Signals Getting Crossed in the Entanglement of Redox and Phosphorylation Pathways: Phosphorylation of Peroxiredoxin Proteins Sparks Cell Signaling


Abstract
:1. Reactive Oxygen and Nitrogen Species in Biology
2. Enzymatic H2O2 Scavengers
3. Catalase and GPx
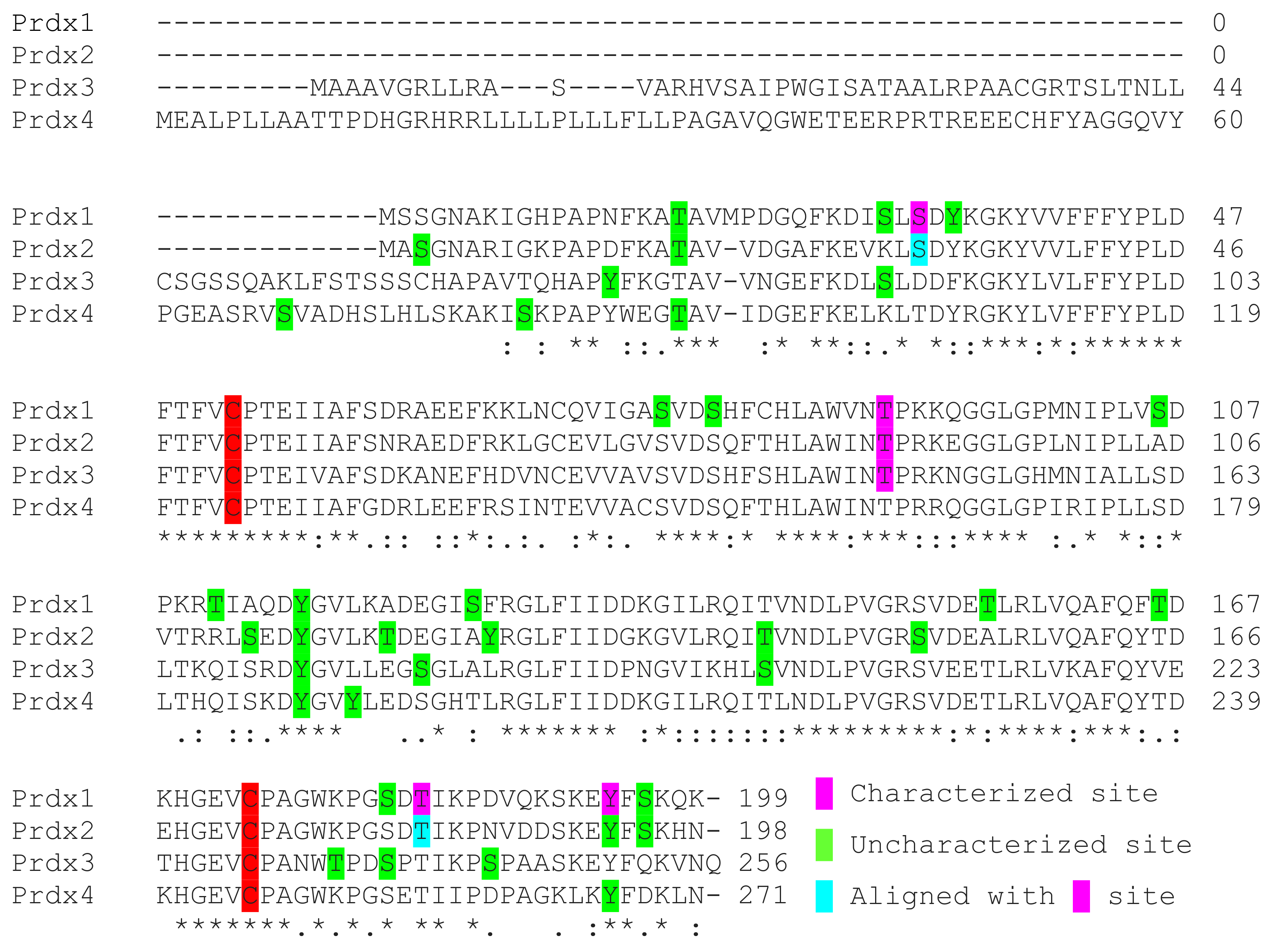
4. Prdx
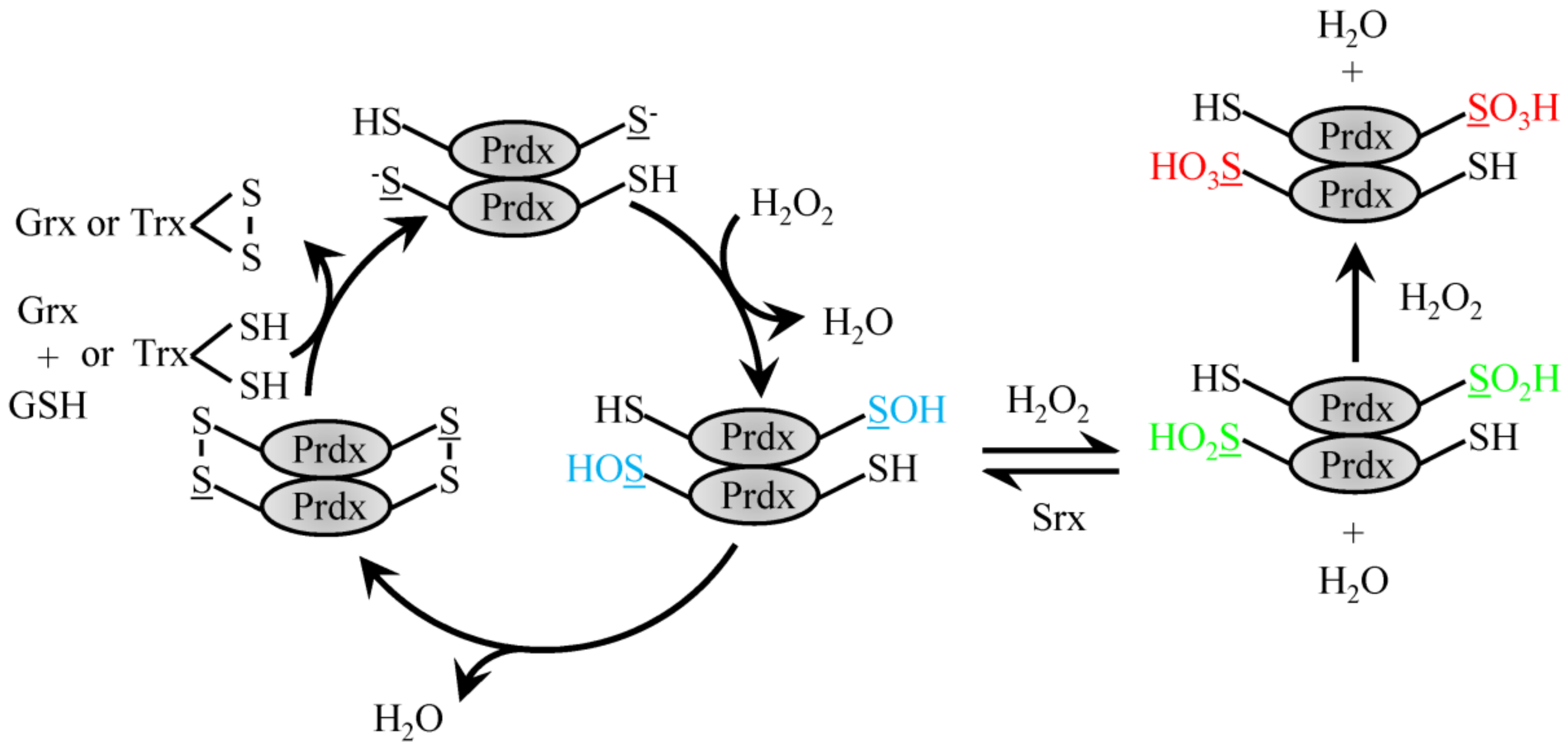
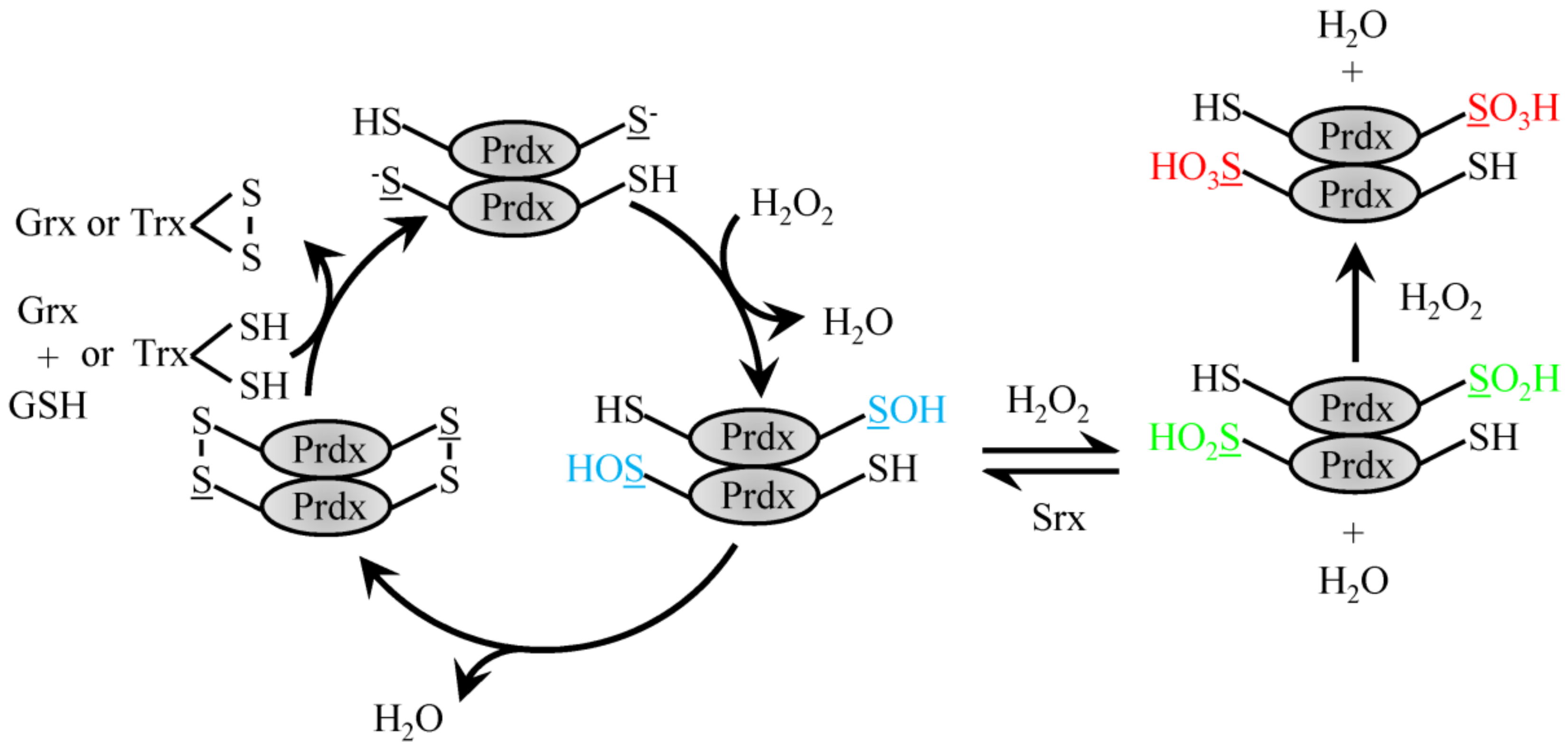
4.1. 2-Cys Typical Prdx
4.2. 2-Cys Atypical Prdx
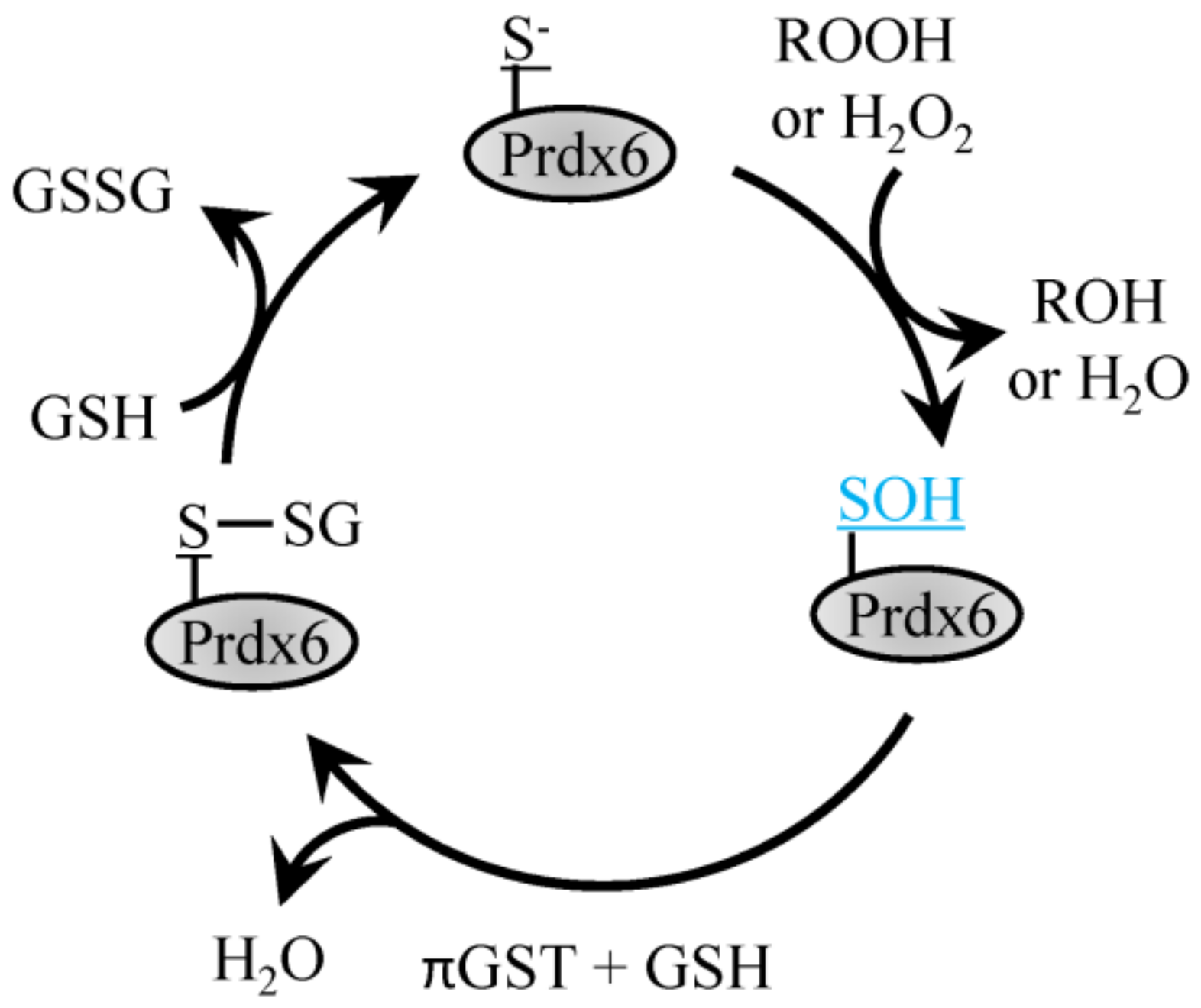
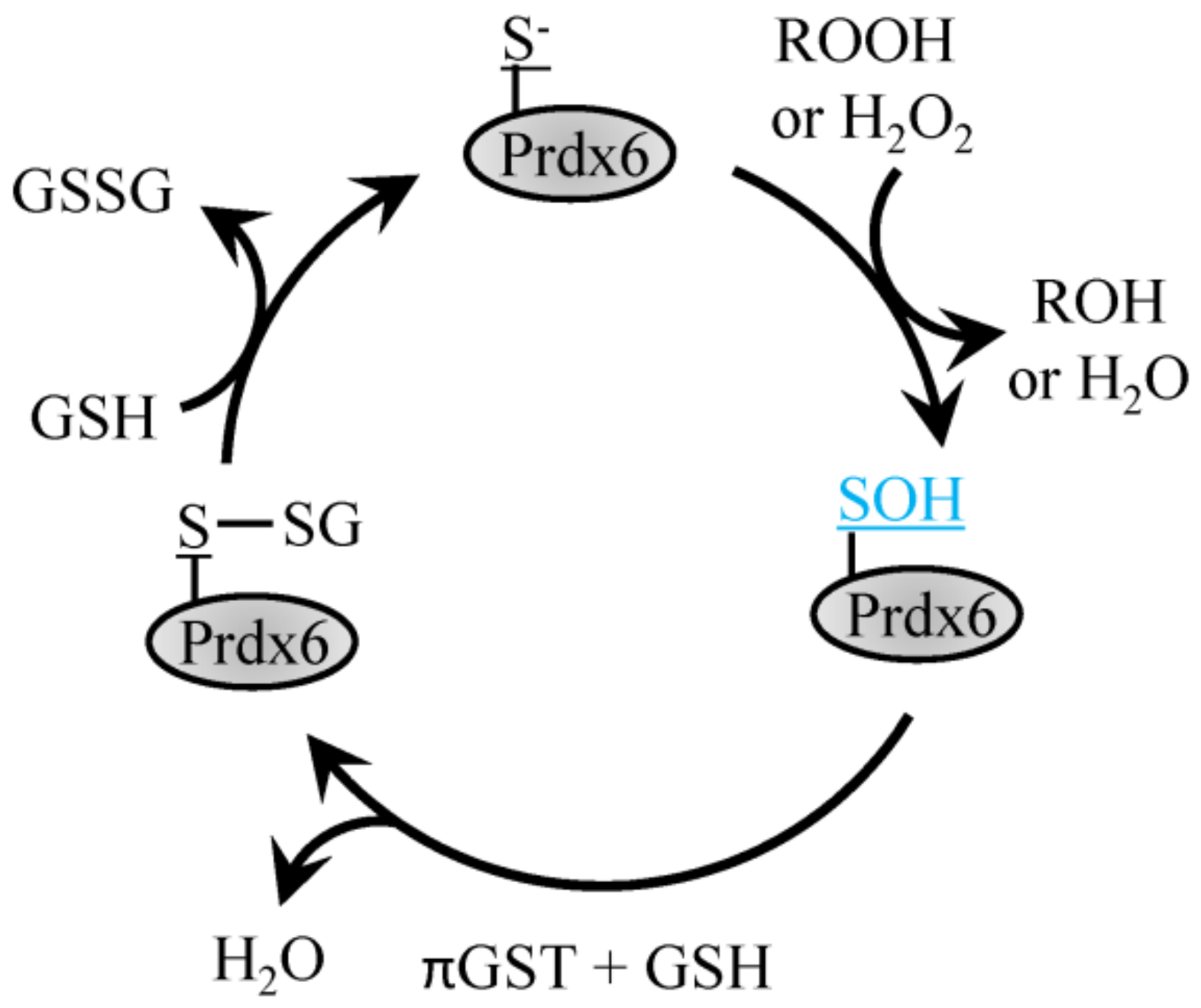
4.3. 1-Cys Prdx
5. The Role of Prdx in Signaling through Redox Relay
6. Phosphorylation of Prdx to Promote Proliferation and Survival
6.1. Protein Tyrosine Kinase Phosphorylation of Prdx1 at the Plasma Membrane
6.2. Cdk1-Cyclin B Phosphorylation of Prdx1 in the Nucleus
6.3. Phosphorylation of Prdx1 to Enhance Peroxidase Activity
6.4. Prdx6 Phosphorylation to Control Phospholipase A2 Activity
7. Phosphorylation of Prdx to Induce Cell Death
7.1. Phosphorylation of Prdx1 by Mst1
7.2. Prdx2 Inactivation by Phosphorylation by Cdk5 Complexes
7.3. Prdx3 Phosphorylation in the Mitochondria by LRRK2
8. Dephosphorylation of Prdx Family Members
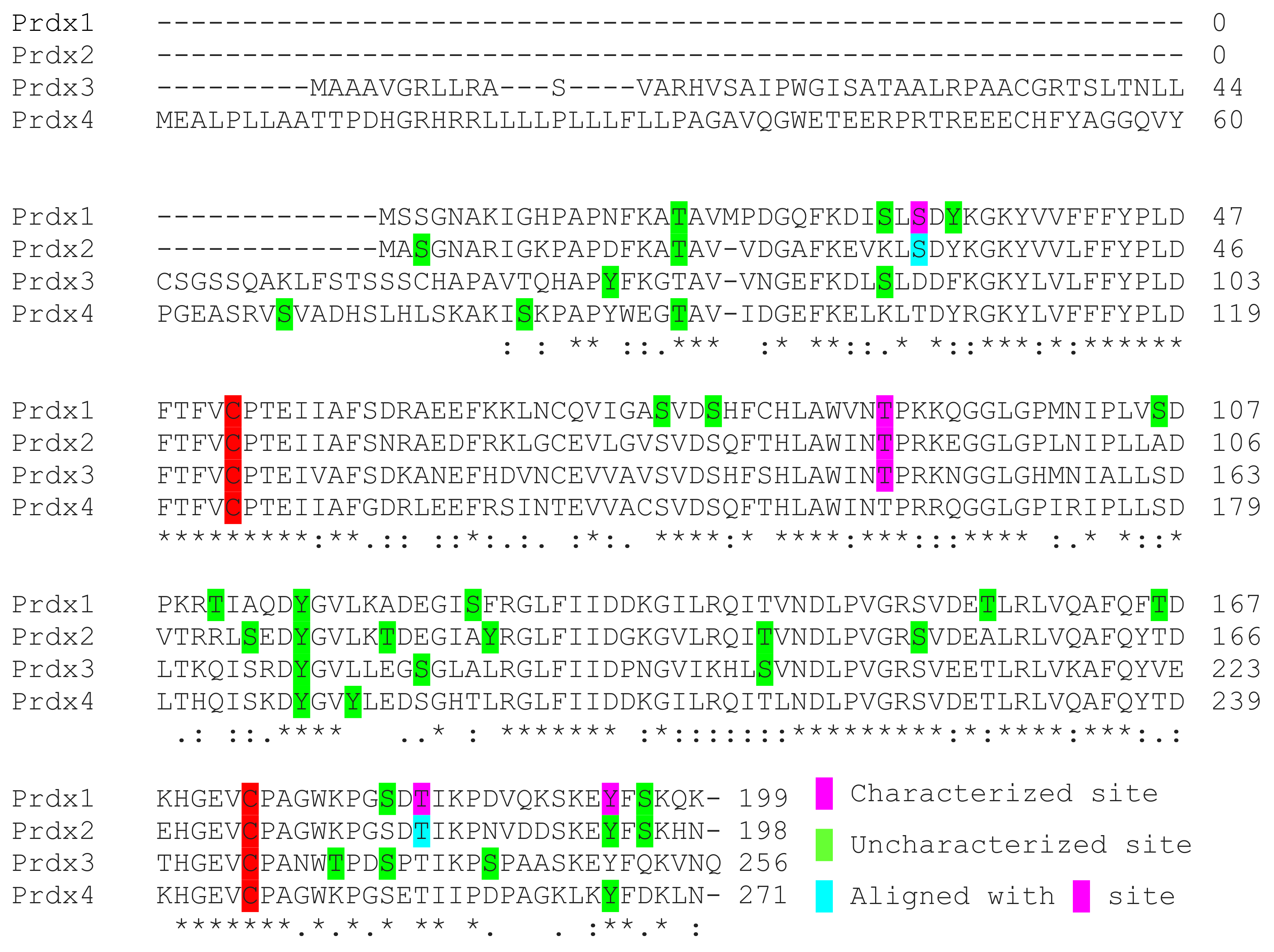
9. Unexplored Regulatory Prdx Phosphorylation
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Vissers, M.C.; Hampton, M.; Kettle, A.J. Hydrogen Peroxide Metabolism in Health and Disease; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by nadh:Ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADPH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef]
- Loschen, G.; Azzi, A.; Richter, C.; Flohe, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Tyler, D.D. Polarographic assay and intracellular distribution of superoxide dismutase in rat liver. Biochem. J. 1975, 147, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef]
- Margittai, E.; Low, P.; Stiller, I.; Greco, A.; Garcia-Manteiga, J.M.; Pengo, N.; Benedetti, A.; Sitia, R.; Banhegyi, G. Production of H2O2 in the endoplasmic reticulum promotes in vivo disulfide bond formation. Antioxid. Redox Signal. 2012, 16, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Nohl, H.; Gille, L. Lysosomal ROS formation. Redox Rep. 2005, 10, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. Nadph oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar] [PubMed]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. 2014, 126, 111–121. [Google Scholar] [CrossRef]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of nadph oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Mueller, S. Sensitive and nonenzymatic measurement of hydrogen peroxide in biological systems. Free Radic. Biol. Med. 2000, 29, 410–415. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2010, 31, 90–99. [Google Scholar] [CrossRef]
- Esposito, L.A.; Kokoszka, J.E.; Waymire, K.G.; Cottrell, B.; MacGregor, G.R.; Wallace, D.C. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic. Biol. Med. 2000, 28, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.N.; Giblin, F.J.; Lin, L.R.; Dang, L.; Unakar, N.J.; Musch, D.C.; Boyle, D.L.; Takemoto, L.J.; Ho, Y.S.; Knoernschild, T.; et al. Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3247–3255. [Google Scholar]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997, 272, 16644–16651. [Google Scholar] [CrossRef]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef]
- Wong, C.M.; Zhou, Y.; Ng, R.W.; Kung Hf, H.F.; Jin, D.Y. Cooperation of yeast peroxiredoxins Tsa1p and Tsa2p in the cellular defense against oxidative and nitrosative stress. J. Biol. Chem. 2002, 277, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Siu, K.L.; Jin, D.Y. Peroxiredoxin-null yeast cells are hypersensitive to oxidative stress and are genomically unstable. J. Biol. Chem. 2004, 279, 23207–23213. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, S.U.; Yu, S.L.; Kim, S.H.; Park, D.S.; Moon, H.B.; Dho, S.H.; Kwon, K.S.; Kwon, H.J.; Han, Y.H.; et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 2003, 101, 5033–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Switala, J.; Loewen, P.C. Diversity of properties among catalases. Arch. Biochem. Biophys. 2002, 401, 145–154. [Google Scholar] [CrossRef]
- Epp, O.; Ladenstein, R.; Wendel, A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur. J. Biochem. 1983, 133, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Herbette, S.; Roeckel-Drevet, P.; Drevet, J.R. Seleno-independent glutathione peroxidases. More than simple antioxidant scavengers. FEBS J. 2007, 274, 2163–2180. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef]
- Lim, J.C.; Choi, H.I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.Z.; Uhm, T.B.; Rhee, S.G. Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc. Natl. Acad. Sci. USA 1994, 91, 7022–7026. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Poole, L.B.; Hantgan, R.R.; Karplus, P.A. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 2002, 41, 5493–5504. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, B.G.; Souchon, H.; Honore, N.; Saint-Joanis, B.; Brosch, R.; Shepard, W.; Cole, S.T.; Alzari, P.M. Structure and mechanism of the alkyl hydroperoxidase Ahpc, a key element of the Mycobacterium tuberculosis defense system against oxidative stress. J. Biol. Chem. 2005, 280, 25735–25742. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 2011, 15, 795–815. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef]
- Lowther, W.T.; Haynes, A.C. Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid. Redox Signal. 2011, 15, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Pace, P.E.; Behring, J.B.; Paton, L.N.; Soethoudt, M.; Bachschmid, M.M.; Winterbourn, C.C. Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J. Biol. Chem. 2016, 291, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Declercq, J.P.; Evrard, C.; Clippe, A.; Stricht, D.V.; Bernard, A.; Knoops, B. Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 a resolution. J. Mol. Biol. 2001, 311, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 2009, 8, 4072–4078. [Google Scholar] [CrossRef] [Green Version]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Flohe, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Morais, M.A.; Giuseppe, P.O.; Souza, T.A.; Alegria, T.G.; Oliveira, M.A.; Netto, L.E.; Murakami, M.T. How ph modulates the dimer-decamer interconversion of 2-Cys peroxiredoxins from the Prx1 subfamily. J. Biol. Chem. 2015, 290, 8582–8590. [Google Scholar] [CrossRef] [PubMed]
- Kitano, K.; Niimura, Y.; Nishiyama, Y.; Miki, K. Stimulation of peroxidase activity by decamerization related to ionic strength: Ahpc protein from amphibacillus xylanus. J. Biochem. 1999, 126, 313–319. [Google Scholar] [CrossRef]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Vick, K.A.; Skoko, J.J.; Neumann, C.A. Peroxiredoxin involvement in the initiation and progression of human cancer. Antioxid. Redox Signal. 2018, 28, 591–608. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Paletta-Silva, R.; Rocco-Machado, N.; Meyer-Fernandes, J.R. NADPH oxidase biology and the regulation of tyrosine kinase receptor signaling and cancer drug cytotoxicity. Int. J. Mol. Sci. 2013, 14, 3683–3704. [Google Scholar] [CrossRef]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.H.; Kim, S.Y.; Park, S.K.; Jeon, H.S.; Lee, Y.M.; Jung, J.H.; Lee, S.Y.; Chae, H.B.; Jung, Y.J.; Lee, K.O.; et al. Phosphorylation and concomitant structural changes in human 2-Cys peroxiredoxin isotype I differentially regulate its peroxidase and molecular chaperone functions. FEBS Lett. 2006, 580, 351–355. [Google Scholar] [CrossRef]
- Lim, J.M.; Lee, K.S.; Woo, H.A.; Kang, D.; Rhee, S.G. Control of the pericentrosomal H2O2 level by peroxiredoxin i is critical for mitotic progression. J. Cell Biol. 2015, 210, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.A.; Zhu, F.; Vakorina, T.I.; Zhang, J.; Higgins, L.A.; Urusova, D.V.; Bode, A.M.; Dong, Z. T-lak cell-originated protein kinase (TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in rpmi7951 melanoma cells through the regulation of Prx1 peroxidase activity. J. Biol. Chem. 2010, 285, 29138–29146. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Cha, E.J.; Lee, H.L.; Han, K.H.; Lee, K.M.; Kim, W.J. Enhanced expression of peroxiredoxin I and VI correlates with development, recurrence and progression of human bladder cancer. J. Urol. 2006, 175, 1512–1516. [Google Scholar] [CrossRef]
- Walsh, B.; Pearl, A.; Suchy, S.; Tartaglio, J.; Visco, K.; Phelan, S.A. Overexpression of Prdx6 and resistance to peroxide-induced death in Hepa1-6 cells: Prdx suppression increases apoptosis. Redox Rep. 2009, 14, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Lehtonen, S.; Sormunen, R.; Kaarteenaho-Wiik, R.; Kang, S.W.; Rhee, S.G.; Soini, Y. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. J. Pathol. 2002, 196, 316–323. [Google Scholar] [CrossRef]
- Lehtonen, S.T.; Svensk, A.M.; Soini, Y.; Paakko, P.; Hirvikoski, P.; Kang, S.W.; Saily, M.; Kinnula, V.L. Peroxiredoxins, a novel protein family in lung cancer. Int. J. Cancer 2004, 111, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.N.; Lee, S.B.; Lee, S.S.; Yoon, S.H.; Kang, G.Y.; Hwang, S.G.; Um, H.D. Phospholipase A2 activity of peroxiredoxin 6 promotes invasion and metastasis of lung cancer cells. Mol. Cancer Ther. 2010, 9, 825–832. [Google Scholar] [CrossRef]
- Wu, Y.; Feinstein, S.I.; Manevich, Y.; Chowdhury, I.; Pak, J.H.; Kazi, A.; Dodia, C.; Speicher, D.W.; Fisher, A.B. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A2 activity. Biochem. J. 2009, 419, 669–679. [Google Scholar] [CrossRef]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Fatma, N.; Singh, D.P. Sumoylation-deficient Prdx6 gains protective function by amplifying enzymatic activity and stability and escapes oxidative stress-induced aberrant sumoylation. Cell Death Dis. 2017, 8, e2525. [Google Scholar] [CrossRef]
- Pozo, K.; Bibb, J.A. The emerging role of Cdk5 in cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Saunders-Pullman, R.; Barrett, M.J.; Stanley, K.M.; Luciano, M.S.; Shanker, V.; Severt, L.; Hunt, A.; Raymond, D.; Ozelius, L.J.; Bressman, S.B. LRRK2 G2019S mutations are associated with an increased cancer risk in Parkinson disease. Mov. Disord. 2010, 25, 2536–2541. [Google Scholar] [CrossRef] [PubMed]
- Waro, B.J.; Aasly, J.O. Exploring cancer in LRRK2 mutation carriers and idiopathic Parkinson’s disease. Brain Behav. 2018, 8, e00858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; et al. Mammalian mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [Green Version]
- Morinaka, A.; Funato, Y.; Uesugi, K.; Miki, H. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate Mst1 kinase and apoptosis. Oncogene 2011, 30, 4208–4218. [Google Scholar] [CrossRef]
- Rawat, S.J.; Creasy, C.L.; Peterson, J.R.; Chernoff, J. The tumor suppressor Mst1 promotes changes in the cellular redox state by phosphorylation and inactivation of peroxiredoxin-1 protein. J. Biol. Chem. 2013, 288, 8762–8771. [Google Scholar] [CrossRef]
- Ura, S.; Masuyama, N.; Graves, J.D.; Gotoh, Y. Caspase cleavage of Mst1 promotes nuclear translocation and chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 10148–10153. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Rashidian, J.; Mount, M.P.; Aleyasin, H.; Parsanejad, M.; Lira, A.; Haque, E.; Zhang, Y.; Callaghan, S.; Daigle, M.; et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 2007, 55, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Szumlinski, K.K.; Lew, J. P10, the n-terminal domain of p35, protects against Cdk5/p25-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 20041–20046. [Google Scholar] [CrossRef]
- Sun, K.H.; de Pablo, Y.; Vincent, F.; Shah, K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 2008, 107, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidian, J.; Rousseaux, M.W.; Venderova, K.; Qu, D.; Callaghan, S.M.; Phillips, M.; Bland, R.J.; During, M.J.; Mao, Z.; Slack, R.S.; et al. Essential role of cytoplasmic Cdk5 and Prx2 in multiple ischemic injury models, in vivo. J. Neurosci. 2009, 29, 12497–12505. [Google Scholar] [CrossRef]
- Wonsey, D.R.; Zeller, K.I.; Dang, C.V. The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc. Natl. Acad. Sci. USA 2002, 99, 6649–6654. [Google Scholar] [CrossRef] [Green Version]
- Angeles, D.C.; Gan, B.H.; Onstead, L.; Zhao, Y.; Lim, K.L.; Dachsel, J.; Melrose, H.; Farrer, M.; Wszolek, Z.K.; Dickson, D.W.; et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 2011, 32, 1390–1397. [Google Scholar] [CrossRef]
- Angeles, D.C.; Ho, P.; Chua, L.L.; Wang, C.; Yap, Y.W.; Ng, C.; Zhou, Z.; Lim, K.L.; Wszolek, Z.K.; Wang, H.Y.; et al. Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in drosophila. Hum. Mol. Genet. 2014, 23, 3157–3165. [Google Scholar] [CrossRef]
- Chu, K.L.; Lew, Q.J.; Rajasegaran, V.; Kung, J.T.; Zheng, L.; Yang, Q.; Shaw, R.; Cheong, N.; Liou, Y.C.; Chao, S.H. Regulation of PRDX1 peroxidase activity by Pin1. Cell Cycle 2013, 12, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Palande, K.; Roovers, O.; Gits, J.; Verwijmeren, C.; Iuchi, Y.; Fujii, J.; Neel, B.G.; Karisch, R.; Tavernier, J.; Touw, I.P. Peroxiredoxin-controlled G-CSF signalling at the endoplasmic reticulum-early endosome interface. J. Cell Sci. 2011, 124, 3695–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebthal, M.; Maynard, D.; Dietz, K.J. Peroxiredoxins and redox signaling in plants. Antioxid. Redox Signal. 2018, 28, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Rigas, J.R.; Gerber, S.A. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteomics 2013, 91, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of aurora and polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [PubMed]
- Robles, M.S.; Humphrey, S.J.; Mann, M. Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 2017, 25, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of tyr and ser/thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Grady, J.T.; Haas, W.; Gygi, S.P. Quantitative comparison of the fasted and re-fed mouse liver phosphoproteomes using lower ph reductive dimethylation. Methods (San Diego, Calif.) 2013, 61, 277–286. [Google Scholar] [CrossRef]
- Wu, X.; Tian, L.; Li, J.; Zhang, Y.; Han, V.; Li, Y.; Xu, X.; Li, H.; Chen, X.; Chen, J.; et al. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol. Cell. Proteomics 2012, 11, 1640–1651. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Demirkan, G.; Yu, K.; Boylan, J.M.; Salomon, A.R.; Gruppuso, P.A. Phosphoproteomic profiling of in vivo signaling in liver by the mammalian target of rapamycin complex 1 (mTORC1). PLoS ONE 2011, 6, e21729. [Google Scholar] [CrossRef]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteomics 2014, 96, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteomics 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed]
- Reinartz, M.; Raupach, A.; Kaisers, W.; Godecke, A. AKT1 and AKT2 induce distinct phosphorylation patterns in Hl-1 cardiac myocytes. J. Proteome Res. 2014, 13, 4232–4245. [Google Scholar] [CrossRef]
- Tsai, C.F.; Wang, Y.T.; Yen, H.Y.; Tsou, C.C.; Ku, W.C.; Lin, P.Y.; Chen, H.Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y.J. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Sacco, F.; Humphrey, S.J.; Cox, J.; Mischnik, M.; Schulte, A.; Klabunde, T.; Schafer, M.; Mann, M. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat. Commun. 2016, 7, 13250. [Google Scholar] [CrossRef] [Green Version]
- Hoffert, J.D.; Pisitkun, T.; Wang, G.; Shen, R.F.; Knepper, M.A. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: Regulation of aquaporin-2 phosphorylation at two sites. Proc. Natl. Acad. Sci. USA 2006, 103, 7159–7164. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Haar Petersen, M.; Ibanez-Vea, M.; Lassen, P.S.; Larsen, M.R.; Palmisano, G. Simultaneous enrichment of cysteine-containing peptides and phosphopeptides using a Cysteine-specific Phosphonate Adaptable Tag (CysPAT) in combination with titanium dioxide (TiO2) chromatography. Mol. Cell. Proteomics 2016, 15, 3282–3296. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Jeong, W.; Choi, S.Y.; Yu, S.; Kang, S.W.; Rhee, S.G. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002, 277, 25370–25376. [Google Scholar] [CrossRef]
- Santamaria, A.; Wang, B.; Elowe, S.; Malik, R.; Zhang, F.; Bauer, M.; Schmidt, A.; Sillje, H.H.; Korner, R.; Nigg, E.A. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol. Cell. Proteomics 2011, 10, M110.004457. [Google Scholar] [CrossRef]
- Gu, T.L.; Deng, X.; Huang, F.; Tucker, M.; Crosby, K.; Rimkunas, V.; Wang, Y.; Deng, G.; Zhu, L.; Tan, Z.; et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS ONE 2011, 6, e15640. [Google Scholar] [CrossRef]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Muller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. Cell. Proteomics 2012, 11, 651–668. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stockli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minard, A.Y.; Tan, S.X.; Yang, P.; Fazakerley, D.J.; Domanova, W.; Parker, B.L.; Humphrey, S.J.; Jothi, R.; Stockli, J.; James, D.E. mTORC1 is a major regulatory node in the FGF21 signaling network in adipocytes. Cell Rep. 2016, 17, 29–36. [Google Scholar] [CrossRef]
- Molina, H.; Horn, D.M.; Tang, N.; Mathivanan, S.; Pandey, A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 2007, 104, 2199–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.M.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005, 23, 94–101. [Google Scholar] [CrossRef]
- Jorgensen, C.; Sherman, A.; Chen, G.I.; Pasculescu, A.; Poliakov, A.; Hsiung, M.; Larsen, B.; Wilkinson, D.G.; Linding, R.; Pawson, T. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science 2009, 326, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tong, J.; Taylor, P.; St-Germain, J.R.; Navab, R.; Moran, M.F.; Tsao, M.S. Quantitative phospho-proteomic profiling of hepatocyte growth factor (HGF)-MET signaling in colorectal cancer. J. Proteome Res. 2011, 10, 3200–3211. [Google Scholar] [CrossRef]
- Bai, Y.; Li, J.; Fang, B.; Edwards, A.; Zhang, G.; Bui, M.; Eschrich, S.; Altiok, S.; Koomen, J.; Haura, E.B. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. 2012, 72, 2501–2511. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, I.M.; Chaerkady, R.; Zhong, J.; Molina, H.; Jacob, H.K.; Herbst-Robinson, K.; Dancy, B.M.; Katju, V.; Bose, R.; Zhang, J.; et al. Identification of targets of c-Src tyrosine kinase by chemical complementation and phosphoproteomics. Mol. Cell. Proteomics 2012, 11, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.M.; Nirujogi, R.S.; Rojas, P.L.; Patil, A.H.; Manda, S.S.; Subbannayya, Y.; Roa, J.C.; Chatterjee, A.; Prasad, T.S.; Pandey, A. Quantitative phosphoproteomic analysis of IL-33-mediated signaling. Proteomics 2015, 15, 532–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigbolt, K.T.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Guest, P.C.; Vanattou-Saifoudine, N.; Rahmoune, H.; Bahn, S. Phosphoproteomic differences in major depressive disorder postmortem brains indicate effects on synaptic function. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Schreiber, T.B.; Daub, H. Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J. Proteomics 2012, 75, 1343–1356. [Google Scholar] [CrossRef]
- Franchin, C.; Cesaro, L.; Salvi, M.; Millioni, R.; Iori, E.; Cifani, P.; James, P.; Arrigoni, G.; Pinna, L. Quantitative analysis of a phosphoproteome readily altered by the protein kinase CK2 inhibitor quinalizarin in HEK-293T cells. Biochim. Biophys. Acta 2015, 1854, 609–623. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Demeyer, S.; Govaerts, I.; Bornschein, S.; Verbeke, D.; Jacobs, K.; Binos, S.; Skerrett-Byrne, D.A.; Murray, H.C.; et al. Mutant JAK3 phosphoproteomic profiling predicts synergism between JAK3 inhibitors and MEK/BCL2 inhibitors for the treatment of T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 788–800. [Google Scholar] [CrossRef]
- Choudhary, C.; Olsen, J.V.; Brandts, C.; Cox, J.; Reddy, P.N.; Bohmer, F.D.; Gerke, V.; Schmidt-Arras, D.E.; Berdel, W.E.; Muller-Tidow, C.; et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 2009, 36, 326–339. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Yu, M.J.; Wang, G.; Boja, E.S.; Hoffert, J.D.; Pisitkun, T.; Knepper, M.A. Quantitative phosphoproteomic analysis reveals vasopressin v2-receptor-dependent signaling pathways in renal collecting duct cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3882–3887. [Google Scholar] [CrossRef]
- Iliuk, A.B.; Martin, V.A.; Alicie, B.M.; Geahlen, R.L.; Tao, W.A. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell. Proteomics 2010, 9, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Reed, J.C.; Kim, H.R.; Chae, H.J. Proteomic profiling of differentially expressed proteins from bax inhibitor-1 knockout and wild type mice. Mol. Cells 2012, 34, 15–23. [Google Scholar] [CrossRef]
- Gramage, E.; Perez-Garcia, C.; Vicente-Rodriguez, M.; Bollen, S.; Rojo, L.; Herradon, G. Regulation of extinction of cocaine-induced place preference by midkine is related to a differential phosphorylation of peroxiredoxin 6 in dorsal striatum. Behav. Brain Res. 2013, 253, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.L.; Yang, G.; Humphrey, S.J.; Chaudhuri, R.; Ma, X.; Peterman, S.; James, D.E. Targeted phosphoproteomics of insulin signaling using data-independent acquisition mass spectrometry. Sci. Signal. 2015, 8, rs6. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Fu, Y.; Huang, X.; Wang, W.; Jiang, X.; Gritsenko, M.A.; Zhao, R.; Monore, M.E.; Pertz, O.C.; et al. Spatial phosphoprotein profiling reveals a compartmentalized extracellular signal-regulated kinase switch governing neurite growth and retraction. J. Biol. Chem. 2011, 286, 18190–18201. [Google Scholar] [CrossRef] [PubMed]
- Bordoli, M.R.; Yum, J.; Breitkopf, S.B.; Thon, J.N.; Italiano, J.E., Jr.; Xiao, J.; Worby, C.; Wong, S.K.; Lin, G.; Edenius, M.; et al. A secreted tyrosine kinase acts in the extracellular environment. Cell 2014, 158, 1033–1044. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Nagaraj, N.; Zougman, A.; Gnad, F.; Mann, M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 2010, 9, 3280–3289. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brill, L.M.; Xiong, W.; Lee, K.B.; Ficarro, S.B.; Crain, A.; Xu, Y.; Terskikh, A.; Snyder, E.Y.; Ding, S. Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell 2009, 5, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Zhai, B.; Yu, Y.; Kiyotsugu, Y.; Raschle, T.; Etzkorn, M.; Seo, H.C.; Nagiec, M.; Luna, R.E.; Reinherz, E.L.; et al. Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A phosphoproteomic comparison of B-RAF (V600E) and MKK1/2 inhibitors in melanoma cells. Mol. Cell. Proteomics 2015, 14, 1599–1615. [Google Scholar] [CrossRef] [PubMed]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygun, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.P.; et al. Multiomic analysis of the UV-induced DNA damage response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prdx1 Residue | Effect | Biochemical Assay References | Proteomic Dataset References |
|---|---|---|---|
| Thr18 | Function unknown | [92] | [104,105] |
| Ser30 | Function unknown | [106,107,108,109,110,111] | |
| Ser32 | Enhanced peroxidase activity | [75] | [106,107,108,109,110,111,112,113,114,115,116,117,118,119,120] |
| Tyr34 | Function unknown | [118,121] | |
| Ser77 | Function unknown | [122] | |
| Ser80 | Function unknown | [122] | |
| Thr90 | Decreased peroxidase activity | [73,74,92,96,123] | [107] |
| Ser106 | Function unknown | [105,124] | |
| Thr111 | Function unknown | [92] | [104,105] |
| Tyr116 | Function unknown | [105,118,125] | |
| Ser126 | Function unknown | [104,119,126] | |
| Thr156 | Function unknown | [92] | |
| Thr166 | Function unknown | [105] | |
| Ser181 | Function unknown | [107,119,127,128,129] | |
| Thr183 | Decreased peroxidase activity | [92] | [107,127,130] |
| Tyr194 | Decreased peroxidase activity | [69] | [131,132,133,134,135,136] |
| Ser196 | Function unknown | [116] |
| Prdx2 Residue | Effect | Biochemical Assay References | Proteomic Dataset References |
|---|---|---|---|
| Ser3 | Function unknown | [137] | |
| Thr18 | Function unknown | [105] | |
| Ser31 | Function unknown | [111,115,116,119] | |
| Thr89 | Decreased peroxidase activity | [94,95,96,97] | [138] |
| Ser112 | Function unknown | [105,115,116,119,126,139,140] | |
| Tyr115 | Function unknown | [118,141] | |
| Thr120 | Function unknown | [111,119,140] | |
| Tyr126 | Function unknown | [140] | |
| Thr142 | Function unknown | [105,142,143] | |
| Ser151 | Function unknown | [104,111] | |
| Thr182 | Function unknown | [115] | |
| Tyr193 | Function unknown | [133,134,135,136,144] | |
| Ser195 | Function unknown | [144] |
| Prdx3 Residue | Effect | Biochemical Assay References | Proteomic Dataset References |
|---|---|---|---|
| Tyr71 | Function unknown | [125] | |
| Ser86 | Function unknown | [105,119] | |
| Thr146 | Decreased peroxidase activity | [99,100] | |
| Tyr172 | Function unknown | [118] | |
| Ser179 | Function unknown | [105,119] | |
| Ser199 | Function unknown | [104,127] | |
| Thr234 | Function unknown | [107,115] | |
| Ser237 | Function unknown | [107,119,147] | |
| Ser243 | Function unknown | [110,147] |
| Prdx4 Residue | Effect | Proteomic Dataset References |
|---|---|---|
| Ser68 | Function unknown | [115] |
| Ser82 | Function unknown | [148] |
| Thr91 | Function unknown | [148] |
| Tyr188 | Function unknown | [118] |
| Tyr191 | Function unknown | [118] |
| Tyr266 | Function unknown | [149] |
| Prdx5 Residue | Effect | Proteomic Dataset References |
|---|---|---|
| Thr97 | Function unknown | [105,107,116,119,150,151] |
| Ser101 | Function unknown | [105,119] |
| Ser168 | Function unknown | [108,112] |
| Ser171 | Function unknown | [111,150] |
| Ser182 | Function unknown | [105,112,116,119] |
| Prdx6 Residue | Effect | Biochemical Assay References | Proteomic Dataset References |
|---|---|---|---|
| Ser32 | Function unknown | [111] | |
| Thr44 | Function unknown | [105,114,116,119,126,128,140,152,153,154,155,156,157] | |
| Tyr89 | Function unknown | [107,111,131] | |
| Ser146 | Function unknown | [104,105] | |
| Thr177 | Enhanced acidic calcium-independent phospholipase A2 activity | [81,82,83] | [115,118,153,156] |
| Ser186 | Function unknown | [105,110,154] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoko, J.J.; Attaran, S.; Neumann, C.A. Signals Getting Crossed in the Entanglement of Redox and Phosphorylation Pathways: Phosphorylation of Peroxiredoxin Proteins Sparks Cell Signaling. Antioxidants 2019, 8, 29. https://doi.org/10.3390/antiox8020029
Skoko JJ, Attaran S, Neumann CA. Signals Getting Crossed in the Entanglement of Redox and Phosphorylation Pathways: Phosphorylation of Peroxiredoxin Proteins Sparks Cell Signaling. Antioxidants. 2019; 8(2):29. https://doi.org/10.3390/antiox8020029
Chicago/Turabian StyleSkoko, John J., Shireen Attaran, and Carola A. Neumann. 2019. "Signals Getting Crossed in the Entanglement of Redox and Phosphorylation Pathways: Phosphorylation of Peroxiredoxin Proteins Sparks Cell Signaling" Antioxidants 8, no. 2: 29. https://doi.org/10.3390/antiox8020029
APA StyleSkoko, J. J., Attaran, S., & Neumann, C. A. (2019). Signals Getting Crossed in the Entanglement of Redox and Phosphorylation Pathways: Phosphorylation of Peroxiredoxin Proteins Sparks Cell Signaling. Antioxidants, 8(2), 29. https://doi.org/10.3390/antiox8020029