 Regulates Translocation to Mitochondria, Dynamin-Related Protein 1 Dephosphorylation, and Neuronal Survival. FEBS J. 2013, 280, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Bossy, B.; Petrilli, A.; Klinglmayr, E.; Chen, J.; Lutz-Meindl, U.; Knott, A.B.; Masliah, E.; Schwarzenbacher, R.; Bossy-Wetzel, E. S-Nitrosylation of DRP1 does Not Affect Enzymatic Activity and is Not Specific to Alzheimer’s Disease. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S513–S526. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates Beta-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-Nitrosylation of Dynamin-Related Protein 1 Mediates Mutant Huntingtin-Induced Mitochondrial Fragmentation and Neuronal Injury in Huntington’s Disease. Antioxid. Redox Signal. 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Kim, J.E. PDI-Mediated S-Nitrosylation of DRP1 Facilitates DRP1-S616 Phosphorylation and Mitochondrial Fission in CA1 Neurons. Cell. Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-Mediated Drp1 Phosphorylation Induced Elongated Mitochondrial Morphology Against Oxidative Stress. PLoS ONE 2012, 7, e49112. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A Novel Mitochondrial Ubiquitin Ligase Plays a Critical Role in Mitochondrial Dynamics. EMBO J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a Novel Mitofusin 2- and Drp1-Binding Protein Able to Change Mitochondrial Morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The Mitochondrial E3 Ubiquitin Ligase MARCH5 is Required for Drp1 Dependent Mitochondrial Division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Hemion, C.; Goldblum, D.; Meyer, P.; Orgul, S.; Frank, S.; Flammer, J.; Neutzner, A. Inactivation of MARCH5 Prevents Mitochondrial Fragmentation and Interferes with Cell Death in a Neuronal Cell Model. PLoS ONE 2012, 7, e52637. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin Ubiquitinates Drp1 for Proteasome-Dependent Degradation: Implication of Dysregulated Mitochondrial Dynamics in Parkinson Disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lammermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of Parkin Or PINK1 Function Increases Drp1-Dependent Mitochondrial Fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., 2nd. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Harder, Z.; Zunino, R.; McBride, H. Sumo1 Conjugates Mitochondrial Substrates and Participates in Mitochondrial Fission. Curr. Biol. 2004, 14, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Cieplak, P.; Cho, D.H.; Godzik, A.; Lipton, S.A. S-Nitrosylation of Drp1 Links Excessive Mitochondrial Fission to Neuronal Injury in Neurodegeneration. Mitochondrion 2010, 10, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.; Lipton, S.A. Aberrant Protein S-Nitrosylation Contributes to the Pathophysiology of Neurodegenerative Diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Nunez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif Affinity and Mass Spectrometry Proteomic Approach for the Discovery of Cellular AMPK Targets: Identification of Mitochondrial Fission Factor as a New AMPK Substrate. Cell. Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-Activated Protein Kinase Mediates Mitochondrial Fission in Response to Energy Stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Cherok, E.; Das, S.; Li, S.; Roelofs, B.A.; Ge, S.X.; Polster, B.M.; Boyman, L.; Lederer, W.J.; Wang, C.; et al. Mitochondrial E3 Ubiquitin Ligase MARCH5 Controls Mitochondrial Fission and Cell Sensitivity to Stress-Induced Apoptosis through Regulation of MiD49 Protein. Mol. Biol. Cell 2016, 27, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A Novel De Novo Dominant Negative Mutation in DNM1L Impairs Mitochondrial Fission and Presents as Childhood Epileptic Encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.J.; Wangler, M.F. Missense Variants in the Middle Domain of DNM1L in Cases of Infantile Encephalopathy Alter Peroxisomes and Mitochondria when Assayed in Drosophila. Hum. Mol. Genet. 2016, 25, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic Mutations in DNM1L are Associated with a Slowly Progressive Infantile Encephalopathy. Hum. Mutat. 2016, 37, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-Related Mitochondrial Fission Defect Presenting as Refractory Epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal Microcephaly and Pain Insensitivity due to a De Novo Heterozygous DNM1L Mutation Causing Impaired Mitochondrial Fission and Function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Cahill, T.J.; Leo, V.; Kelly, M.; Stockenhuber, A.; Kennedy, N.W.; Bao, L.; Cereghetti, G.M.; Harper, A.R.; Czibik, G.; Liao, C.; et al. Resistance of Dynamin-Related Protein 1 Oligomers to Disassembly Impairs Mitophagy, Resulting in Myocardial Inflammation and Heart Failure. J. Biol. Chem. 2015, 290, 25907–25919. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Docherty, L.; Leo, V.; Towlson, C.; Neilan, M.; Steeples, V.; Lygate, C.A.; Hough, T.; Townsend, S.; Williams, D.; et al. A Mutation in the Mitochondrial Fission Gene Dnm1l Leads to Cardiomyopathy. PLoS Genet. 2010, 6, e1001000. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fernandez-Sanz, C.; Sheu, S.S. Regulation of Mitochondrial Bioenergetics by the Non-Canonical Roles of Mitochondrial Dynamics Proteins in the Heart. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased Production of Reactive Oxygen Species in Hyperglycemic Conditions Requires Dynamic Change of Mitochondrial Morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Hoque, A.; Sivakumaran, P.; Bond, S.T.; Ling, N.X.Y.; Kong, A.M.; Scott, J.W.; Bandara, N.; Hernandez, D.; Liu, G.S.; Wong, R.C.B.; et al. Mitochondrial Fission Protein Drp1 Inhibition Promotes Cardiac Mesodermal Differentiation of Human Pluripotent Stem Cells. Cell. Death Discov. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Rosdah, A.A.; K Holien, J.; Delbridge, L.M.; Dusting, G.J.; Lim, S.Y. Mitochondrial Fission—A Drug Target for Cytoprotection Or Cytodestruction? Pharmacol. Res. Perspect. 2016, 4, e00235. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor Mdivi-1 is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell. 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Czarnowska, E.; Bierla, J.B.; Toczek, M.; Tyrankiewicz, U.; Pajak, B.; Domal-Kwiatkowska, D.; Ratajska, A.; Smolenski, R.T.; Mende, U.; Chlopicki, S. Narrow Time Window of Metabolic Changes Associated with Transition to Overt Heart Failure in Tgaq*44 Mice. Pharmacol. Rep. 2016, 68, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., 2nd; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial Oxidative Stress Mediates Angiotensin II-Induced Cardiac Hypertrophy and Galphaq Overexpression-Induced Heart Failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Wang, F.; Yang, P.; Wang, X.; Xu, R.; Chen, J.; Yuan, Y.; Lu, Z.; Duan, J. Mitochondrial Fission is Required for Angiotensin II-Induced Cardiomyocyte Apoptosis Mediated by a Sirt1-p53 Signaling Pathway. Front. Pharmacol. 2018, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gong, G.; Wang, X.; Wei-LaPierre, L.; Cheng, H.; Dirksen, R.; Sheu, S.S. Mitochondrial Flash: Integrative Reactive Oxygen Species and pH Signals in Cell and Organelle Biology. Antioxid. Redox Signal. 2016, 25, 534–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Del Re, D.P.; Xiang, S.Y.; Zhao, X.; Florholmen, G.; Brown, J.H. Revisited and Revised: Is RhoA always a Villain in Cardiac Pathophysiology? J. Cardiovasc. Transl. Res. 2010, 3, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Rothermel, B.A. Calcineurin Signaling in the Heart: The Importance of Time and Place. J. Mol. Cell. Cardiol. 2017, 103, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Wang, X.; Ke, Y.; Solaro, R.J. Regulation of Ca2+ Transient by PP2A in Normal and Failing Heart. Front. Physiol. 2015, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Gotz, J.; Feany, M.B. Tau Promotes Neurodegeneration Via DRP1 Mislocalization in Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM Kinase-2 Induces Programmed Necrotic Neuronal Death Via Dysfunction of DRP1-Mediated Mitochondrial Fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial Calcium and the Regulation of Metabolism in the Heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O-Uchi, J.; Jhun, B.S.; Hurst, S.; Bisetto, S.; Gross, P.; Chen, M.; Kettlewell, S.; Park, J.; Oyamada, H.; Smith, G.L.; et al. Overexpression of Ryanodine Receptor Type 1 Enhances Mitochondrial Fragmentation and Ca2+-Induced ATP Production in Cardiac H9c2 Myoblasts. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1736. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Mitochondrial Membrane Potential and Aging. Aging Cell. 2004, 3, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-Optimized ROS Balance: A Unifying Hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.Q.; Akhtar, M.S.; Akhtar, M.; Ansari, S.H.; Ali, J.; Haque, S.E.; Najmi, A.K. Benidipine Prevents Oxidative Stress, Inflammatory Changes and Apoptosis Related Myofibril Damage in Isoproterenol-Induced Myocardial Infarction in Rats. Toxicol. Mech. Methods 2015, 25, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basanez, G.; Meda, P.; et al. Membrane Remodeling Induced by the Dynamin-Related Protein Drp1 Stimulates Bax Oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhaszova, M.; Zorov, D.B.; Yaniv, Y.; Nuss, H.B.; Wang, S.; Sollott, S.J. Role of Glycogen Synthase Kinase-3beta in Cardioprotection. Circ. Res. 2009, 104, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Sips, P.Y.; Kai, S.; Kida, K.; Ikeda, K.; Hirai, S.; Moazzami, K.; Jiramongkolchai, P.; Bloch, D.B.; Doulias, P.T.; et al. S-Nitrosylation of Calcium-Handling Proteins in Cardiac Adrenergic Signaling and Hypertrophy. Circ. Res. 2015, 117, 793–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, C.R.; Foster, C.R.; Yakoob, S.; Dalal, S.; Joyner, W.L.; Singh, M.; Singh, K. Exogenous Ubiquitin Modulates Chronic Beta-Adrenergic Receptor-Stimulated Myocardial Remodeling: Role in Akt Activity and Matrix Metalloproteinase Expression. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1459–H1468. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart Against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
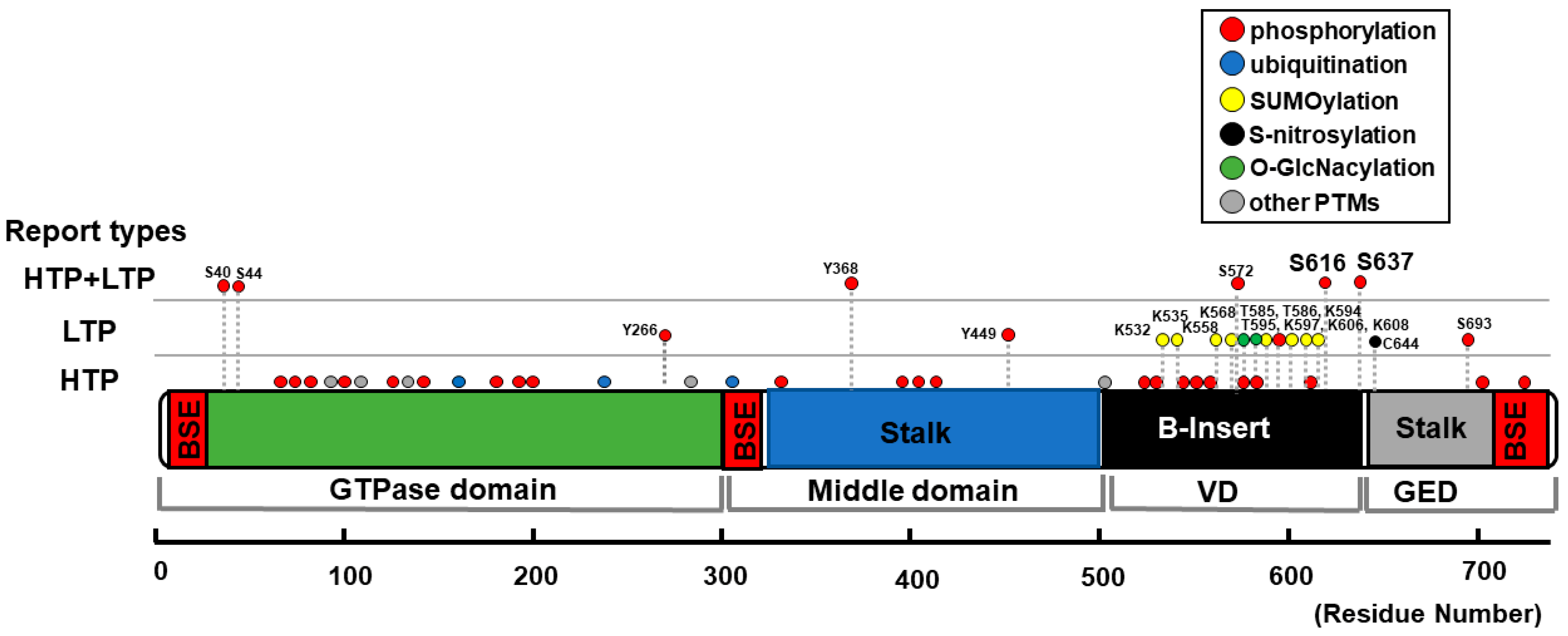
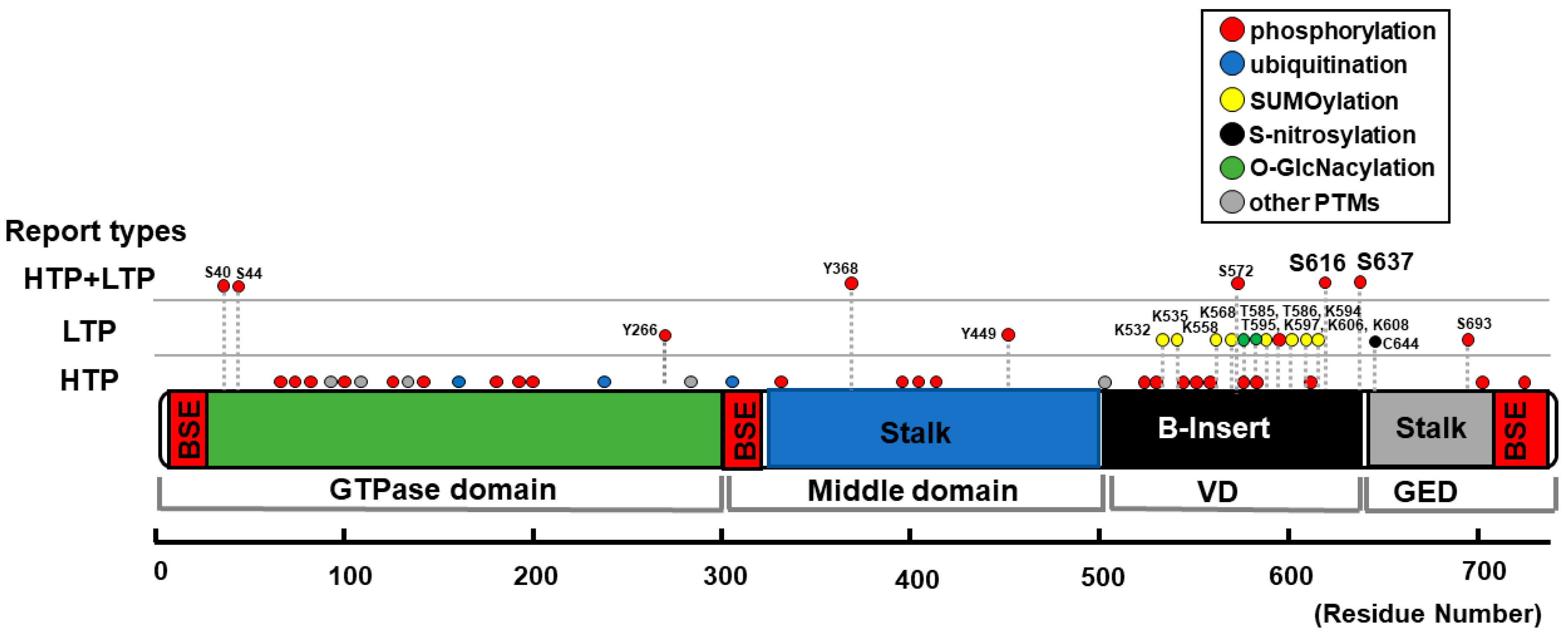
| Position | Type of Modification | Type of Reports | Upstream Molecules | Effect on Mitochondrial Morphology | Detected/Tested in CMs? |
|---|---|---|---|---|---|
| S40 S44 | phosphorylation | HTP + LTP | GSK3β [120] | phosphorylation → fragmentation [120] | No (primary cultured hippocampus neurons [120]) |
| Y266 Y368, Y449 | phosphorylation | LTP only or HTP + LTP | c-Abl [121] | phosphorylation → fragmentation [121] | No (neuron-specific c-Abl knockout mice [121]) |
| K532 K535 K558 K568 K594 K597 K606 K608 | SUMOylation | LTP | Ubc9 [122] MAPL [123,124] SENP5 [125,126,127] | SUMOylation → fragmentation [122,123,124] deSUMOylation → elongation [125] → enlarged and swollen mitochondria [127] → fragmentation [126] | Yes [127] |
| S572 | phosphorylation | HTP + LTP | CDK5 [128] | phosphorylation → fragmentation [128] dephosphorylation → elongation [128] | No (cerebellar granule neurons [128]) |
| T585 T586 | O-GlcNAcylation | LTP | N.D. (O-GlcNAc-transferase?) | O-GlcNAcylation → fragmentation [129] | Yes [129] |
| T595 | phosphorylation | LTP | LRRK2-G2019S [130] | phosphorylation → fragmentation [130] dephosphorylation → elongation [130] | No (HeLa cells, HEK293 cells, human fibroblasts [130]) |
| S616 | phosphorylation | HTP + LTP | CDK1/cyclin B [131,132,133,134] CDK5 [135] ERK1/2 [103,136,137,138]. PKCδ [133,139] CaMKII [57,140] ROCK [141] PP2A [142] Rip1 [143] | phosphorylation → fragmentation [103,133,136,137,138,142] → elongation [135] dephosphorylation → elongation [133,139] → fragmentation [135] S616D → increased fission event rate [140] → no significant changes [144] → elongation [135] S616A → elongation [131] → fragmentation [56,135] → no significant changes [132,137,139,141,144] | Yes [56,57,141,143] (See also foot notes for 1 [57], 2 [103], 3 [133], and 4 [56]) |
| S637 | phosphorylation | HTP + LTP | PKA [58,145,146,147] CaMKIα [148] ROCK1 [149] PKD [56] Pim-1 [150] CaN [58,60,133,144,151,152] PP2A [153,154] | phosphorylation → fragmentation [56,148,149] → elongation [58,145,146,147,152,153,154] dephosphorylation → fragmentation [60,133,151,153,154] S637D → elongation [58,144,145,152] → fragmentation [148] S637A → fragmentation [58,144,152] → elongation [148] → no significant changes [56] | Yes [56,58,60,150] (see also foot notes for 3 [133], and 5 [150]) |
| C644 | S-Nitrosylation | LTP | NO [155,156,157] | S-Nitrosylation → fragmentation [156,157,158] → no significant changes [155] | No (Neurons [156,157,158], human brain tissue [155]) |
| S693 | phosphorylation | HTP + LTP | GSK3β [159] | phosphorylation → elongation [159] dephosphorylation → fragmentation [159] | No (HeLa cells, HEK293 cells [159]) |
| N.D. | ubiquitination | HTP + LTP | MARCH5 [160,161,162,163] Parkin [164,165] | ubiquitination → degradation of DLP1, followed by elongation [160,161,164,165] → recruitment of DLP1 to the OMM followed by fragmentation [162,163] | No (HeLa cells [160,162,164], COS7 cells [161], RGC5 cells [163], SH-SY5Y cells [164,165]. (See also foot note for 6 [166]) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jhun, B.S.; O-Uchi, J.; Adaniya, S.M.; Cypress, M.W.; Yoon, Y. Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology. Antioxidants 2018, 7, 195. https://doi.org/10.3390/antiox7120195
Jhun BS, O-Uchi J, Adaniya SM, Cypress MW, Yoon Y. Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology. Antioxidants. 2018; 7(12):195. https://doi.org/10.3390/antiox7120195
Chicago/Turabian StyleJhun, Bong Sook, Jin O-Uchi, Stephanie M. Adaniya, Michael W. Cypress, and Yisang Yoon. 2018. "Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology" Antioxidants 7, no. 12: 195. https://doi.org/10.3390/antiox7120195
APA StyleJhun, B. S., O-Uchi, J., Adaniya, S. M., Cypress, M. W., & Yoon, Y. (2018). Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology. Antioxidants, 7(12), 195. https://doi.org/10.3390/antiox7120195