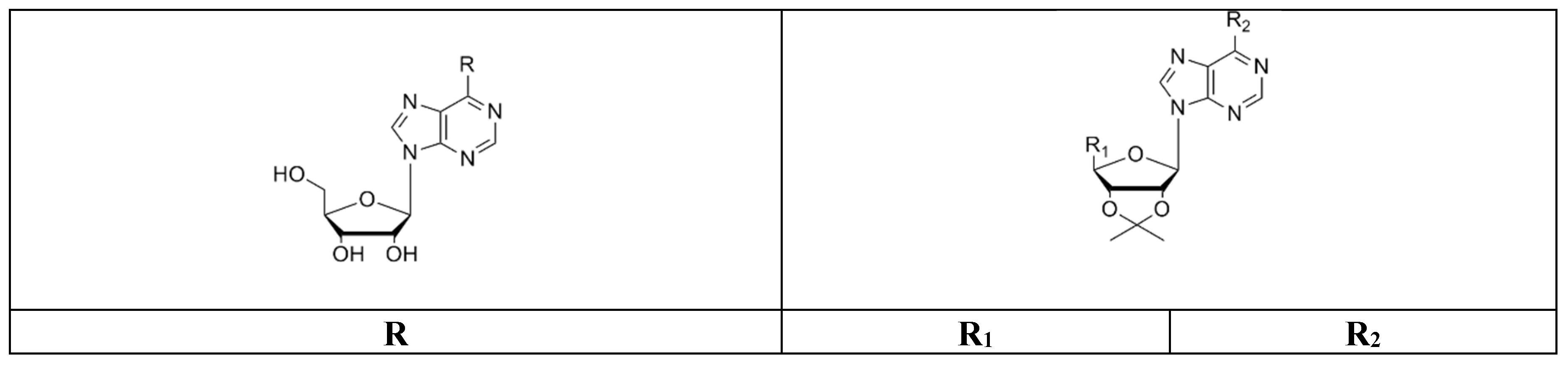
2.2. Synthetic Procedures
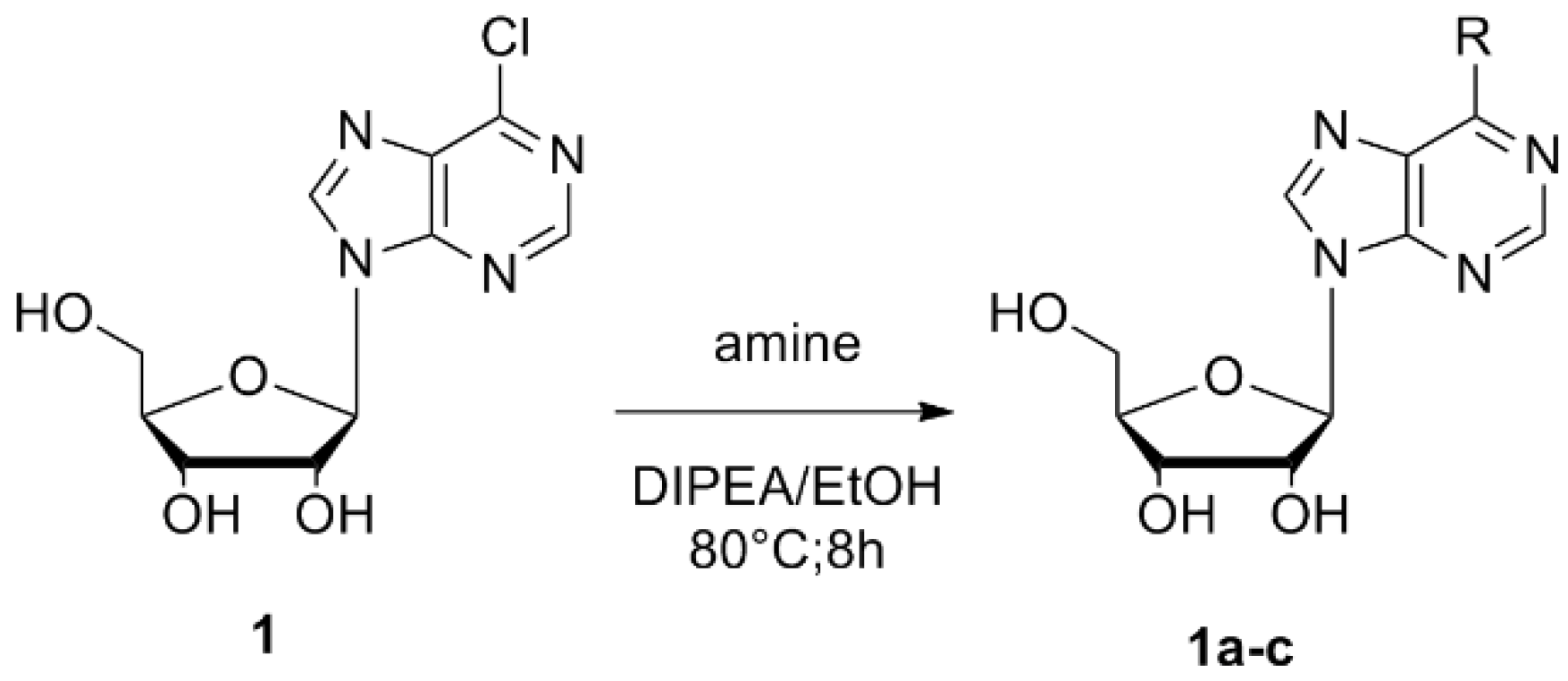
Compounds were prepared by condensation of 6-chloropurine riboside (6-chloro-9-b-d-ribofuranosyl-9H-purine) (1) with amines containing linear and branched chains as well as aromatic amines by nucleophilic aromatic substitution. Using this approach, we obtained the following compounds:
Compounds
1a–
c: these compounds were obtained by nucleophilic attack of the respective amine over the adenosine starting reagent using
N,
N-diisopropylethylamine (DIPEA) as a Lewis base (
Scheme 1). The nucleoside derivatives were obtained through semisynthetic methods described previously by Ottria et al. in 2010 [
24]. In a round-bottomed flask, the 6-chloropurine riboside (0.35 mmol,
1) was dissolved in absolute EtOH or dimethylformamide (DMF) followed by the addition DIPEA (1.05 mmol) and the appropriate amine (4.5 mmol). The mixture was refluxed at 80 °C with stirring for 8 h. The reaction mixture was cooled down to room temperature. The solvent was removed by filtration (once we had a solid precipitate) or under vacuum to leave a residue that was further analyzed by TLC. The residue was washed with hexane, dried, and purified by SiO
2 column chromatography (CH
2Cl
2-MeOH, 97:3). In some cases, the addition of dry Et
2O was used to precipitate DIPEACl (
N,
N-diisopropylethylamine chloride), which was then filtered off. The crude residue obtained after evaporation was purified by column chromatography.
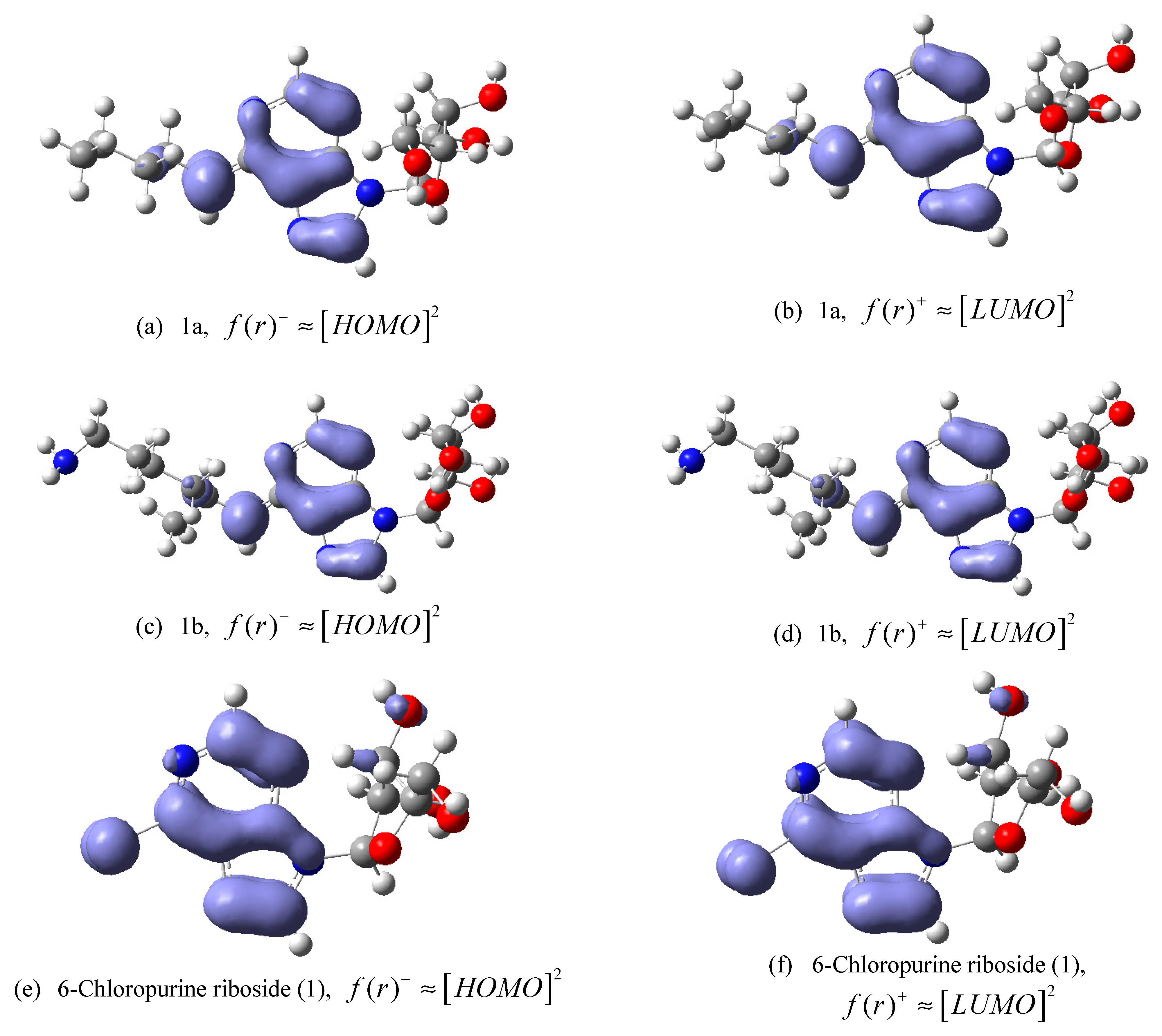
2-(6-butylamino-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol (
1a). Reagents: 6-chloropurine riboside (
1) and butylamine. Amorphous white solid with 43.17% yield; mp 162–165 °C.
1H NMR (DMSO-
d6, 400 MHz); δ: 8.36 (s, 1H, CH-Ar purine); 8.15 (s, 1H, CH-Ar purine);7.83 (s, 1H, NH); 5.90 (d,
J = 6.11 Hz, 1H, CH-1′); 5.44 (s, 2H, 3′-OH, 5′-OH); 5.17 (s, 1H, 2′-OH); 4.61 (s, 1H, CH-2′); 4.18 (s, 1H, CH-3′); 3.93 (d,
J = 3.93, 1H, CH-4′); 3.64 (m, 2H, CH
2-5′);3.40 (s, 2H, CH
2-R); 1.56 (m, 2H, CH
2-R); 1.29 (m, 2H, CH
2-R); 0.87 (t,
J = 7.15 Hz,3H, CH
3-R).
13C NMR (DMSO-
d6, 100 MHz) δ: 155.16, 152.83, 148.64, 140.16, 120.18, 86.39, 83.43, 73.93, 71.15, 62.14, 31.63, 29.47, 20.03, 14.20. IR (KBr) λ/cm
−1: 3444, 3414, 2955, 2926, 1630, 1485, 1340, 1218. Anal. Cal. C
14H
21N
5O
4: C = 51.92%, H = 6.49%, N = 21.63%. Compound
1a was reported previously [
24].
2-[6-(5-amino-2-methyl-pentylamino)-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol (1b). Reagents: 6-chloropurine riboside (1) and 1,5-diamine-2-methylpentane. Yellow solid, 48.09% yield; mp 145–148 °C. 1H NMR (DMSO-d6, 400 MHz, δ) 8.32 (m, 1H, CH-Ar purine), 8.17 (d, J = 7.83 Hz, 1H, CH-Ar purine), 7.86 (s, 1H, NH); 5.88 (d, J = 6.11 Hz, 1H, CH-1′), 5.48 (m, 1H, 2′-OH), 5.45 (m, 1H, 3′-OH), 5.22 (m, 1H, 5′-OH), 4.60 (t, J = 5.38 Hz, 1H, CH-2′), 4.14 (pseudo t, J = 4.65 Hz, J = 3.18 Hz, 1H, CH-3′), 3.95 (q, J = 3.18 Hz, 1H, CH-4′), 3.65 (m, 2H, CH2-5′), 3.54 (dd, J = 11.98 Hz, J = 3.42 Hz, 2H, CH2-R), 3.15 (s, 2H, CH2-R), 1.77 (s, 2H, NH2-R), 1.62 (m, 1H, CH-R), 1.15 (m, 2H, CH2-R), 0.88 (m, 3H, CH3-R). 13C NMR (DMSO-d6, 100 MHz, δ) 152.83, 148.78, 140.53, 140.08, 120.11, 88.35, 86.35, 73.99, 71.10, 62.10, 46.98, 45.18, 31.88, 31.20, 26.60, 17.83. IR (KBr) λ/cm−1: 3384, 2921, 2850, 1626, 1466, 1232. Anal. Cal. C16H26N6O4: C = 52.40%, H = 7.1%, N = 22.92%.
2-[6-(3-propylamino)-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol (1c). Reagents: 6-chloropurine riboside (1) and 1,3-diaminepropane. Yellow solid, 61.47% yield; mp 184–186 °C. 1H NMR (DMSO-d6, 400 MHz, δ) 8.33 (s, 1H, CH-Ar purine); 8.18 (s, 1H, CH-Ar purine); 7.96 (s, 1H, NH), 5.88 (d, J = 6.11 Hz, 1H, CH-1′), 4.61 (t, J = 5.50 Hz, 1H, CH-2′), 4.16 (dd, J = 4.52 Hz, J = 3.30 Hz, 1H, CH-3′), 3.96 (m, 1H, CH-4′), 3.66 (m, 1H, CH-5′), 3.56 (d, J = 3.67 Hz, 1H, CH-5′), 3.52 (d, J = 3.42 Hz, 2H, CH2-R), 2.62 (t, J = 6.36 Hz, 2H, CH2-R), 1.66 (m, 2H, CH2-R). 13C NMR (DMSO-d6, 100 MHz, δ) 155.14, 152.83, 148.65, 140.12, 120.22, 88.46, 86.38, 74.04, 71.08, 62.11, 39.27, 38.92, 32.03. IR (KBr) λ/cm−1: 3372, 3340, 3146, 2940, 2860, 1629, 1587, 1477, 1297, 1219. Anal. Cal. C13H20N6O4: C = 48.10%, H = 6.17%, N = 25.90%.
Compounds
2 to
5: These derivatives of adenosine were obtained by successive modifications of the commercial precursor 6-chloropurine riboside (
1) in the ribose ring and by nucleophilic substitution at the
N6-position of the purine ring, protection of vicinal diols 2′-OH and 3′-OH (compound
2), and then total oxidation of the 5′-OH group to an acid group, forming compound
3. After the carboxyl group (COOH group) formation, the vicinal diols were deprotected by the action of formic acid (HCOOH) to obtain compound
4. Immediately, an amidation reaction was carried out to generate an uronamide group at the 5′-carbon position of the furanose ring. This compound was subjected to nucleophilic substitution at the
N6-position of the purine ring with 5-amino-1,3,4-thiadiazole-2-thiol, forming compound
5.
Scheme 2 shows the general procedure and the reaction conditions.
[6-(6-Chloro-purin-9-yl)-2,2-dimethyl-tetrahydro-furo[3,4-d][1,3]dioxol-4-yl]-methanol (
2): 200 mg (0.7 mmol) of the commercial precursor 6-chloropurine riboside (
1) and acetone (10 mL) were stirred at room temperature for 30 min, after which
p-toluensulfonic acid (5.57 mmol) was added. The reaction was stirred at room temperature for 3 h. The reaction progress was monitored by TLC. Sodium bicarbonate (1.5 g) was added, and the mixture was maintained under agitation. Once the reaction was finished, the solid phase was removed by filtration and washed with ethyl acetate (×2). The product was then purified by column chromatography with mixtures of CH
2Cl
2-MeOH, obtaining the compound
2. Yellow solid, 78.1% yield; mp 155–158 °C
1H NMR (CDCl
3, 400 MHz, δ) 8.72 (s, 1H, CH-Ar purine); 8.31 (s, 1H, CH-Ar purine); 6.01 (d,
J = 8.0 Hz, 1H, CH-1′); 5.16 (m, 1H, CH-2′); 4.97 (d,
J = 7.83 Hz, 1H, CH-3′); 4.52 (d,
J = 1.22 Hz, 1H, CH-4′); 3.83 (m, 2H, CH
2-5′); 5.06 (m, 1 OH); 1.62 (s, 3H, ketal); 1.35 (s, 3H, ketal).
13C NMR (CDCl
3, 100 MHz, δ) 151.6, 151.4, 148.8, 144.4, 132.5, 114.0, 93.4, 86.2, 83.2, 81.1, 62.7, 27.1, 24.8. IR (KBr) λ/cm
−1 3320, 2906, 2863, 959, 733. Anal. Cal. C
13H
15ClN
4O
4: C = 47.75%, H = 4.59%, Cl = 10.85%, N = 17.14%. Compound
2 was reported previously [
23].
6-(6-Chloro-purin-9-yl)-2,2-dimethyl-tetrahydro-furo[3,4-d][1,3]dioxole-4-carboxylic acid (
3): For oxidation of the 5′-OH, 100 mg of compound
2 (0.31 mmol) were dissolved in H
2O/CH
3CN 1:1 and placed in an ultrasound bath for 30 min. The solvent was removed by vacuum, and the residue obtained was stirred with diethyl ether (50 mL), filtered, and then dried before being purified in a SiO
2 column chromatography with mixtures of CH
2Cl
2-MeOH, obtaining the compound
3. Yellow solid, 83.5% yield; mp 209–211 °C.
1H NMR (DMSO-
d6, 400 MHz, δ) 9.20 (s, 1H, CH-Ar purine); 8.73 (s, 1H, CH-Ar purine); 6.31 (s, 1H, CH-1′); 5.28 (d,
J = 5.62, 1H, CH-2′); 5.20 (d,
J = 5.62 Hz, 1H, CH-3′); 4.54 (s, 1H, CH-4′); 1.52 (s, 3 H, ketal); 1.31 (s, 3H, ketal).
13C NMR (DMSO-
d6, 100 MHz) δ: 173.1, 151.9, 150.9, 149.1, 147.3, 131.4, 112.8, 91.7, 88.2, 84.9, 84.4, 27.2, 25.5. IR (KBr) λ/cm
−1 3409, 2990, 2937, 1592, 1336, 1206, 1086, 635. Anal. Cal. C
13H
13ClN
4O
5: C = 45.79%, H = 3.82%, Cl = 10.40%, N = 16.44%. The compound
3 was reported previously [
23].
1’-deoxy-1’-(6-chloro-9H-purin-9-yl)-β-d-ribofuranuronic acid (4): The deprotection of diols present at the 2′ and the 3′ position of the ribose was carried out by an acid hydrolysis of the cyclic ketal group. For this reaction, compound 3 was mixed in 50% formic acid (HCOOH) at 80 °C, and the product was washed and dried under vacuum. Then, the compound was purified, and a brown amorphous solid was formed with a yield of 53.02%. mp 135–140 °C. 1H NMR (DMSO-d6, 400 MHz, δ) 12.40 (s, 1H, –COOH); 8.38 (d, J = 13.94 Hz, 1H, CH-Ar purine); 8.04 (d, J = 16.00 Hz, 1H, CH-Ar purine); 6.02 (d, J = 6.36 Hz, 1H, CH-1′); 4.55 (m, 1H, CH-2′); 4.48 (m, 1H, CH-3′); 4.33 (s, 1H, CH-4′). 13C-NMR (DMSO-d6, 100 MHz, δ:) 171.23, 156.95, 155.33, 146.61, 146.10, 138.79, 87.57, 82.94, 74.34, 73.60. IR (KBr) λ/cm−1 3492, 2956, 2867, 1683, 1206, 607. Anal. Cal. C10H9ClN4O5: C = 39.91%, H = 2.99%, Cl = 11.79%, N = 18.63%.
6-[(4-amino-2,3,5-triazole) thio]-β-
d-ribofuranosyl-9
H-purine-5′-
N-ethyluronamide (
5): Compound
5 was synthesized in a reaction involving compound
3, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and hydroxybenzotriazole (HBOt). Ethylamine (EtNH
2) was immediately added, and the mixture was left stirring for 24 h at room temperature in DMF. Subsequently,
N,
N-diisopropylethylamine (DIPEA) and 5-amino-1,3,4-thiadiazole-2-thiol were added in situ to carry out the aromatic nucleophilic substitution reaction. The reaction was refluxed at 80 °C and under stirring in DMF for 8 h according to the modified procedures of Ottria et al., 2010 [
24]. The resulting product was purified and dried, forming a brown solid with 47% yield. mp 135–138 °C.
1H NMR (DMSO-
d6, 400 MHz, δ) 8.97 (s, 1H, CH-Ar purine), 8.59 (s,1H, CH-Ar purine), 7.80 (s, 1H, NH), 7.56 (s, 2H, NH
2); 6.20 (s, 1H, CH-1′), 5.21 (d,
J = 5.87 Hz, 1H, CH-2′), 5.07 (d,
J = 5.87 Hz, 1H, CH-3′), 4.38 (s, 1H, CH-4′), 2.70 (m, 2H, CH-5′), 1.42 (s, 3H, CH
3-cetal), 1.21 (s, 3H, CH
3-cetal), 1.00 (d,
J = 6.60, 3H, CH
3-R).
13C NMR (DMSO-
d6, 100 MHz, δ) 173.56, 162.79, 155.65, 151.95, 149.58, 145.97, 141.10, 130.75, 112.79, 91.34, 88.14, 84.86, 84.51, 36.26, 27.27, 25.55, 18.78. IR (KBr) λ/cm
−1 3430, 3146, 2984, 2932, 1633, 1560, 1494, 1074 cm
−1. Anal. Cal. C
17H
20N
8O
4S
2: C = 43.92%, H = 4.31%, N = 24.11%, O = 13.78%, S = 13.78%.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}