Targeting G-quadruplexes with Organic Dyes: Chelerythrine–DNA Binding Elucidated by Combining Molecular Modeling and Optical Spectroscopy

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
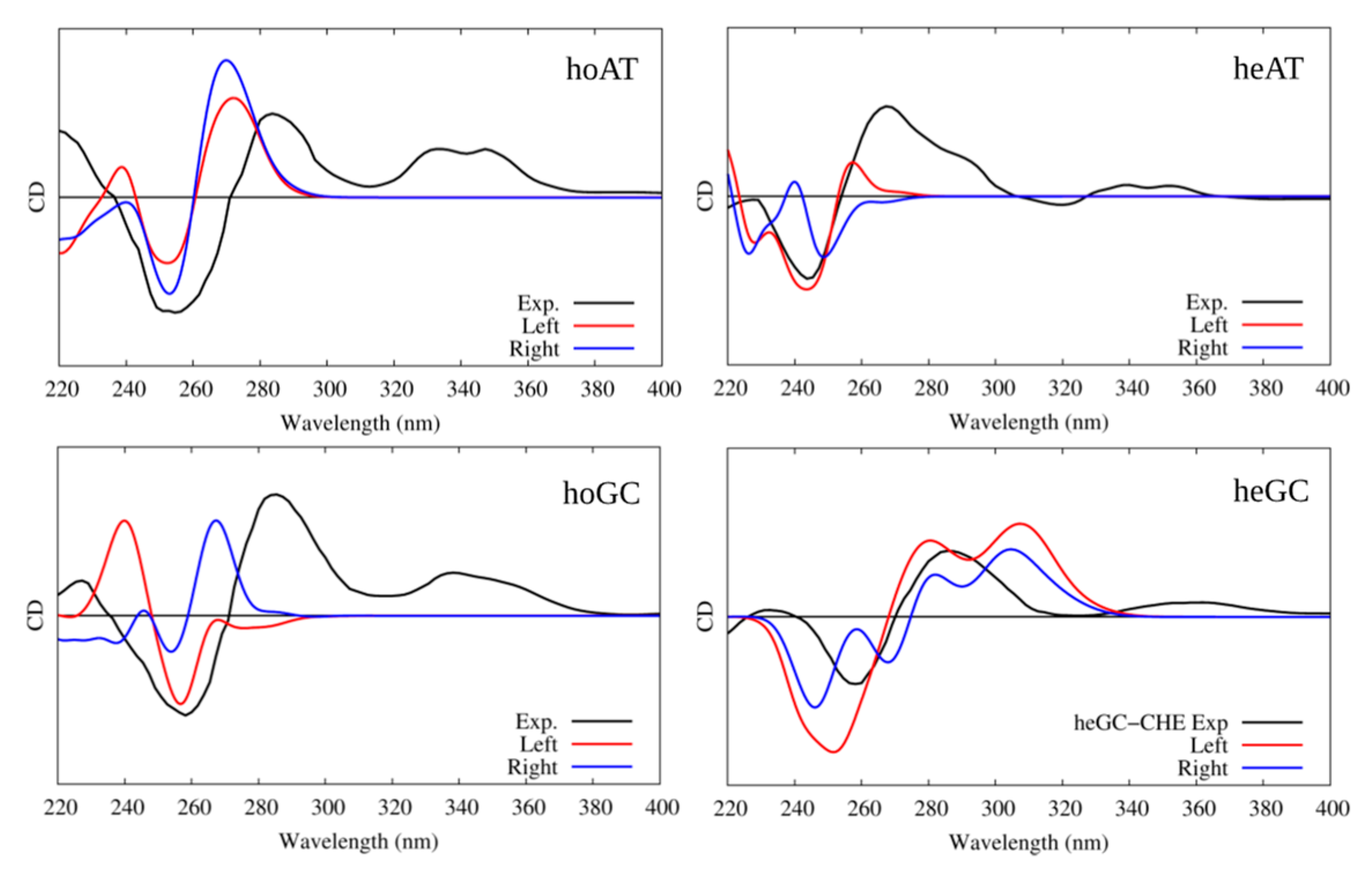
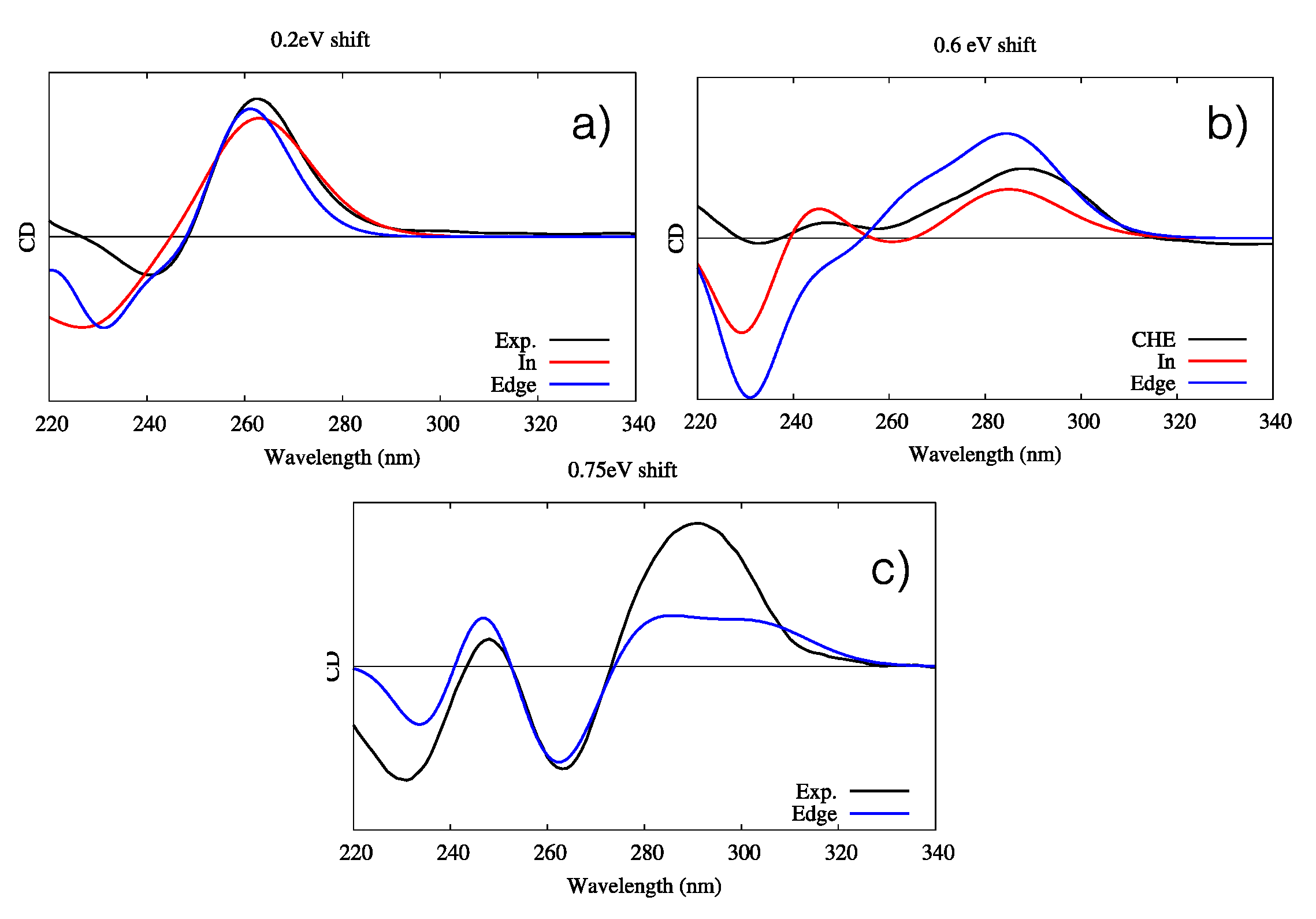
2.1. Circular Dichroism and UV-Vis Spectroscopy
2.2. Modeling and Simulation
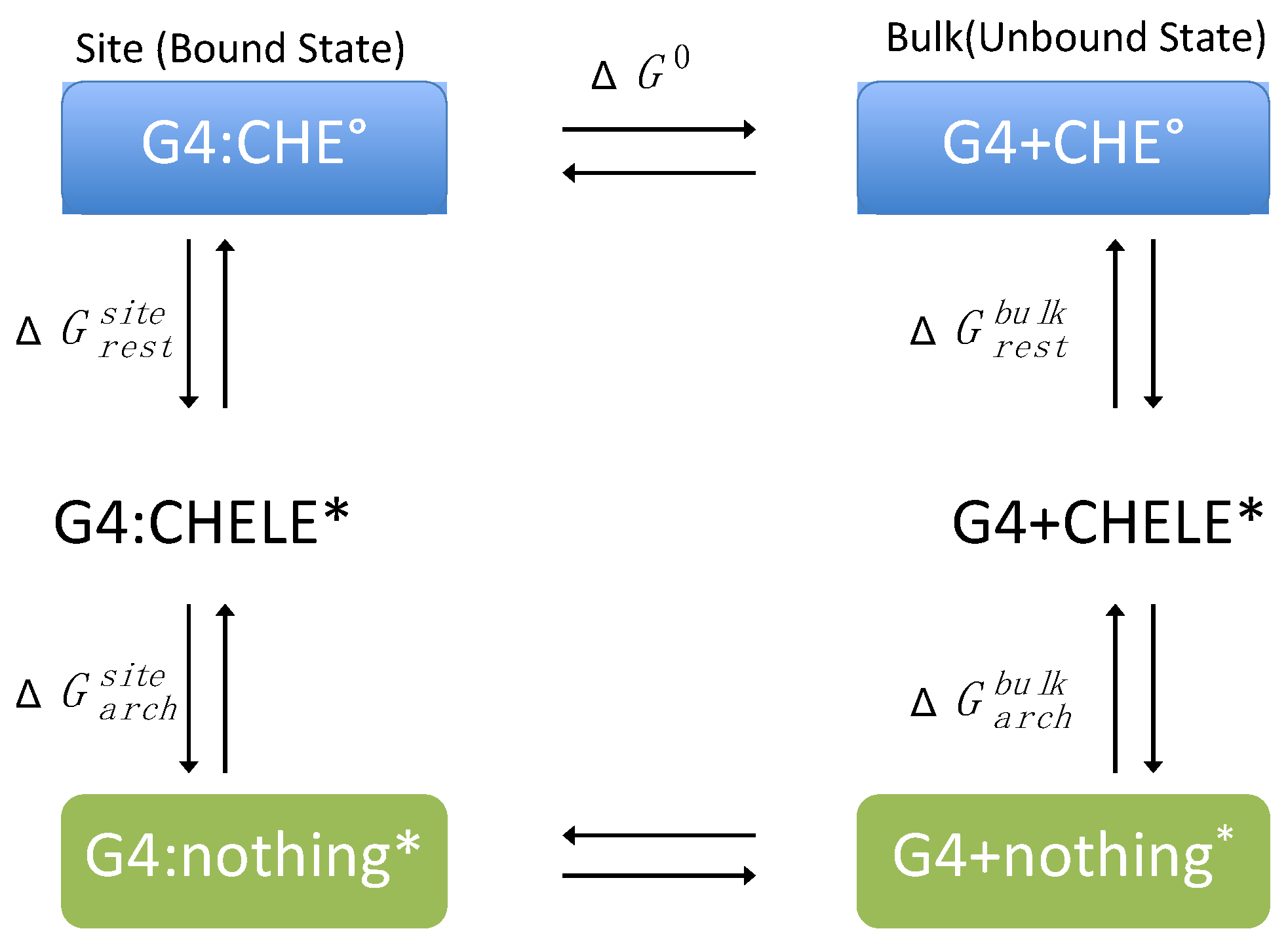
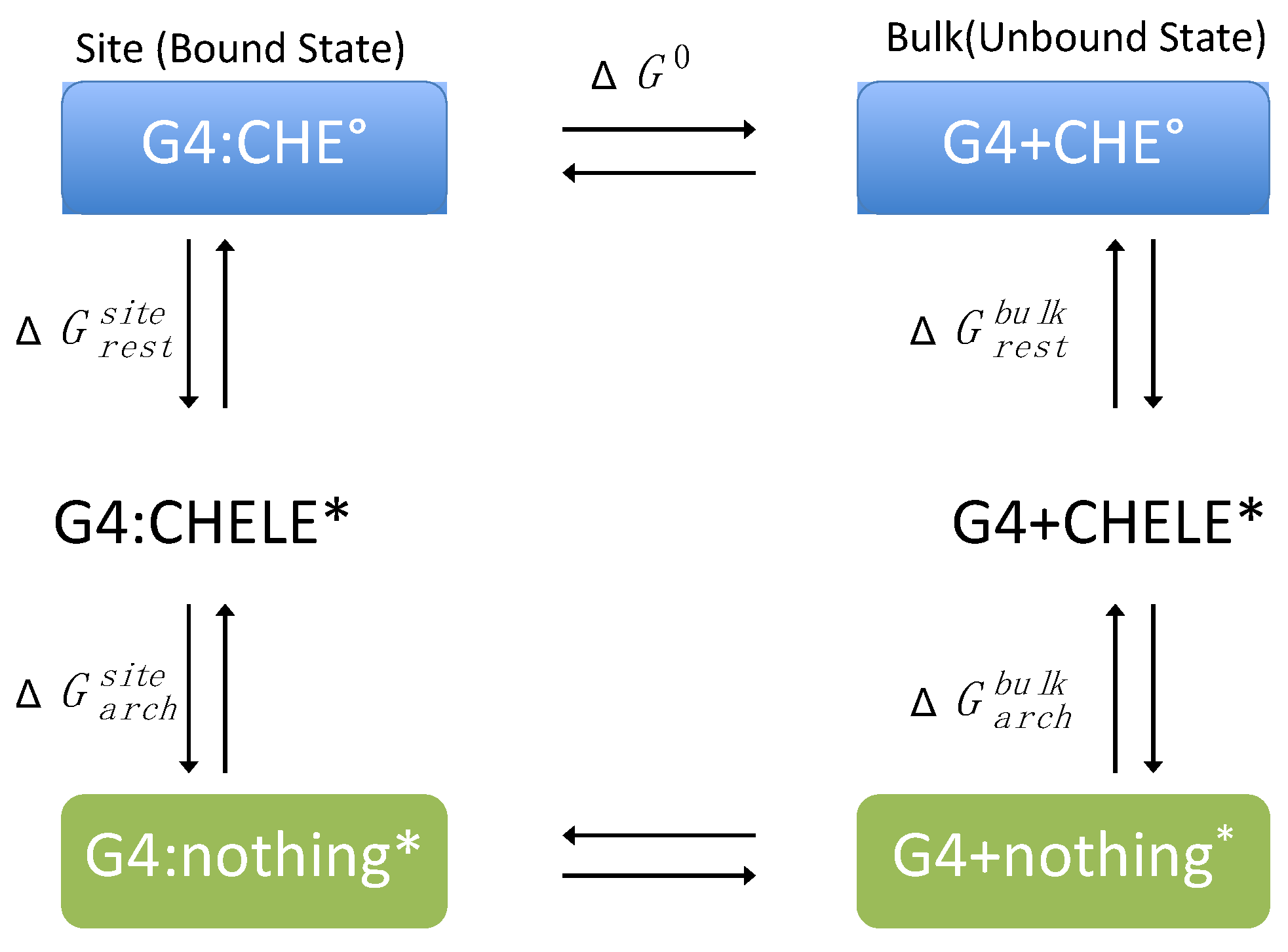
2.3. Binding Free Energy Calculation
3. Results
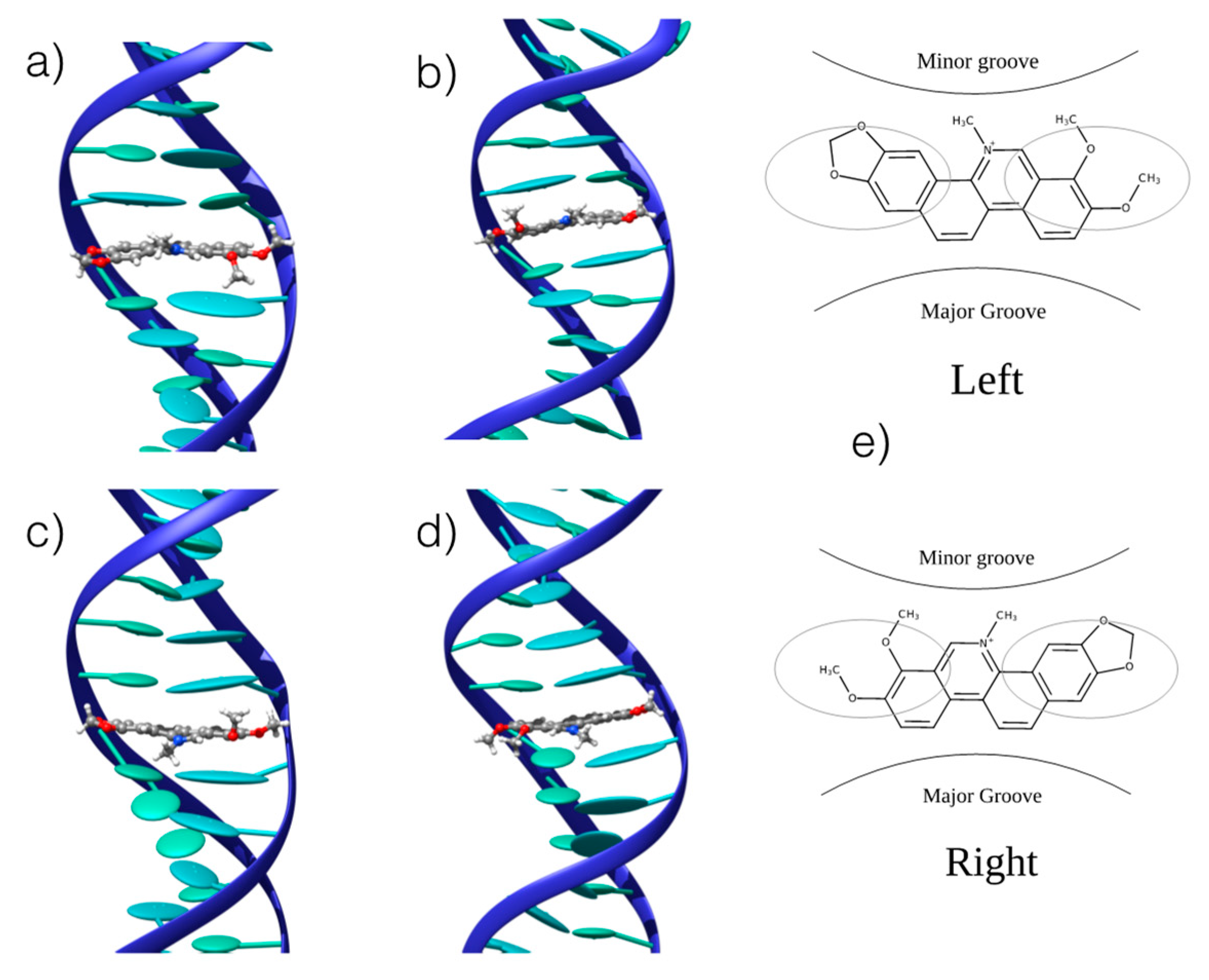
3.1. Interaction with B-DNA
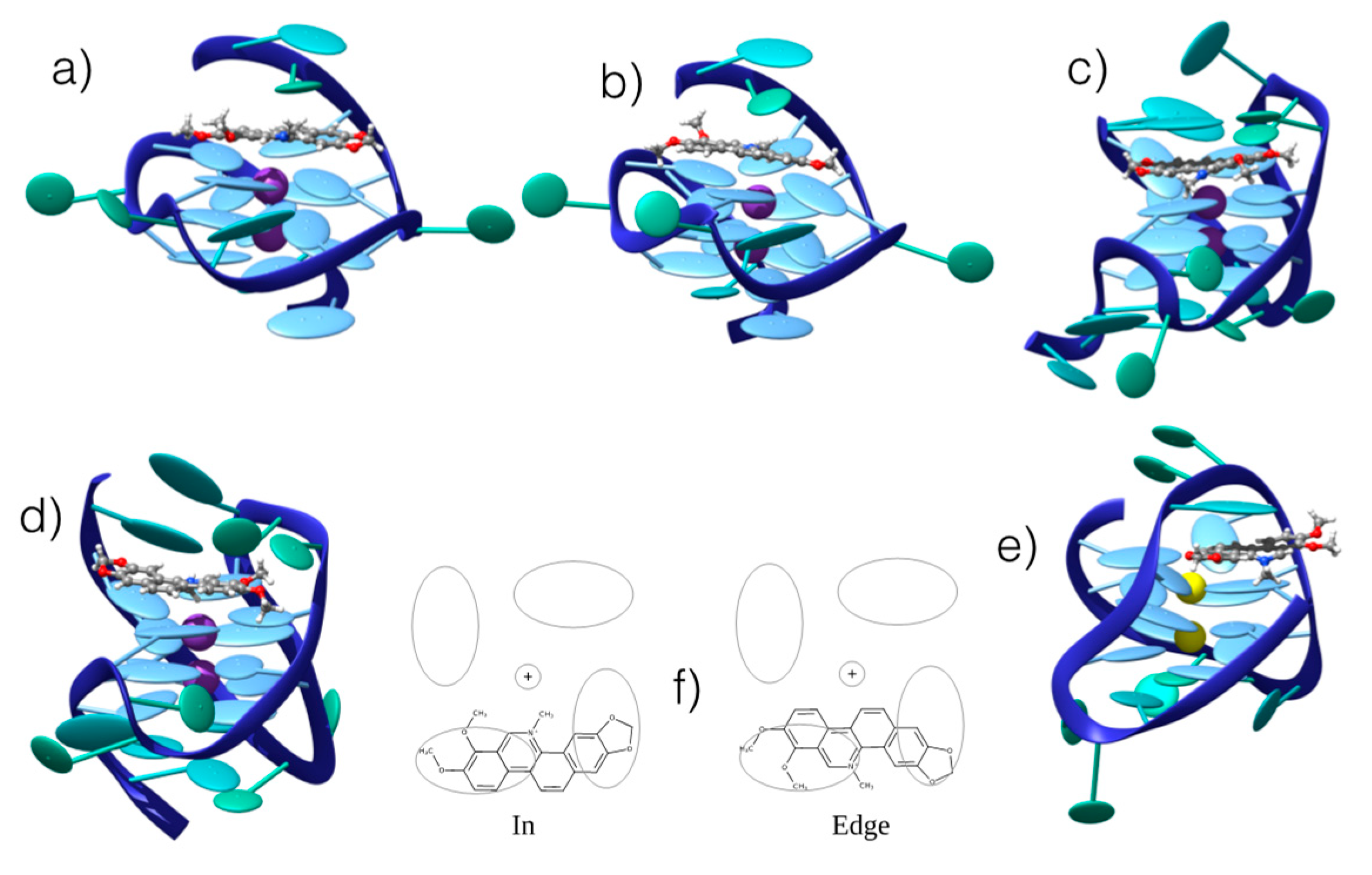
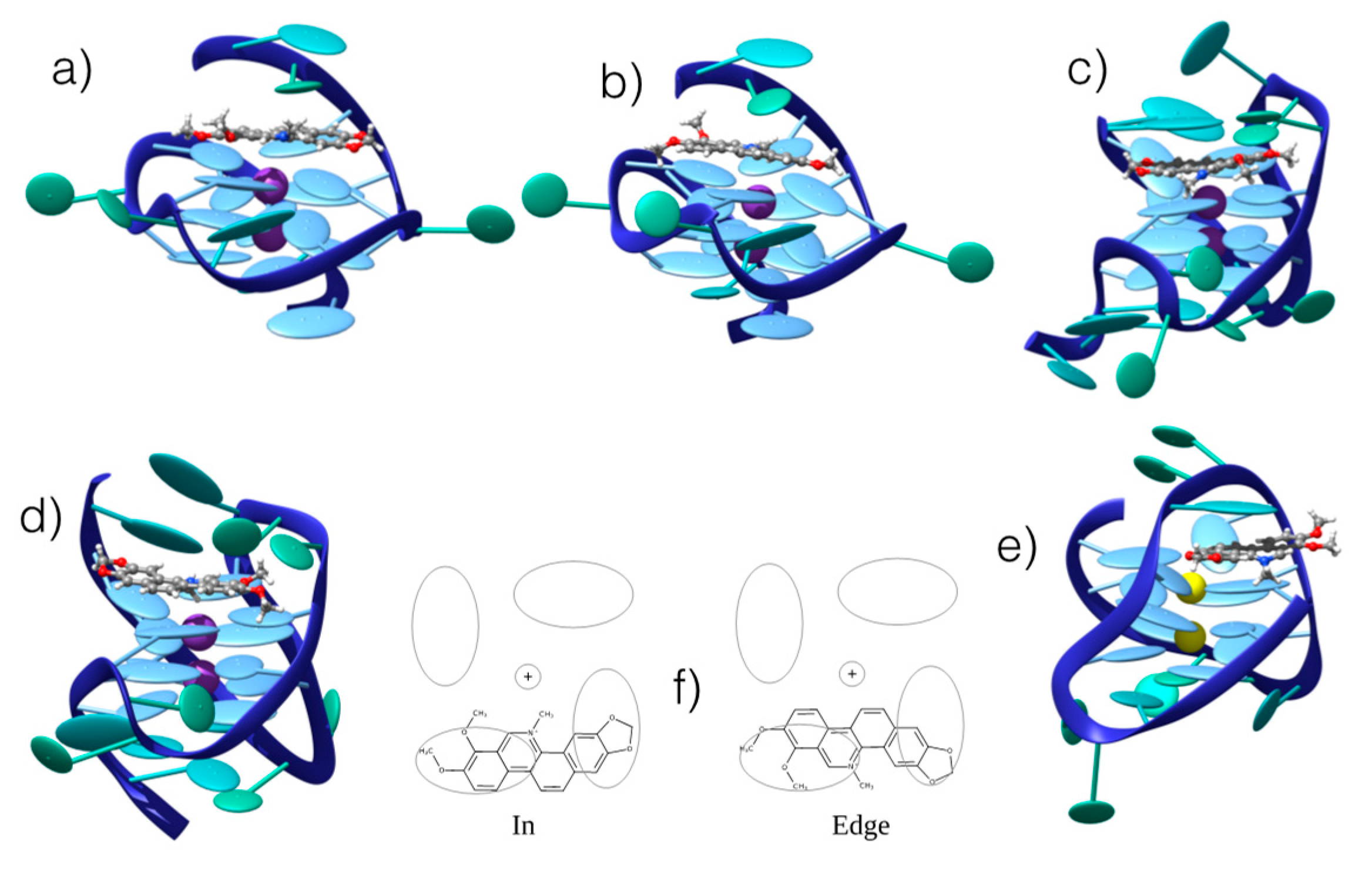
3.2. Interaction with G4 DNA
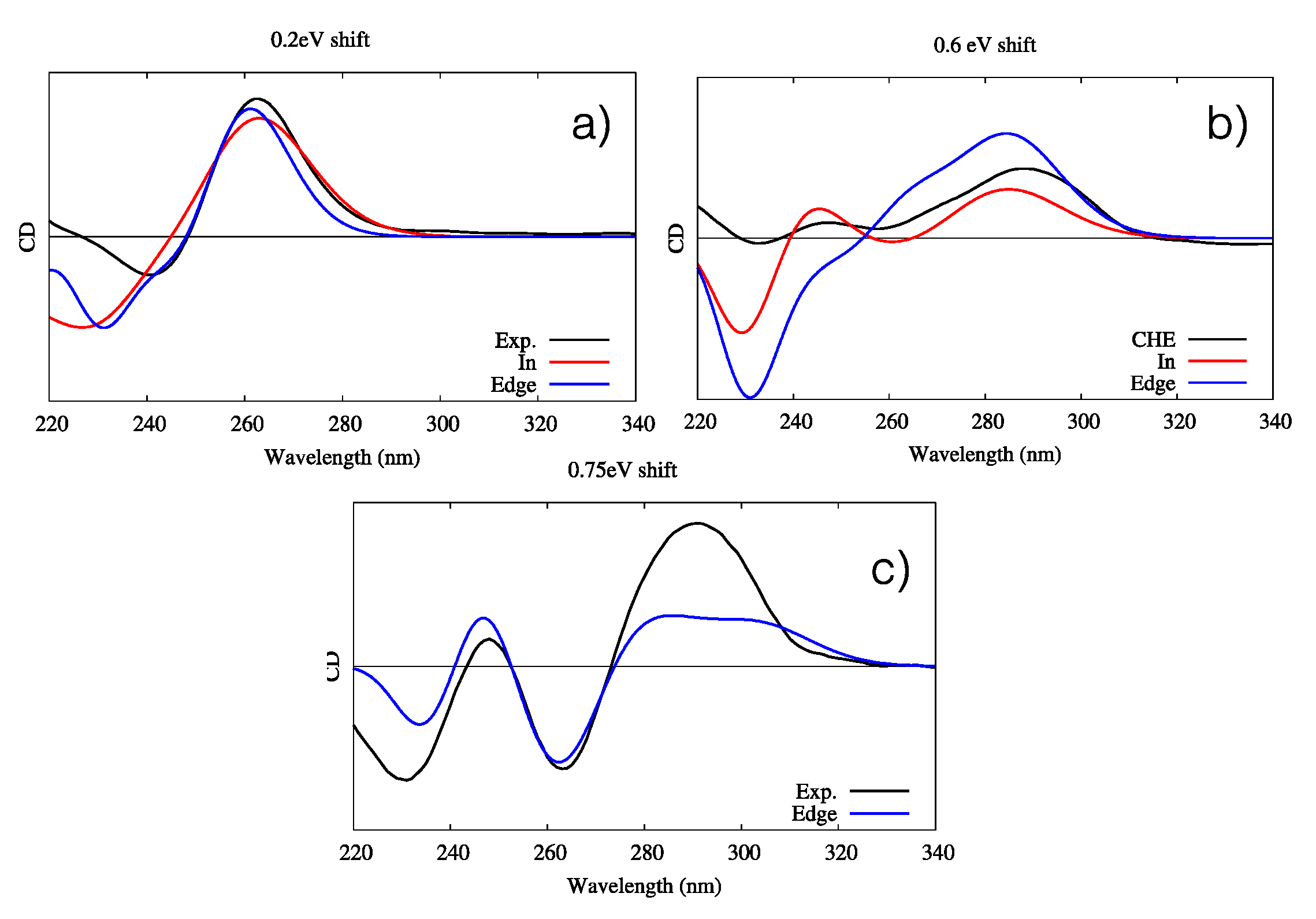
3.3. Determination of Binding Constants and Free Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Magnander, K.; Elmroth, K. Biological consequences of formation and repair of complex DNA damage. Cancer Lett. 2012, 327, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, E.; Girard, P.-M.; Francesconi, S. Unravelling UVA-induced mutagenesis. Photochem. Photobiol. Sci. 2012, 11, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Rünger, T.M.; Kappes, U.P. Mechanisms of mutation formation with long-wave ultraviolet light (UVA). Photodermatol. Photoimmunol. Photomed. 2008, 24, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Sedletska, Y.; Radicella, J.P.; Sage, E. Replication fork collapse is a major cause of the high mutation frequency at three-base lesion clusters. Nucleic Acids Res. 2013, 41, 9339–9348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.X.; Bernerd, F.; Halliday, G.M. Ultraviolet A within sunlight induces mutations in the epidermal basal layer of engineered human skin. Am. J. Pathol. 2009, 174, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef]
- Epe, B. DNA damage spectra induced by photosensitization. Photochem. Photobiol. Sci. 2012, 11, 98–106. [Google Scholar] [CrossRef]
- Viswesh, V.; Gates, K.; Sun, D. Characterization of DNA damage induced by a natural product antitumor antibiotic leinamycin in human cancer cells. Chem. Res. Toxicol. 2010, 23, 99–107. [Google Scholar] [CrossRef]
- Dumont, E.; Monari, A. Understanding DNA under oxidative stress and sensitization: The role of molecular modeling. Front. Chem. 2015, 3, 43. [Google Scholar] [CrossRef]
- Cadet, J.; Mouret, S.; Ravanat, J.L.; Douki, T. Photoinduced damage to cellular DNA: Direct and photosensitized reactions. In Proceedings of the Photochemistry and Photobiology; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2012; pp. 1048–1065. [Google Scholar]
- Tarantini, A.; Douki, T.; Personnaz, M.B.; Besombes, J.L.; Jafrezzo, J.L.; Maîtrea, A. Effect of the chemical composition of organic extracts from environmental and industrial atmospheric samples on the genotoxicity of polycyclic aromatic hydrocarbons mixtures. Toxicol. Environ. Chem. 2011, 93, 941–954. [Google Scholar] [CrossRef]
- Brinkmann, J.; Stolpmann, K.; Trappe, S.; Otter, T.; Genkinger, D.; Bock, U.; Liebsch, M.; Henkler, F.; Hutzler, C.; Luch, A. Metabolically competent human skin models: Activation and genotoxicity of benzo[a]pyrene. Toxicol. Sci. 2013, 131, 351–359. [Google Scholar] [CrossRef] [PubMed]
- De Rainho, C.R.; MacHado Corrêa, S.; Mazzei, J.-L.; Fortes Aiub, C.A.; Felzenszwalb, I. Genotoxicity of polycyclic aromatic hydrocarbons and nitro-derived in respirable airborne particulate matter collected from urban areas of Rio de Janeiro (Brazil). Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Cuquerella, M.C.; Lhiaubet-Vallet, V.; Cadet, J.; Miranda, M.A. Benzophenone photosensitized DNA damage. Acc. Chem. Res. 2012, 45, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Wibowo, M.; Roca-Sanjuán, D.; Garavelli, M.; Assfeld, X.; Monari, A. Resolving the benzophenone DNA-photosensitization mechanism at QM/MM level. J. Phys. Chem. Lett. 2015, 6, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Monari, A. Interaction of palmatine with DNA: An environmentally controlled phototherapy drug. J. Phys. Chem. B 2015, 119, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Mutlu, E.; Sharma, V.; Collins, L.; Bodnar, W.; Yu, R.; Lai, Y.; Moeller, B.; Lu, K.; Swenberg, J. The endogenous exposome. DNA Rep. 2014, 19, 3–13. [Google Scholar] [CrossRef]
- Kostova, I. Ruthenium Complexes as Anticancer Agents. Curr. Med. Chem. 2006, 13, 1085–1107. [Google Scholar] [CrossRef]
- Véry, T.; Ambrosek, D.; Otsuka, M.; Gourlaouen, C.; Assfeld, X.; Monari, A.; Daniel, C. Photophysical properties of ruthenium(ii) polypyridyl DNA intercalators: Effects of the molecular surroundings investigated by theory. Chem. Eur. J. 2014, 20, 12901–12909. [Google Scholar] [CrossRef]
- Chantzis, A.; Very, T.; Daniel, C.; Monari, A.; Assfeld, X. Theoretical evidence of photo-induced charge transfer from DNA to intercalated ruthenium (II) organometallic complexes. Chem. Phys. Lett. 2013, 578, 133–137. [Google Scholar] [CrossRef]
- Friedman, A.E.; Barton, J.K.; Chambron, J.C.; Sauvage, J.P.; Turro, N.J.; Barton, J.K. Molecular “Light Switch” for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc. 1990, 112, 4960–4962. [Google Scholar] [CrossRef]
- Regulus, P.; Duroux, B.; Bayle, P.-A.; Favier, A.; Cadet, J.; Ravanat, J.-L. Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion. Proc. Natl. Acad. Sci. USA 2007, 104, 14032–14037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Arnaiz, C.; Busto, N.; Leal, J.M.; García, B. New insights into the mechanism of the DNA/doxorubicin interaction. J. Phys. Chem. B 2014, 118, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, A.; Pirker, C.; Keppler, B.K.; Berger, W. Anticancer metal drugs and immunogenic cell death. J. Inorg. Biochem. 2016, 165, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Grant, K.B. DNA photo-cleaving agents in the far-red to near-infrared range—A review. RSC Adv. 2016, 6, 24617–24634. [Google Scholar] [CrossRef]
- Fan, W.; Huang, P.; Chen, X. Overcoming the Achilles’ heel of photodynamic therapy. Chem. Soc. Rev. 2016, 45, 6488–6519. [Google Scholar] [CrossRef] [PubMed]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Fernandez, J.M.; Bilgin, M.D.; Grossweiner, L.I. Singlet oxygen generation by photodynamic agents. J. Photochem. Photobiol. B Biol. 1997, 37, 131–140. [Google Scholar] [CrossRef]
- Zou, Q.; Zhao, H.; Zhao, Y.; Fang, Y.; Chen, D.; Ren, J.; Wang, X.; Wang, Y.; Gu, Y.; Wu, F. Effective Two-Photon Excited Photodynamic Therapy of Xenograft Tumors Sensitized by Water-Soluble Bis(arylidene)cycloalkanone Photosensitizers. J. Med. Chem. 2015, 58, 7949–7958. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; Dumont, E.; Marazzi, M.; Monari, A. Two-photon-absorption DNA sensitization via solvated electrons production: Unraveling the photochemical pathways by molecular modeling and simulation. Phys. Chem. Chem. Phys. 2016, 18, 18598–18606. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, N.G.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Parkinson, G. Telomere maintenance as a target for anticancer drug discovery. Nat. Rev. Drug Discov. 2002, 1, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex nucleic acids as targets for anticancer therapeutics. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Alenko, A.; Burrows, C.J. Zika Virus Genomic RNA Possesses Conserved G-Quadruplexes Characteristic of the Flaviviridae Family. ACS Infect. Dis. 2016, 2, 674–681. [Google Scholar] [CrossRef]
- Hognon, C.; Gebus, A.; Barone, G.; Monari, A. Human DNA Telomeres in Presence of Oxidative Lesions: The Crucial Role of Electrostatic Interactions on the Stability of Guanine Quadruplexes. Antioxidants 2019, 8, 337. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J. Howpay Manage, S.A.; Burrows, C.J. Human NEIL3 Gene Expression Regulated by Epigenetic-Like Oxidative DNA Modification. J. Am. Chem. Soc. 2019, 141, 11036–11049. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Correia, J.J.; Wang, L.; Trent, J.O.; Chaires, J.B. Not so crystal clear: The structure of the human telomere G-quadruplex in solution differs from that present in a crystal. Nucleic Acids Res. 2005, 33, 4649–4659. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Kan, Z.Y.; Wang, Q.; Yao, Y.; Liu, J.; Hao, Y.H.; Tan, Z. Human telomeric DNA forms parallel-stranded intramolecular G-quadruplex in K+ solution under molecular crowding condition. J. Am. Chem. Soc. 2007, 129, 11185–11191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-F.; Chang, T.-C. Molecular engineering of G-quadruplex ligands based on solvent effect of polyethylene glycol. Nucleic Acids Res. 2012, 40, 8711–8720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.F.; Li, M.H.; Chen, W.W.; Hsu, S.T.D.; Chang, T.C. A novel transition pathway of ligand-induced topological conversion from hybrid forms to parallel forms of human telomeric G-quadruplexes. Nucleic Acids Res. 2016, 44, 3958–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, K.S. An Overview of Chemical Processes that Damage Cellular DNA: Spontaneous Hydrolysis, Alkylation, and Reactions with Radicals; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1747–1760. [Google Scholar]
- Gattuso, H.; Spinello, A.; Terenzi, A.; Assfeld, X.; Barone, G.; Monari, A. Circular Dichroism of DNA G-Quadruplexes: Combining Modeling and Spectroscopy to Unravel Complex Structures. J. Phys. Chem. B 2016, 120, 3113–3121. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; García-Iriepa, C.; Sampedro, D.; Monari, A.; Marazzi, M. Simulating the Electronic Circular Dichroism Spectra of Photoreversible Peptide Conformations. J. Chem. Theory Comput. 2017, 13, 3290–3296. [Google Scholar] [CrossRef]
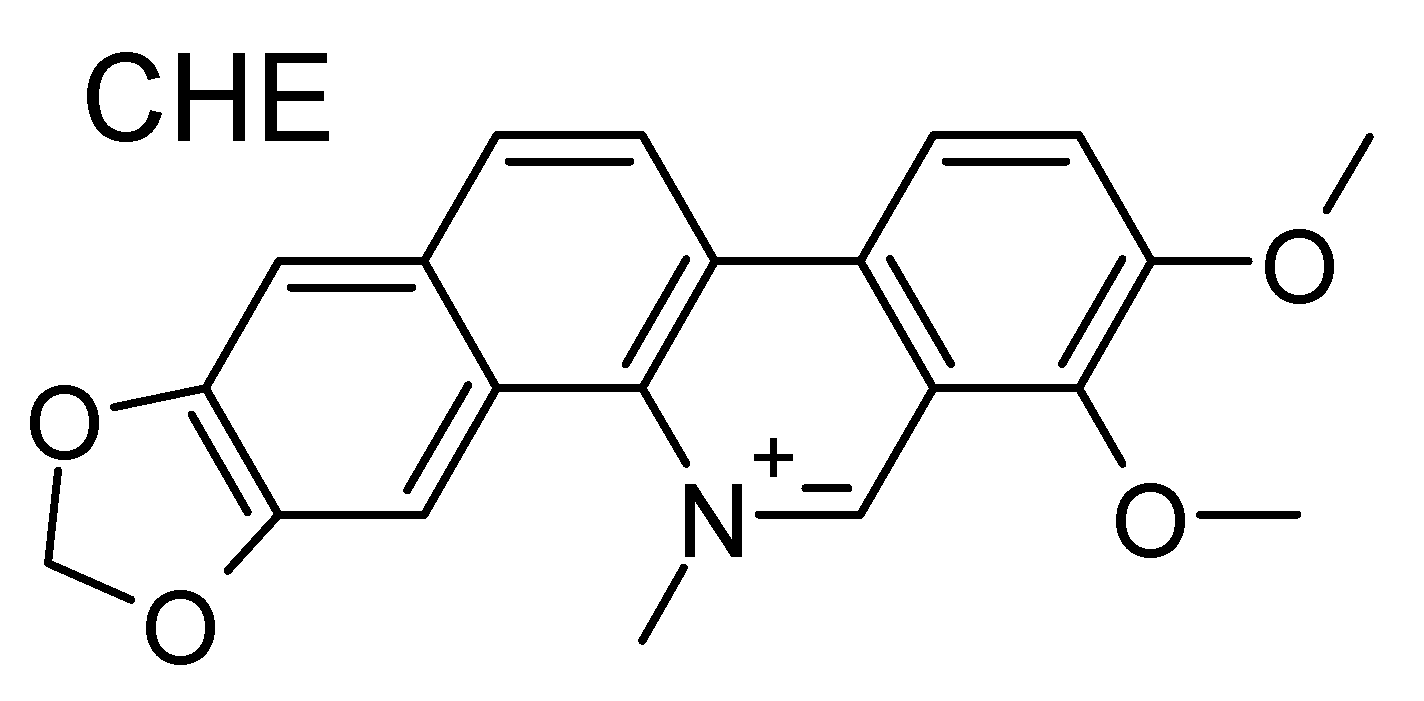
- Bai, L.P.; Hagihara, M.; Nakatani, K.; Jiang, Z.H. Recognition of chelerythrine to human telomeric DNA and RNA G-quadruplexes. Sci. Rep. 2014, 4, 6767. [Google Scholar] [CrossRef]
- Kim, J.; Lin, J.; Adam, R.M.; Lamb, C.; Shively, S.B.; Freeman, M.R. An oxidative stress mechanism mediates chelerythrine-induced heparin-binding EGF-like growth factor ectodomain shedding. J. Cell. Biochem. 2005, 94, 39–49. [Google Scholar] [CrossRef]
- He, N.; Wang, P.; Wang, P.; Ma, C.; Kang, W. Antibacterial mechanism of chelerythrine isolated from root of Toddalia asiatica (Linn) Lam. BMC Complement. Altern. Med. 2018, 18, 261. [Google Scholar] [CrossRef]
- Wu, S.; Yang, Y.; Li, F.; Huang, L.; Han, Z.; Wang, G.; Yu, H.; Li, H. Chelerythrine induced cell death through ROS-dependent ER stress in human prostate cancer cells. OncoTargets Ther. 2018, 11, 2593–2601. [Google Scholar] [CrossRef] [PubMed]
- Plazas, E.A.; Avila, M.C.; Delgado, W.A.; Patino, O.J.; Cuca, L.E. In vitro Antioxidant and Anticholinesterase Activities of Colombian Plants as Potential Neuroprotective Agents. Res. J. Med. Plants 2018, 12, 9–18. [Google Scholar] [CrossRef]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, A.E.; Zhong, Z.; Sessler, J.L.; Anslyn, E.V. Algorithms for the determination of binding constants and enantiomeric excess in complex host: Guest equilibria using optical measurements. New J. Chem. 2010, 34, 348. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of alphaγConformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Gattuso, H.; Assfeld, X.; Monari, A. Modeling DNA electronic circular dichroism by QM/MM methods and Frenkel Hamiltonian. Theor. Chem. Acc. 2015, 134, 36. [Google Scholar] [CrossRef]
- Ferré, N.; Assfeld, X.; Rivail, J.-L. Application of the local self-consistent-field method to core-ionized and core-excited molecules, polymers, and proteins: True orthogonality between ground and excited states. J. Chem. Phys. 2002, 117, 4119–4125. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revis. D.01, Gaussian Inc.: Wallingford, CT, USA, 2010.
- TINKER Molecular Modeling Software. Available online: http://dasher.wustl.edu/ffe/Tinker (accessed on 10 August 2019).
- Monari, A.; Rivail, J.-L.L.; Assfeld, X. Theoretical modeling of large molecular systems. Advances in the local self consistent field method for mixed quantum mechanics/molecular mechanics calculations. Acc. Chem. Res. 2013, 46, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Loco, D.; Jurinovich, S.; Di Bari, L.; Mennucci, B. A fast but accurate excitonic simulation of the electronic circular dichroism of nucleic acids: How can it be achieved? Phys. Chem. Chem. Phys. 2016, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipot, C.; Pohorille, A. Free Energy Calculations: Theory and Applications in Chemistry and Biology; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Gumbart, J.C.; Roux, B.; Chipot, C. Standard binding free energies from computer simulations: What is the best strategy? J. Chem. Theory Comput. 2013, 9, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Ducani, C.; Bernardinelli, G.; Högberg, B.; Keppler, B.K.; Terenzi, A. Interplay of Three G-Quadruplex Units in the KIT Promoter. J. Am. Chem. Soc. 2019, 141, 10205–10213. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA | Experimental Kb (M−1) | Experimental ΔG (kcal/mol) | Calculated ΔG (kcal/mol) |
|---|---|---|---|
| B-DNA | (1.3 ± 0.2) × 104 | −5.6 ± 0.1 | |
| Hybrid G4 (2Y9A) | (1.1 ± 0.1) × 105 | −6.9 ± 0.1 | −12.3 |
| Parallel G4 (c-MYC) | (1.0 ± 0.1) × 105 | −6.8 ± 0.02 | −13.5 |
| Antiparallel G4 (hTelo) | (2.69 ± 0.06) × 105 | −7.4 ± 0.2 | |
| Parallel G4 (hTelo + PEG) | (3.5 ± 0.9) × 105 | −7.6 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terenzi, A.; Gattuso, H.; Spinello, A.; Keppler, B.K.; Chipot, C.; Dehez, F.; Barone, G.; Monari, A. Targeting G-quadruplexes with Organic Dyes: Chelerythrine–DNA Binding Elucidated by Combining Molecular Modeling and Optical Spectroscopy. Antioxidants 2019, 8, 472. https://doi.org/10.3390/antiox8100472
Terenzi A, Gattuso H, Spinello A, Keppler BK, Chipot C, Dehez F, Barone G, Monari A. Targeting G-quadruplexes with Organic Dyes: Chelerythrine–DNA Binding Elucidated by Combining Molecular Modeling and Optical Spectroscopy. Antioxidants. 2019; 8(10):472. https://doi.org/10.3390/antiox8100472
Chicago/Turabian StyleTerenzi, Alessio, Hugo Gattuso, Angelo Spinello, Bernhard K. Keppler, Christophe Chipot, François Dehez, Giampaolo Barone, and Antonio Monari. 2019. "Targeting G-quadruplexes with Organic Dyes: Chelerythrine–DNA Binding Elucidated by Combining Molecular Modeling and Optical Spectroscopy" Antioxidants 8, no. 10: 472. https://doi.org/10.3390/antiox8100472
APA StyleTerenzi, A., Gattuso, H., Spinello, A., Keppler, B. K., Chipot, C., Dehez, F., Barone, G., & Monari, A. (2019). Targeting G-quadruplexes with Organic Dyes: Chelerythrine–DNA Binding Elucidated by Combining Molecular Modeling and Optical Spectroscopy. Antioxidants, 8(10), 472. https://doi.org/10.3390/antiox8100472