Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction-What is Oxidative Stress?
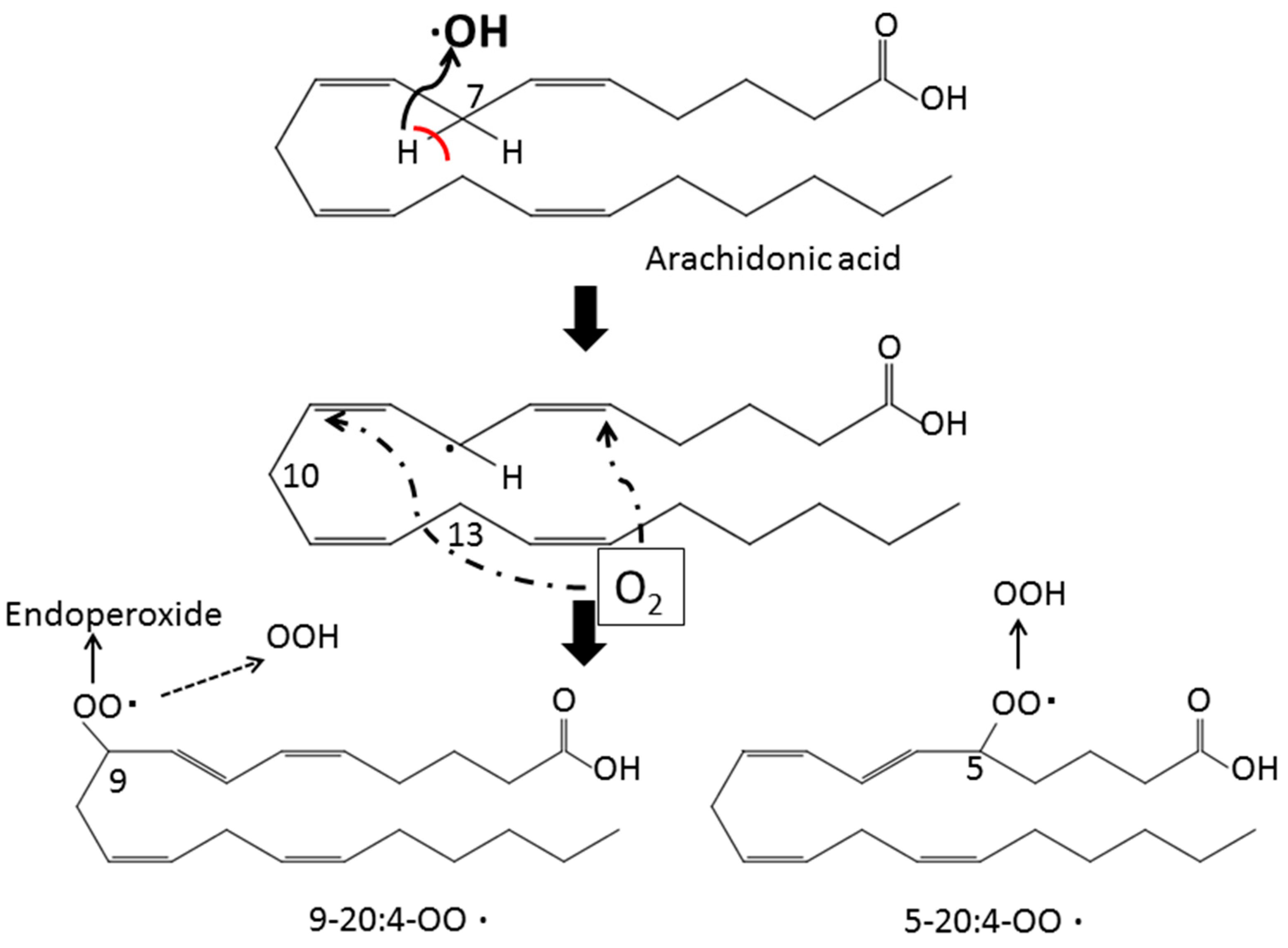
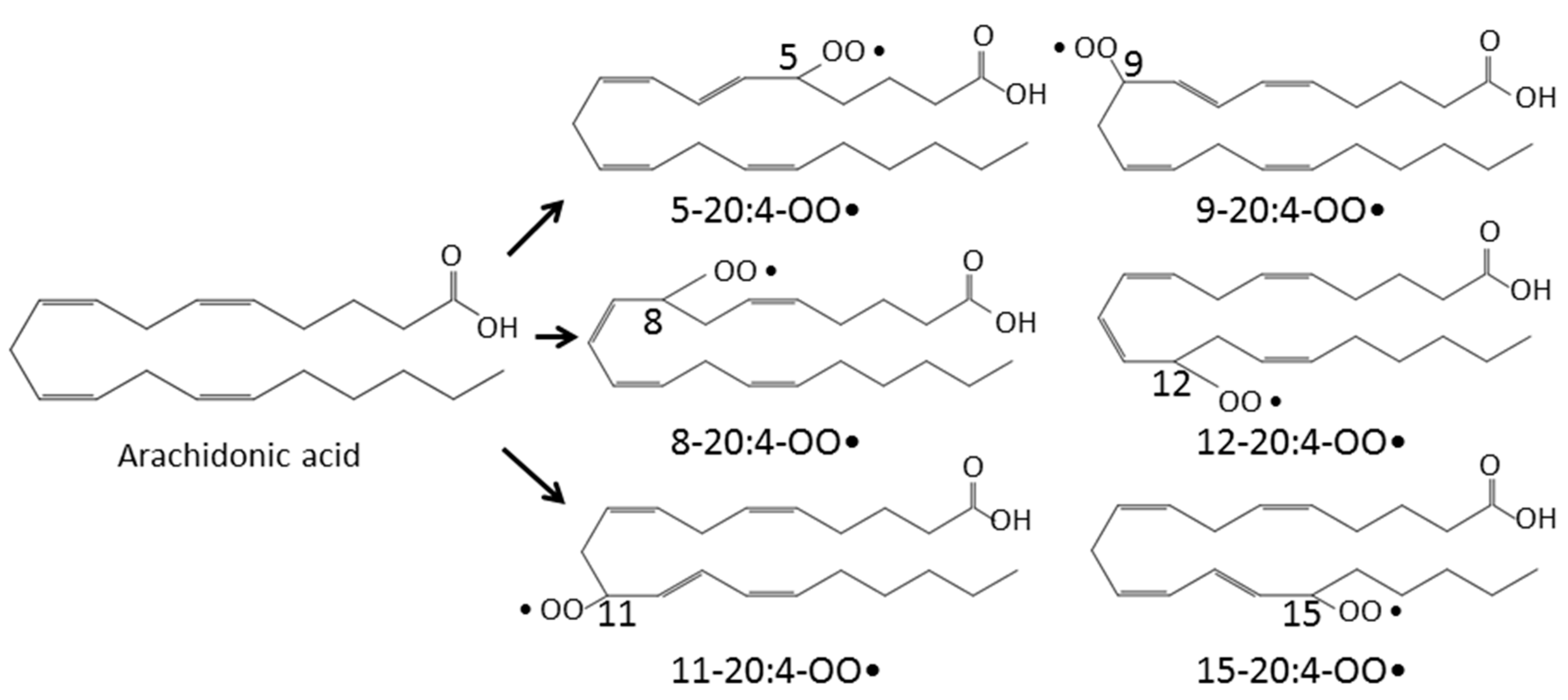
2. Lipid Peroxidation
3. Assay of Lipid Peroxidation
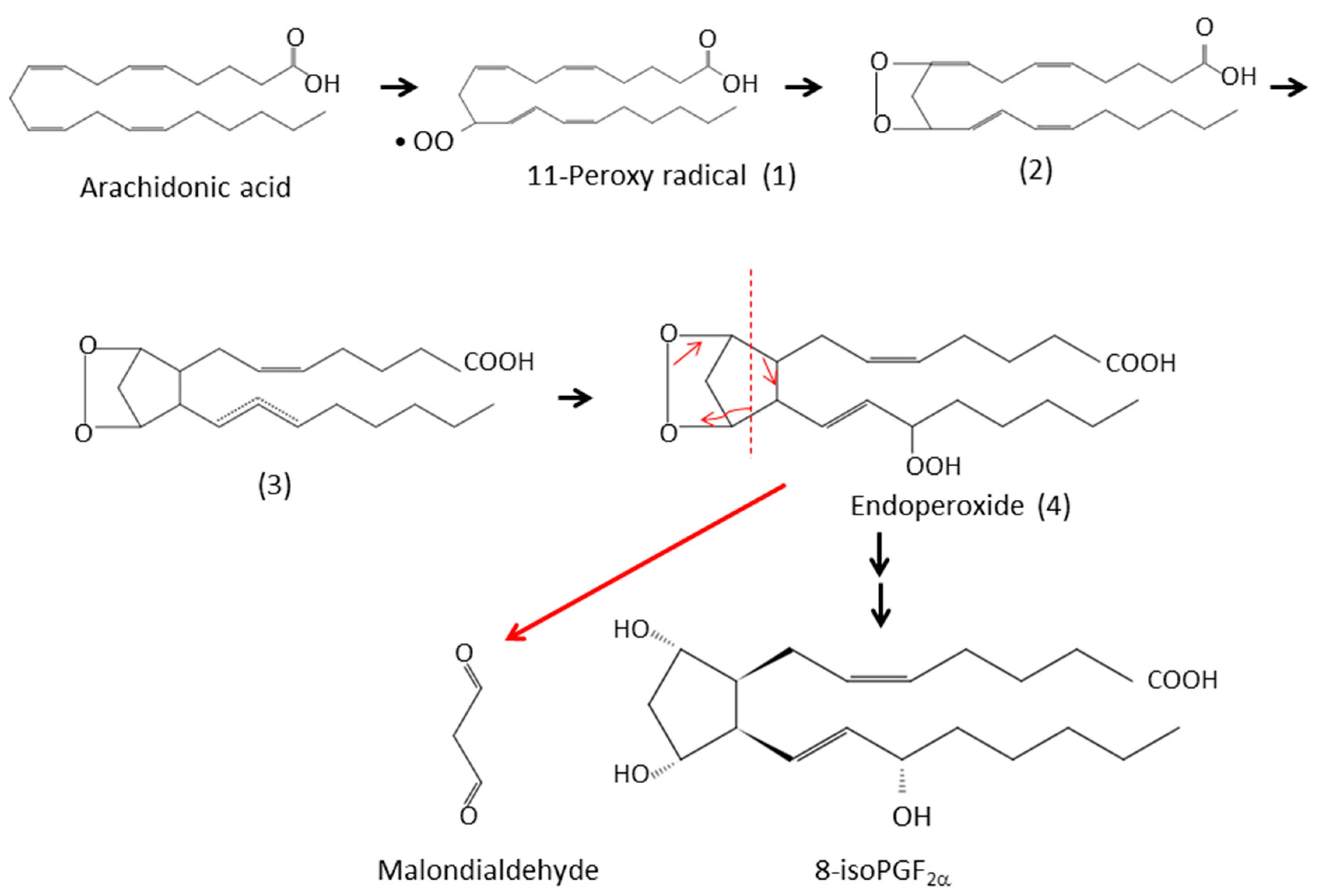
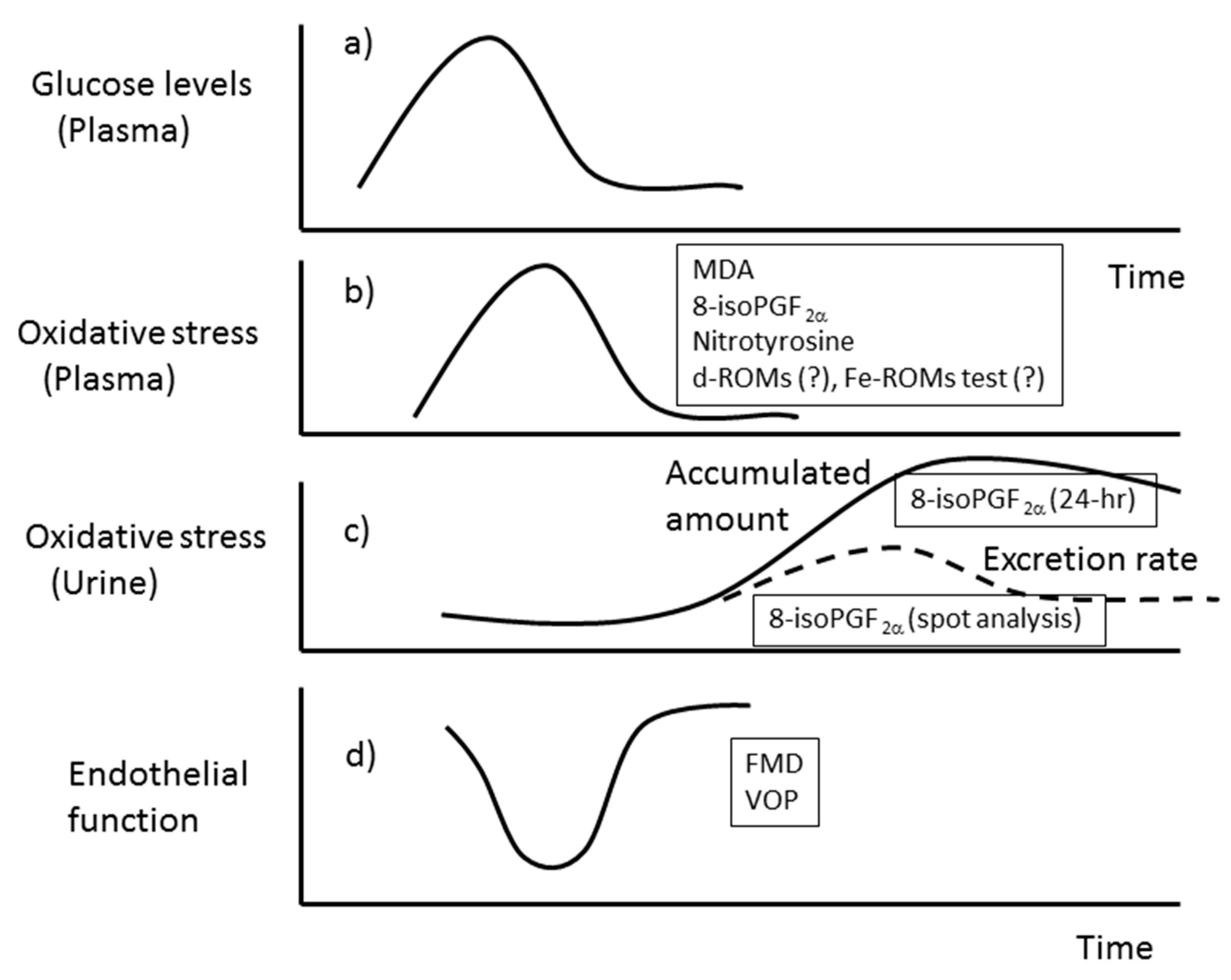
3.1. Isoprostanes
3.2. Malondialdehyde (MDA)
3.3. d-ROMs Test
3.4. Fe-ROMs Test
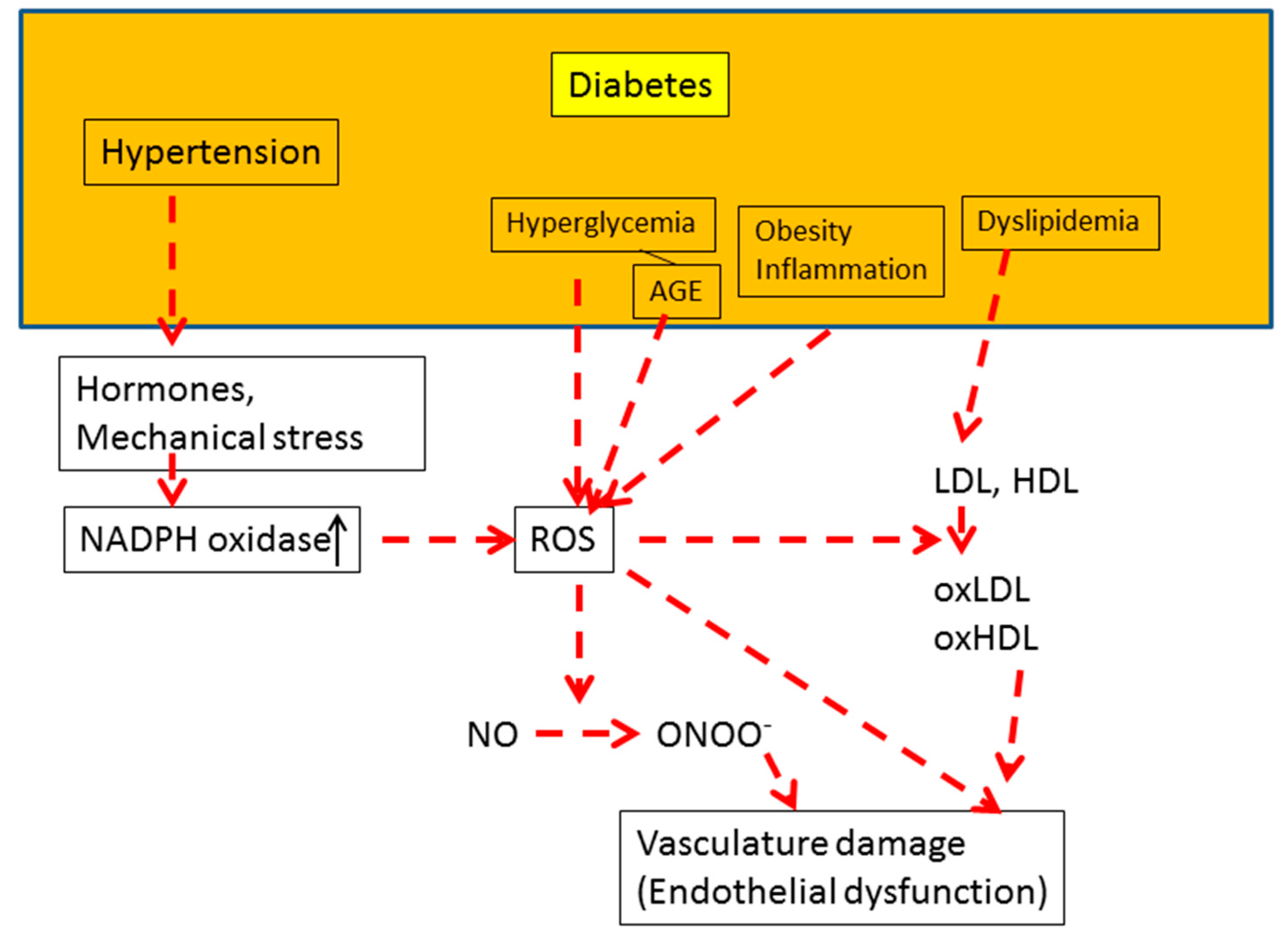
4. Diabetes and Oxidative Stress
4.1. Diabetes Complications
4.2. Hyperglycemia and Oxidative Stress
5. Oxidative Stress and Endothelial Function in Diabetes
5.1. Underlying Mechanism of Endothelial Dysfunction
5.2. Control Mechanisms of Endothelial Function
5.3. Hyperglycemia and Endothelial Function
5.4. Dyslipidemia and Endothelial Function
5.5. Hypertension, AGE, and Endothelial Function
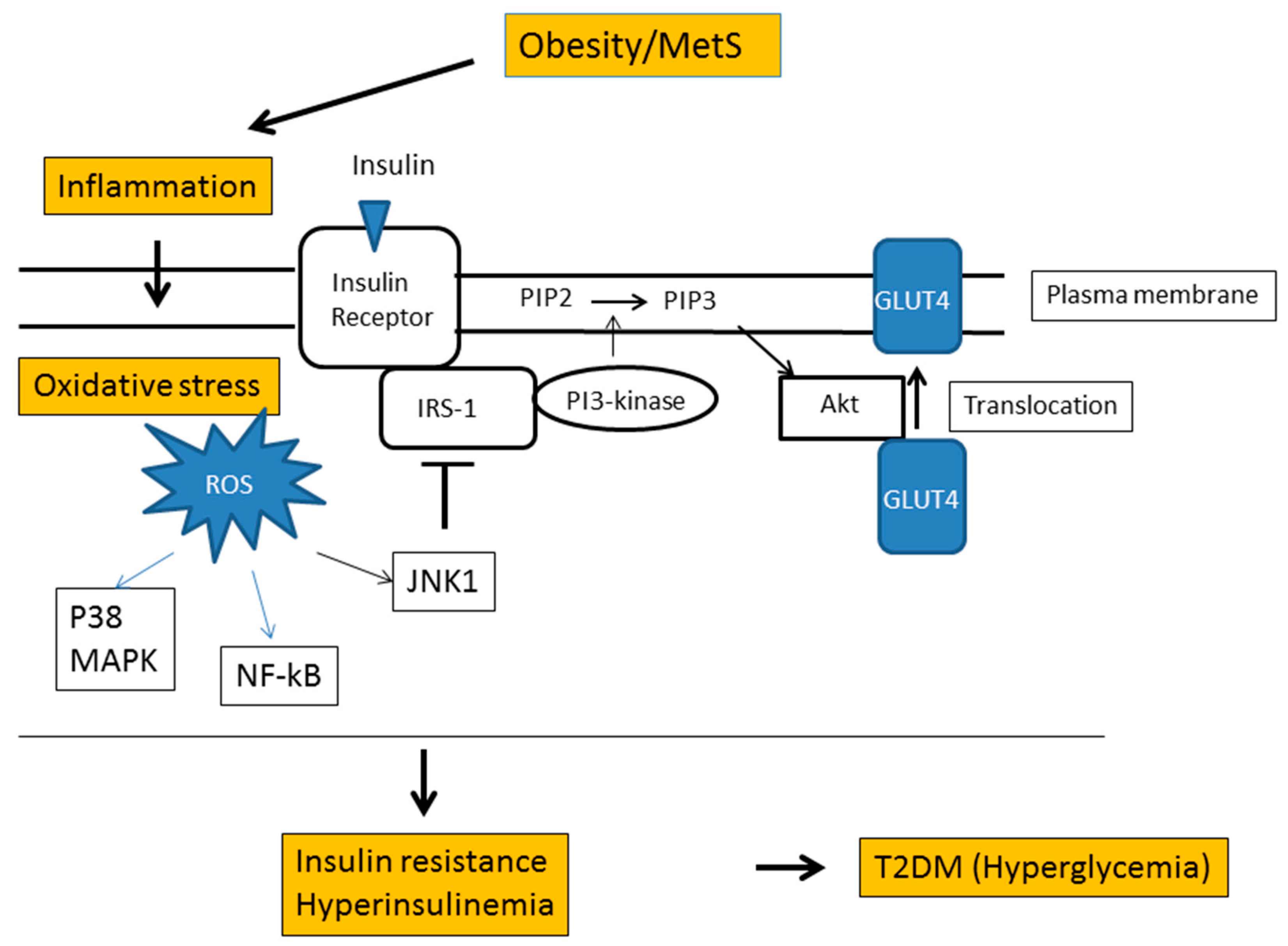
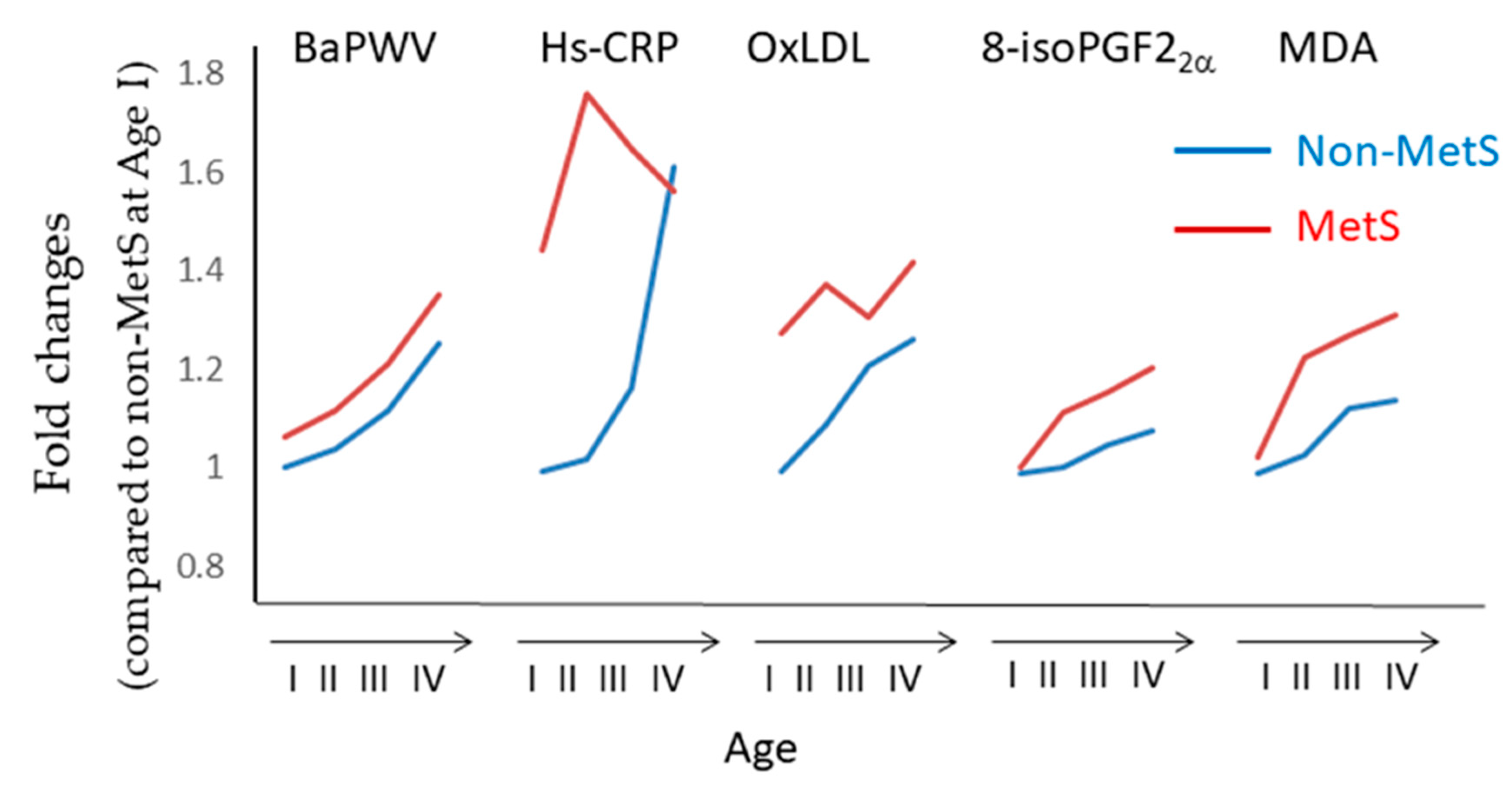
6. Obesity and Oxidative Stress
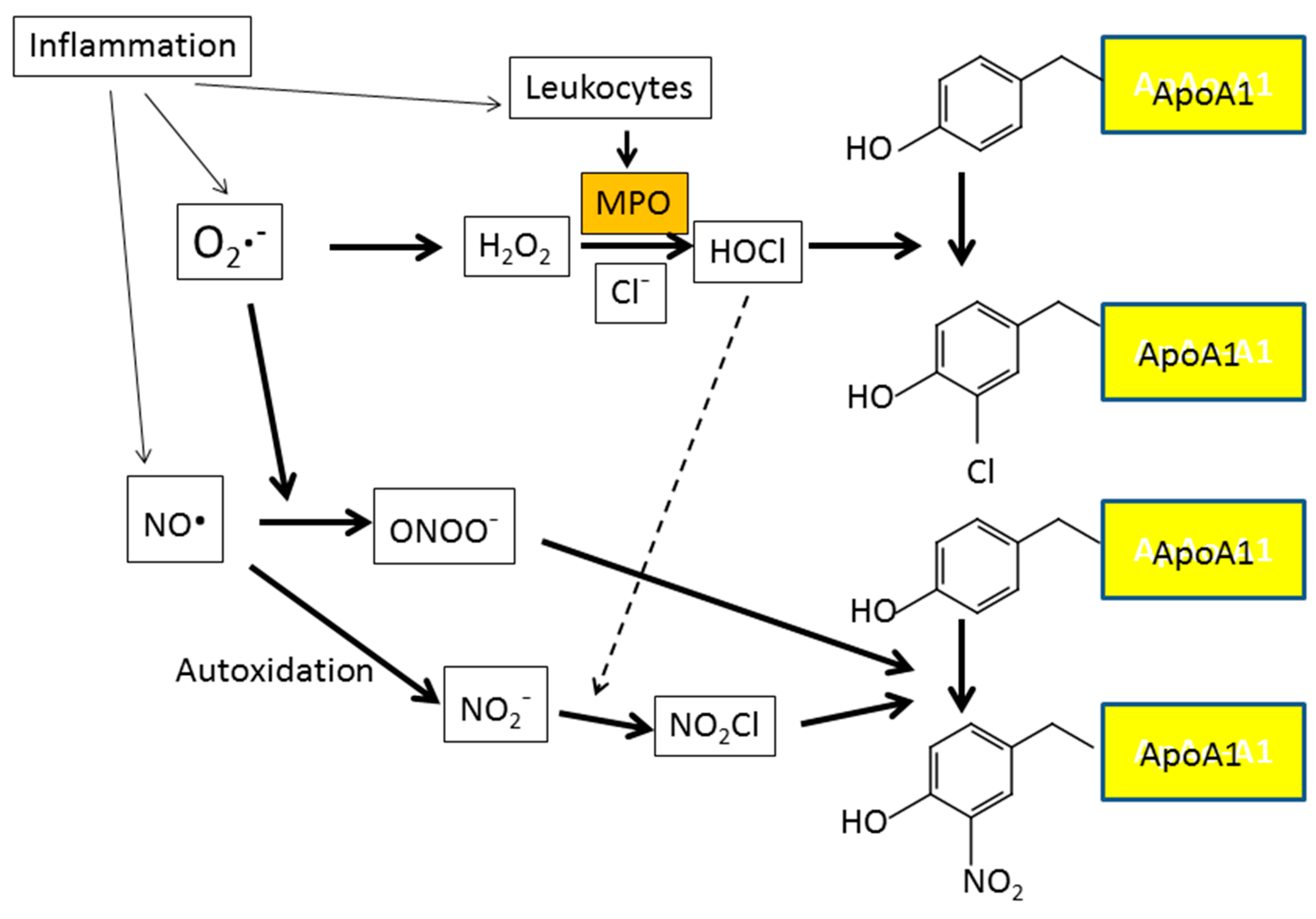
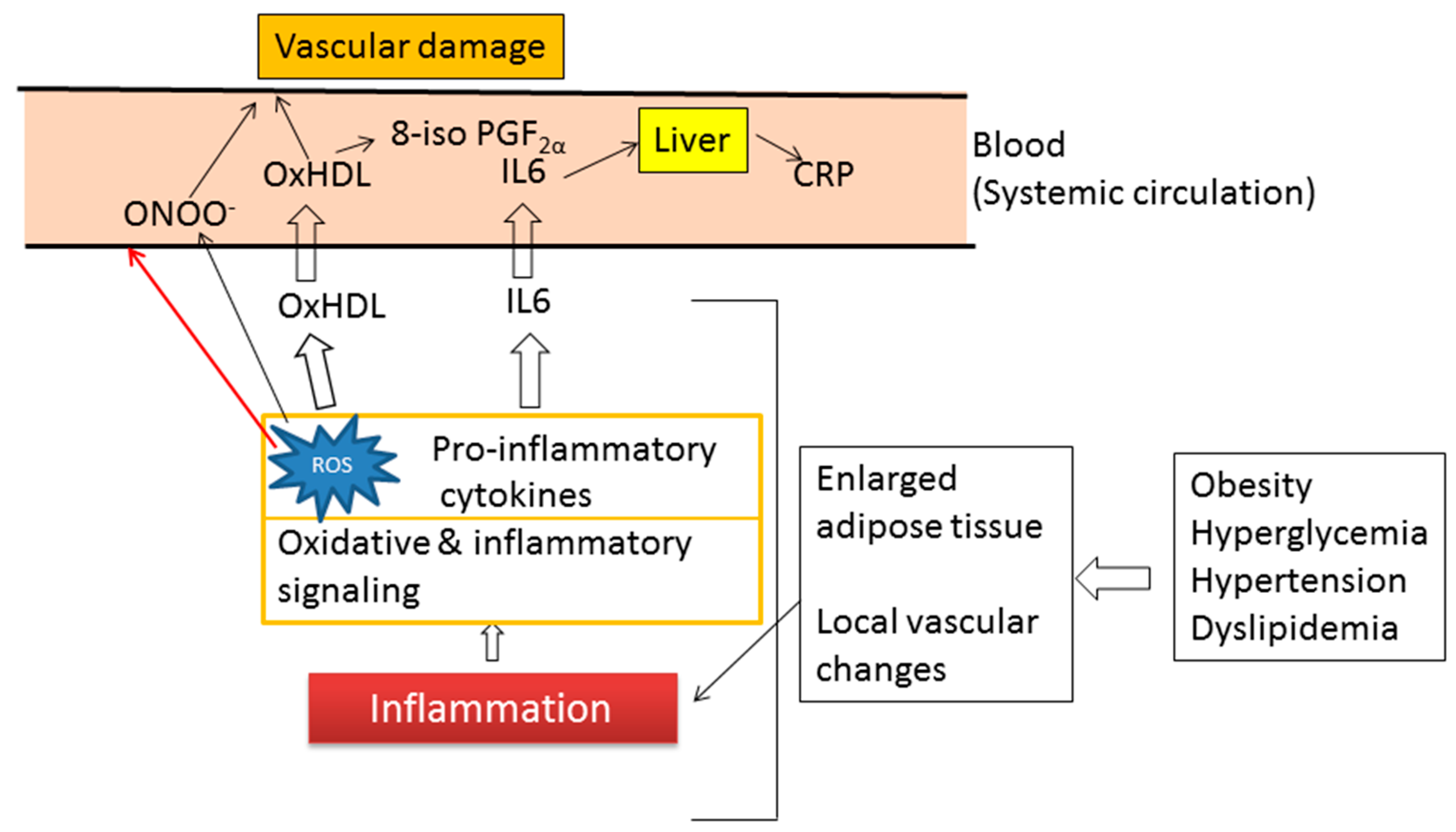
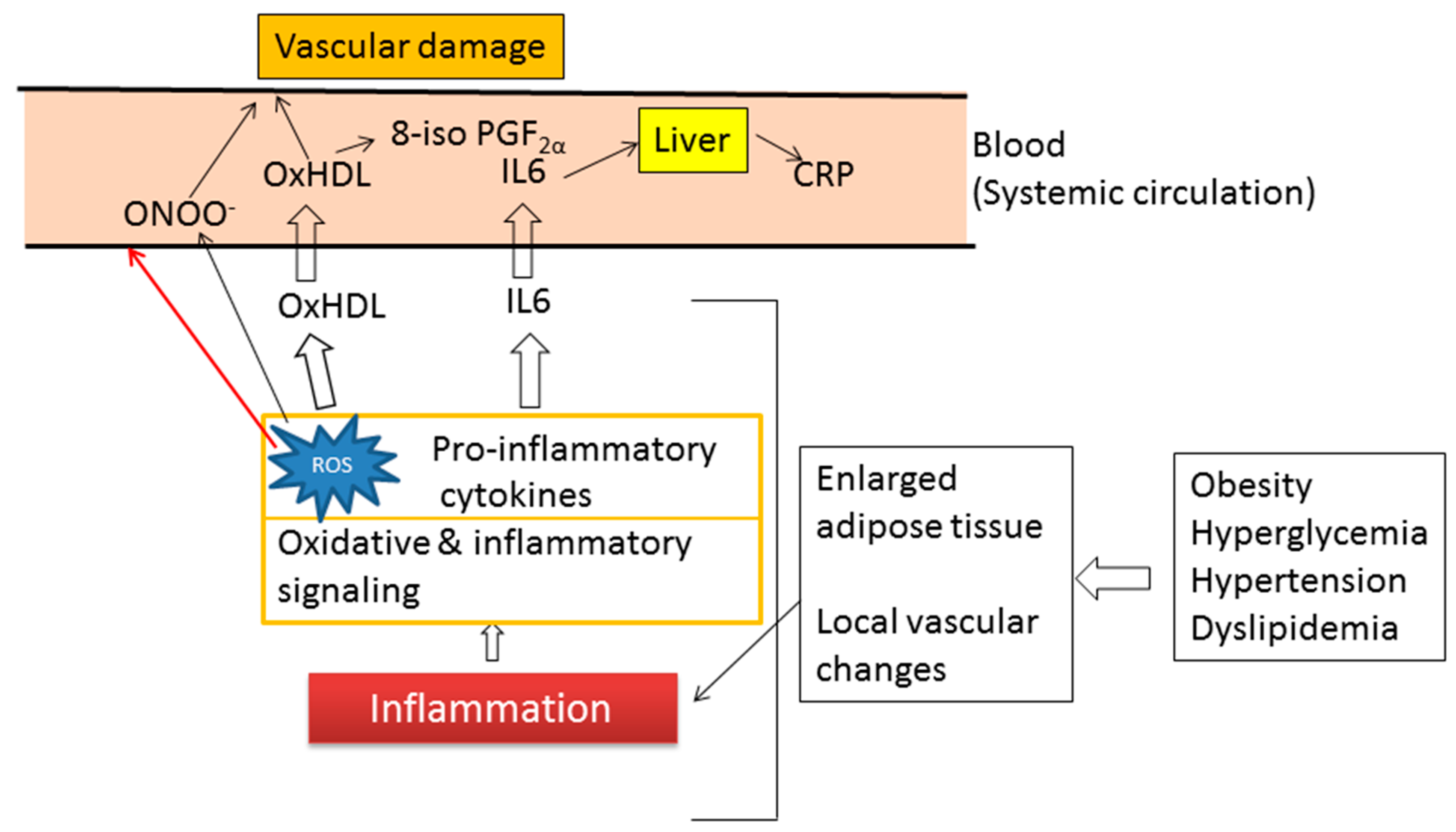
7. Inflammation and Oxidative Stress
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem.-Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [PubMed]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B., 3rd. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef]
- Bonomini, F.; Tengattini, S.; Fabiano, A.; Bianchi, R.; Rezzani, R. Atherosclerosis and oxidative stress. Histol. Histopathol. 2008, 23, 381–390. [Google Scholar] [PubMed]
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005, 338, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Petersen, D.R. An overview of the chemistry and biology of reactive aldehydes. Free Radic. Biol. Med. 2013, 59, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Adachi, T.; Matsui, R.; Xu, S.; Jiang, B.; Zou, M.H.; Kirber, M.; Lieberthal, W.; Cohen, R.A. Quantitative assessment of tyrosine nitration of manganese superoxide dismutase in angiotensin II-infused rat kidney. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Brennand, D.M.; Bradley, N.; Gao, B.; Bruckdorfer, R.; Jacobs, M. 3-Nitrotyrosine in the proteins of human plasma determined by an ELISA method. Biochem. J. 1998, 330, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotan, Y.; Lichtenberg, D.; Pinchuk, I. Lipid peroxidation cannot be used as a universal criterion of oxidative stress. Prog. Lipid Res. 2004, 43, 200–227. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, D.; Pinchuk, I. Oxidative stress, the term and the concept. Biochem. Biophys. Res. Commun. 2015, 461, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Le, N.A. Lipoprotein-associated oxidative stress: A new twist to the postprandial hypothesis. Int. J. Mol. Sci. 2014, 16, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Roberts, L.J.; Daniel, V.C.; Awad, J.A.; Mirochnitchenko, O.; Swift, L.L.; Burk, R.F. Comparison of formation of D2/E2-isoprostanes and F2-isoprostanes in vitro and in vivo--effects of oxygen tension and glutathione. Arch. Biochem. Biophys. 1998, 353, 160–171. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef]
- Slatter, D.A.; Murray, M.; Bailey, A.J. Formation of a dihydropyridine derivative as a potential cross-link derived from malondialdehyde in physiological systems. FEBS Lett. 1998, 421, 180–184. [Google Scholar] [CrossRef] [Green Version]
- Lamore, S.D.; Azimian, S.; Hom, D.; Anglin, B.L.; Uchida, K.; Cabello, C.M.; Wondrak, G.T. The malondialdehyde-derived fluorophore DHP-lysine is a potent sensitizer of UVA-induced photooxidative stress in human skin cells. J. Photochem. Photobiol. B 2010, 101, 251–264. [Google Scholar] [CrossRef]
- Palinski, W.; Ord, V.A.; Plump, A.S.; Breslow, J.L.; Steinberg, D.; Witztum, J.L. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. 1994, 14, 605–616. [Google Scholar] [CrossRef]
- Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Cylooxygenase-dependent formation of the isoprostane, 8-epi prostaglandin F2α. J. Biol. Chem. 1995, 270, 9800–9808. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ciabattoni, G.; Créminon, C.; Lawson, J.; Fitzgerald, G.A.; Patrono, C.; Maclouf, J. Immunological characterization of urinary 8-epi-prostaglandin F2 alpha excretion in man. J. Pharmacol. Exp. Ther. 1995, 275, 94–100. [Google Scholar] [PubMed]
- Lawson, J.A.; Li, H.; Rokach, J.; Adiyaman, M.; Hwang, S.W.; Khanapure, S.P.; FitzGerald, G.A. Identification of two major F2 isoprostanes, 8,12-iso- and 5-epi-8,12-iso-isoprostane F2α-VI, in human urine. J. Biol. Chem. 1998, 273, 29295–29301. [Google Scholar] [CrossRef]
- Praticò, D.; Barry, O.P.; Lawson, J.A.; Adiyaman, M.; Hwang, S.W.; Khanapure, S.P.; Iuliano, L.; Rokach, J.; FitzGerald, G.A. IPF2α-I: An index of lipid peroxidation in humans. Proc. Natl. Acad. Sci. USA 1998, 95, 3449–3454. [Google Scholar] [CrossRef]
- Li, H.; Lawson, J.A.; Reilly, M.; Adiyaman, M.; Hwang, S.W.; Rokach, J.; FitzGerald, G.A. Quantitative high performance liquid chromatography/tandem mass spectrometric analysis of the four classes of F2-isoprostanes in human urine. Proc. Natl. Acad. Sci. USA 1999, 96, 13381–13386. [Google Scholar] [CrossRef] [PubMed]
- Bessard, J.; Cracowski, J.L.; Stanke-Labesque, F.; Bessard, G. Determination of isoprostaglandin F2alpha type III in human urine by gas chromatography-electronic impact mass spectrometry. Comparison with enzyme immunoassay. J. Chromatogr. B Biomed. Sci. Appl. 2001, 754, 333–343. [Google Scholar] [CrossRef]
- Davi, G.; Falco, A.; Patrono, C. Lipid peroxidation in diabetes mellitus. Antioxid. Redox Signal. 2005, 7, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.M.; Barden, A.E.; Loke, W.M.; Croft, K.D.; Puddey, I.B.; Mori, T.A. HDL is the major lipoprotein carrier of plasma F2-isoprostanes. J. Lipid Res. 2009, 50, 716–722. [Google Scholar] [CrossRef]
- Proudfoot, J.M.; Barden, A.E.; Croft, K.D.; Galano, J.M.; Durand, T.; Bultel-Poncé, V.; Giera, M.; Mori, T.A. F2-isoprostanes in HDL are bound to neutral lipids and phospholipids. Free Radic. Res. 2016, 50, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Stafforini, D.M.; Sheller, J.R.; Blackwell, T.S.; Sapirstein, A.; Yull, F.E.; McIntyre, M.; Bonventre, J.V.; Prescott, S.M.; Roberts, L.J., 2nd. Release of free F2-isoprostanes from esterified phospholipids is catalyzed by intracellular and plasmaplatelet-activating factor acetylhydrolases. J. Biol. Chem. 2006, 281, 4616–4623. [Google Scholar] [CrossRef]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef]
- Halliwell, B.; Lee, C.Y. Using isoprostanes as biomarkers of oxidative stress: Some rarely considered issues. Antioxid. Redox Signal. 2010, 13, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Stiegel, M.A.; Pleil, J.D.; Sobus, J.R.; Angrish, M.M.; Morgan, M.K. Kidney injury biomarkers and urinary creatinine variability in nominally healthy adults. Biomarkers 2015, 20, 436–452. [Google Scholar] [CrossRef]
- Bauer, J.; Ripperger, A.; Frantz, S.; Ergün, S.; Schwedhelm, E.; Benndorf, R.A. Pathophysiology of isoprostanes in the cardiovascular system: Implications of isoprostane-mediated thromboxane A2 receptor activation. Br. J. Pharmacol. 2014, 171, 3115–3131. [Google Scholar] [CrossRef] [PubMed]
- Praticó, D.; FitzGerald, G.A. Generation of 8-epiprostaglandin F2alpha by human monocytes. Discriminate production by reactive oxygen species and prostaglandin endoperoxide synthase-2. J. Biol. Chem. 1996, 271, 8919–8924. [Google Scholar] [CrossRef] [PubMed]
- Delannoy, E.; Courtois, A.; Freund-Michel, V.; Leblais, V.; Marthan, R.; Muller, B. Hypoxia-induced hyperreactivity of pulmonary arteries: Role of cyclooxygenase-2, isoprostanes, and thromboxane receptors. Cardiovasc. Res. 2010, 85, 582–592. [Google Scholar] [CrossRef]
- Reilly, M.; Delanty, N.; Lawson, J.A.; FitzGerald, G.A. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation 1996, 94, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D.; Suchy, M.T.; Niemann, J.; Tossios, P.; Schneider, Y.; Rothmann, S.; Gutzki, F.M.; Frölich, J.C.; Stichtenoth, D.O. Glutathione promotes prostaglandin H synthase (cyclooxygenase)-dependent formation of malondialdehyde and 15(S)-8-iso-prostaglandin F2α. FEBS Lett. 2012, 586, 3723–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilz, J.; Meineke, I.; Gleiter, C.H. Measurement of free and bound malondialdehyde in plasma by high-performance liquid chromatography as the 2,4-dinitrophenylhydrazine derivative. J. Chromatogr. B Biomed. Sci. Appl. 2000, 742, 315–325. [Google Scholar] [CrossRef]
- Wade, C.R.; Jackson, P.G.; Van Rij, A.M. Quantitation of malondialdehyde (MDA) in plasma, by ion-pairing reverse phase high performance liquid chromatography. Biochem. Med. 1985, 33, 291–296. [Google Scholar] [CrossRef]
- Yu, L.W.; Latriano, L.; Duncan, S.; Hartwick, R.A.; Witz, G. High-performance liquid chromatography analysis of the thiobarbituric acid adducts of malonaldehyde and trans,trans-muconaldehyde. Anal. Biochem. 1986, 156, 326–333. [Google Scholar] [CrossRef]
- Moselhy, H.F.; Reid, R.G.; Yousef, S.; Boyle, S.P. A specific, accurate, and sensitive measure of total plasma malondialdehyde by HPLC. J. Lipid Res. 2013, 54, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Cordis, G.A.; Das, D.K.; Riedel, W. High-performance liquid chromatographic peak identification of 2,4-dinitrophenylhydrazine derivatives of lipid peroxidation aldehydes by photodiode array detection. J. Chromatogr. A 1998, 798, 117–123. [Google Scholar] [CrossRef]
- Sobsey, C.A.; Han, J.; Lin, K.; Swardfager, W.; Levitt, A.; Borchers, C.H. Development and evaluation of a liquid chromatography–mass spectrometry method for rapid, accurate quantitation of malondialdehyde in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1029–1030, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D.; Rothmann, S.; Schneider, J.Y.; Suchy, M.T.; Trettin, A.; Modun, D.; Stuke, N.; Maassen, N.; Frölich, J.C. Development, validation and biomedical applications of stable-isotope dilution GC-MS and GC-MS/MS techniques for circulating malondialdehyde (MDA) after pentafluorobenzyl bromide derivatization: MDA as a biomarker of oxidative stress and its relation to 15(S)-8-iso-prostaglandin F2α and nitric oxide (NO). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1019, 95–111. [Google Scholar] [PubMed]
- Zelzer, S.; Oberreither, R.; Bernecker, C.; Stelzer, I.; Truschnig-Wilders, M.; Fauler, G. Measurement of total and free malondialdehyde by gas–chromatography mass spectrometry—Comparison with high-performance liquid chromatography methology. Free Radic. Res. 2013, 47, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Verde, V.; Fogliano, V.; Ritieni, A.; Maiani, G.; Morisco, F.; Caporaso, N. Use of N,N-dimethyl-p-phenylenediamine to evaluate the oxidative status of human plasma. Free Radic. Res. 2002, 36, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Ito, T.; Suzuki, C.; Yahata, T.; Ikeda, K.; Hamaoka, K. The Application of a modified d-ROMs test for measurement of oxidative stress and oxidized high-density lipoprotein. Int. J. Mol. Sci. 2017, 18, 454. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.; Qian, Z.; Shen, X. The mechanism of Fe2+-initiated lipid peroxidation in liposomes: The dual function of ferrous ions, the roles of the pre-existing lipid peroxides and the lipid peroxyl radical. Biochem. J. 2000, 352, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B.; Chapman, J.; Fazio, S.; Hussain, M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. High-density lipoprotein subfractions—What the clinicians need to know. Cardiology 2013, 124, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Soran, H.; Schofield, J.D.; Durrington, P.N. Antioxidant properties of HDL. Front. Pharmacol. 2015, 6, 222. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont-Rousselot, D.; Motta, C.; Khalil, A.O.; Sola, R.; La Ville, A.E.; Delattre, J.; Gardes-Albert, M. Physicochemical changes in human high-density lipoprotein (HDL) oxidized by gamma radiolysis-generated oxyradicals. Effect on their cholesterol effluxing capacity. Biochim. Biophys. Acta 1995, 1255, 23–30. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Antiatherogenic function of HDL particle subpopulations: Focus on antioxidative activities. Curr. Opin. Lipidol. 2010, 21, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Lűscher, T.F.; Landmesser, U.; von Eckardstein, A.; Fogelman, A.M. High-density lipoprotein: Vascular protective effects, dysfunction, and potential as therapeutic target. Circ. Res. 2014, 114, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Rasmiena, A.A.; Barlow, C.K.; Ng, T.W.; Tull, D.; Meikle, P.J. High density lipoprotein efficiently accepts surface but not internal oxidized lipids from oxidized low density lipoprotein. Biochim. Biophys. Acta 2016, 1861, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Biomarkers of oxidative stress in red blood cells. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2011, 155, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Kiko, T.; Hatade, K.; Sookwong, P.; Arai, H.; Miyazawa, T. Antioxidant effect of lutein towards phospholipid hydroperoxidation in human erythrocytes. Br. J. Nutr. 2009, 102, 1280–1284. [Google Scholar] [CrossRef] [Green Version]
- Ito, F.; Ito, T. Whole blood assay of oxidative stress by Fe-ROMs test. unpublished; manuscript in preparation.
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular risk factors: The Framingham study. Circulation 1979, 59, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes control and complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Postprandial hyperglycemia and cardiovascular disease is the HEART2D study the answer? Diabetes Care 2009, 32, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Similarity of the impact of type 1 and type 2 diabetes on cardiovascular mortality in middle-aged subjects. Diabetes Care 2008, 31, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In vivo formation of 8-iso prostaglandin F2α and platelet activation in diabetes mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar] [CrossRef]
- Sampson, M.J.; Gopaul, N.; Davies, I.R.; Hughes, D.A.; Carrier, M.J. Plasma F2 isoprostanes: Direct evidence of increased free radical damage during acute hyperglycemia in type 2 diabetes. Diabetes Care 2002, 25, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Chiarelli, F.; Santilli, F.; Pomilio, M.; Vigneri, S.; Falco, A.; Basili, S.; Ciabattoni, G.; Patrono, C. Enhanced lipid peroxidation and platelet activation in the early phase of type 1 diabetes mellitus: Role of interleukin-6 and disease duration. Circulation 2003, 107, 3199–3203. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Falco, A.; Davi, G. Isoprostane formation and inhibition in atherothrombosis. Curr. Opin. Pharmacol. 2005, 5, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Saisho, Y. Glycemic variability and oxidative stress: A link between diabetes and cardiovascular disease? Int. J. Mol. Sci. 2014, 15, 18381–18406. [Google Scholar] [CrossRef]
- Dailey, G. Assessing glycemic control with self-monitoring of blood glucose and hemoglobin A1c measurements. Mayo Clin. Proc. 2007, 82, 229–235. [Google Scholar] [CrossRef]
- DECODE Study Group, the European Diabetes Epidemiology Group. Glucose tolerance and cardiovascular mortality: Comparison of fasting and 2-hour diagnostic criteria. Arch. Intern. Med. 2001, 161, 397–405. [Google Scholar] [CrossRef]
- Cavalot, F.; Petrelli, A.; Traversa, M.; Bonomo, K.; Fiora, E.; Conti, M.; Anfossi, G.; Costa, G.; Trovati, M. Postprandial blood glucose is a stronger predictor of cardiovascular events than fasting blood glucose in type 2 diabetes mellitus, particularly in women: Lessons from the San Luigi Gonzaga Diabetes Study. J. Clin. Endocrinol. Metab. 2006, 91, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Kodani, N.; Saisho, Y.; Tanaka, K.; Kawai, T.; Itoh, H. Effects of mitiglinide, a short-acting insulin secretagogue, on daily glycemic variability and oxidative stress markers in Japanese patients with type 2 diabetes mellitus. Clin. Drug Investig. 2013, 33, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-W.; Park, K.-H.; Zhou, Y.-J. The Impact of Hypoglycemia on the Cardiovascular System: Physiology and Pathophysiology. Angiology 2016, 67, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Feldman-Billard, S.; Massin, P.; Meas, T.; Guillausseau, P.J.; Héron, E. Hypoglycemia-induced blood pressure elevation in patients with diabetes. Arch. Intern. Med. 2010, 170, 829–831. [Google Scholar] [CrossRef]
- Assaloni, R.; Da Ros, R.; Quagliaro, L.; Piconi, L.; Maier, A.; Zuodar, G.; Motz, E.; Ceriello, A. Effects of S21403 (mitiglinide) on postprandial generation of oxidative stress and inflammation in type 2 diabetic patients. Diabetologia 2005, 48, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, M.; Fukui, T.; Ouchi, M.; Watanabe, K.; Suzuki, T.; Yamamoto, S.; Yamamoto, T.; Hayashi, T.; Oba, K.; Hirano, T. Relationship between daily and day-to-day glycemic variability and increased oxidative stress in type 2 diabetes. Diabetes Res. Clin. Pract. 2016, 122, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–32. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Stabler, T.; Kenjale, A.A.; Ham, K.L.; Robbins, J.L.; Duscha, B.D.; Kraus, W.E.; Annex, B.H. Diabetes status differentiates endothelial function and plasma nitrite response to exercise stress in peripheral arterial disease following supervised training. J. Diabetes Complic. 2014, 28, 219–225. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Valls, N.; Brito, R.; Rodrigo, R. Essential hypertension and oxidative stress: New insights. World J. Cardiol. 2014, 6, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Alessandrini, P.; Mezzetti, A.; Minotti, G.; Bucciarelli, T.; Costantini, F.; Cipollone, F.; Bon, G.B.; Ciabattoni, G.; Patrono, C. In vivo formation of 8-epi prostaglandin F2α is increased in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3230–3235. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J.; Framingham Study. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham Study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflug. Arch. 2010, 459, 793–806. [Google Scholar] [CrossRef]
- Bauer, P.M.; Fulton, D.; Boo, Y.C.; Sorescu, G.P.; Kemp, B.E.; Jo, H.; Sessa, W.C. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 14841–14849. [Google Scholar] [CrossRef]
- Montagnani, M.; Chen, H.; Barr, V.A.; Quon, M.J. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser1179. J. Biol. Chem. 2001, 276, 30392–30398. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.D.; Palmer, R.M.; Schulz, R.; Hodson, H.F.; Moncada, S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990, 101, 746–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, H.; Timimi, F.K.; Boles, K.S.; Creager, S.J.; Ganz, P.; Creager, M.A. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1996, 97, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Tesfamariam, B.; Brown, M.L.; Cohen, R.A. Elevated glucose impairs endothelium-dependent relaxation by activating protein kinase C. J. Clin. Investig. 1991, 87, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.B.; Goldfine, A.B.; Timimi, F.K.; Ting, H.H.; Roddy, M.A.; Simonson, D.C.; Creager, M.A. Acute hyperglycemia attenuates endothelium-dependent vasodilation in humans in vivo. Circulation 1998, 97, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Ito, H. Assessment of arterial stiffness using the cardio-ankle vascular index. Pulse (Basel) 2016, 4, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Mattace-Raso, F.U.; van der Cammen, T.J.; Hofman, A.; van Popele, N.M.; Bos, M.L.; Schalekamp, M.A.; Asmar, R.; Reneman, R.S.; Hoeks, A.P.; Breteler, M.M.; et al. Arterial stiffness and risk of coronary heart disease and stroke: The Rotterdam Study. Circulation 2006, 113, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Motoyama, T.; Hirashima, O.; Hirai, N.; Miyao, Y.; Sakamoto, T.; Kugiyama, K.; Ogawa, H.; Yasue, H. Hyperglycemia rapidly suppresses flow-mediated endothelium-dependent vasodilation of brachial artery. J. Am. Coll. Cardiol. 1999, 34, 146–154. [Google Scholar] [CrossRef]
- Beckman, J.A.; Goldfine, A.B.; Gordon, M.B.; Creager, M.A. Ascorbate restores endothelium-dependent vasodilation impaired by acute hyperglycemia in humans. Circulation 2001, 103, 1618–1623. [Google Scholar] [CrossRef]
- Ceriello, A.; Taboga, C.; Tonutti, L.; Quagliaro, L.; Piconi, L.; Bais, B.; Da Ros, R.; Motz, E. Evidence for an independent and cumulative effect of postprandial hypertriglyceridemia and hyperglycemia on endothelial dysfunction and oxidative stress generation: Effects of short- and long-term simvastatin treatment. Circulation 2002, 106, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008, 57, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Risso, A.; Mercuri, F.; Quagliaro, L.; Damante, G.; Ceriello, A. Intermittent high glucose enhances apoptosis in human umbilical vein endothelial cells in culture. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E924–E930. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Kawamori, R.; Toyofuku, Y.; Kitahara, Y.; Sato, F.; Shimizu, T.; Miura, K.; Mine, T.; Tanaka, Y.; Mitsumata, M.; et al. Repetitive fluctuations in blood glucose enhance monocyte adhesion to the endothelium of rat thoracic aorta. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2275–2280. [Google Scholar] [CrossRef] [PubMed]
- Manzato, E.; Zambon, A.; Lapolla, A.; Zambon, S.; Braghetto, L.; Crepaldi, G.; Fedele, D. Lipoprotein abnormalities in well-treated type II diabetic patients. Diabetes Care 1993, 16, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.J.; Jenkins, A.J. Glycation, oxidation, and lipoxidation in the development of the complications of diabetes: A carbonyl stress hypothesis. Diabetes Rev. (Alex) 1997, 5, 365–391. [Google Scholar] [PubMed]
- Njajou, O.T.; Kanaya, A.M.; Holvoet, P.; Connelly, S.; Strotmeyer, E.S.; Harris, T.B.; Cummings, S.R.; Hsueh, W.C.; Health ABC Study. Association between oxidized LDL, obesity and type 2 diabetes in a population-based cohort, the Health, Aging and Body Composition Study. Diabetes Metab. Res. Rev. 2009, 25, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Nakhjavani, M.; Khalilzadeh, O.; Khajeali, L.; Esteghamati, A.; Morteza, A.; Jamali, A.; Dadkhahipour, S. Serum oxidized-LDL is associated with diabetes duration independent of maintaining optimized levels of LDL-cholesterol. Lipids 2010, 45, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Clopton, P.; Brilakis, E.S.; Marcovina, S.M.; Khera, A.; Miller, E.R.; de Lemos, J.A.; Witztum, J.L. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race, apolipoprotein(a) isoform size and cardiovascular risk factors: Results from the Dallas Heart Study. Circulation 2009, 119, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.E.; Thelle, D.S.; Forde, O.H.; Mjos, O.D. The Tromsø Heart Study: High-density lipoprotein and coronary heart disease: A prospective case-control study. Lancet 1977, 1, 965–968. [Google Scholar] [CrossRef]
- Ashen, M.D.; Blumenthal, R.S. Clinical practice: Low HDL cholesterol levels. N. Engl. J. Med. 2005, 353, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tall, A.R. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Ragbir, S.; Farmer, J.A. Dysfunctional high-density lipoprotein and atherosclerosis. Curr. Atheroscler. Rep. 2010, 12, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. Ther. 2012, 10, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Duell, P.B.; Oram, J.F.; Bierman, E.L. Nonenzymatic glycosylation of HDL and impaired HDL-receptor-mediated cholesterol efflux. Diabetes 1991, 40, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Tan, K.C.; Shiu, S.W.; Wong, Y. Increased serum advanced glycation end products are associated with impairment in HDL antioxidative capacity in diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 927–933. [Google Scholar] [CrossRef]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, F.; Sono, Y.; Kondo, K.; Ugi, S.; Matsumoto, M.; Maegawa, H.; Morino, K. Oxidized high-density lipoprotein is associated with vascular endothelial dysfunction in patients with type 2 diabetes mellitus. unpublished; manuscript in preparation.
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Kihara, Y.; Noma, K. Endothelial dysfunction and hypertension in aging. Hypertens. Res. 2012, 35, 1039–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular noxs, and hypertension: Focus on translational and clinical research. Antioxid. Redox Signal. 2014, 20, 164–182. [Google Scholar] [CrossRef]
- Koya, D.; King, G.L. Protein kinase C activation and the development of diabetic complications. Diabetes 1998, 47, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomino, Y.; Hagiwara, S.; Gohda, T. AGE–RAGE interaction and oxidative stress in obesity related renal dysfunction. Kidney Int. 2011, 80, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Lazzerini, G.; Del Turco, S.; Ratto, G.M.; Schmidt, A.M.; De Caterina, R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1401–1407. [Google Scholar] [CrossRef]
- Tan, K.C.; Chow, W.S.; Ai, V.H.; Metz, C.; Bucala, R.; Lam, K.S. Advanced glycation end products and endothelial dysfunction in type 2 diabetes. Diabetes Care 2002, 25, 1055–1059. [Google Scholar] [CrossRef]
- Bae, J.P.; Lage, M.J.; Mo, D.; Nelson, D.R.; Hoogwerf, B.J. Obesity and glycemic control in patients with diabetes mellitus: Analysis of physician electronic health records in the US from 2009–2011. J. Diabetes Complic. 2016, 30, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Flock, M.R.; Green, M.H.; Kris-Etherton, P.M. Effects of adiposity on plasma lipid response to reductions in dietary saturated fatty acids and cholesterol. Adv. Nutr. 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Farb, M.G.; Ganley-Leal, L.; Mott, M.; Liang, Y.; Ercan, B.; Widlansky, M.E.; Bigornia, S.J.; Fiscale, A.J.; Apovian, C.M.; Carmine, B.; et al. Arteriolar function in visceral adipose tissue is impaired in human obesity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K.; Nishizawa, H.; Funahashi, T.; Shimomura, I.; Shimabukuro, M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ. J. 2006, 70, 1437–1442. [Google Scholar] [CrossRef]
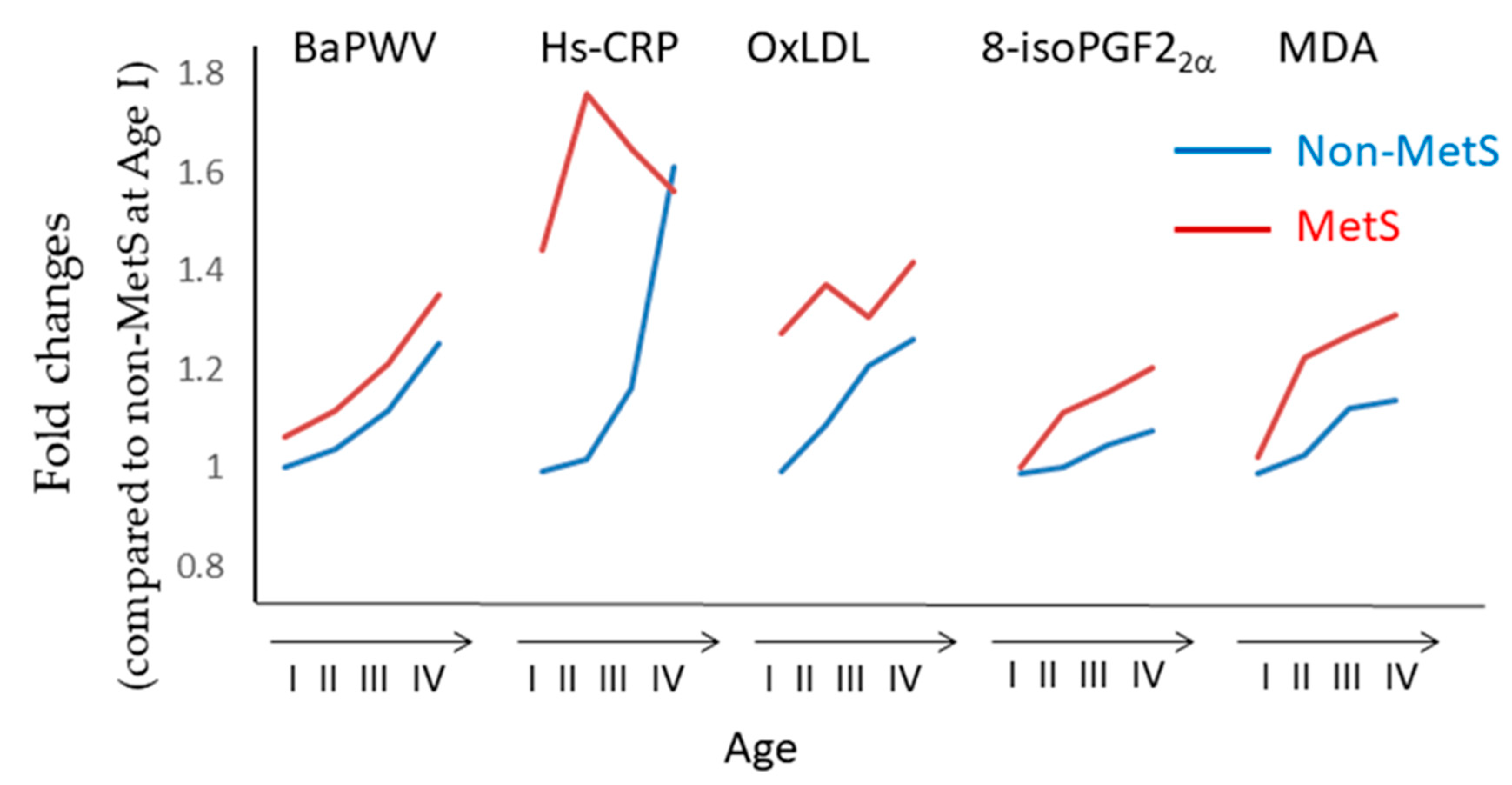
- Morelli, N.R.; Scavuzzi, B.M.; Miglioranza, L.H.D.S.; Lozovoy, M.A.B.; Simão, A.N.C.; Dichi, I. Metabolic syndrome components are associated with oxidative stress in overweight and obese patients. Arch. Endocrinol. Metab. 2018, 62, 309–318. [Google Scholar] [CrossRef]
- Yokota, T.; Kinugawa, S.; Yamato, M.; Hirabayashi, K.; Suga, T.; Takada, S.; Harada, K.; Morita, N.; Oyama-Manabe, N.; et al. Systemic oxidative stress is associated with lower aerobic capacity and impaired skeletal muscle energy metabolism in patients with metabolic syndrome. Diabetes Care 2013, 36, 1341–1346. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S. Body mass index, diabetes, and C-reactive protein among U.S. adults. Diabetes Care 1999, 22, 1971–1977. [Google Scholar] [CrossRef]
- Festa, A.; D’Agostino, R., Jr.; Howard, G.; Mykkanen, L.; Tracy, R.P.; Haffner, S.M. Chronic subclinical inflammation as part of the insulin resistance syndrome: The Insulin Resistance Atherosclerosis Study (IRAS). Circulation 2000, 102, 42–47. [Google Scholar] [CrossRef]
- Sesso, H.D.; Buring, J.E.; Rifai, N.; Blake, G.J.; Gaziano, J.M.; Ridker, P.M. C-reactive protein and the risk of developing hypertension. JAMA 2003, 290, 2945–2951. [Google Scholar] [CrossRef] [PubMed]
- Pearle, A.D.; Scanzello, C.R.; George, S.; Mandl, L.A.; DiCarlo, E.F.; Peterson, M.; Sculco, T.P.; Crow, M.K. Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarthr. Cartil. 2007, 15, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Daghestani, H.N.; Kraus, V.B. Inflammatory biomarkers in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1890–1896. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid. Med. Cell Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Fröhlich, M.; Imhof, A.; Berg, G.; Hutchinson, W.L.; Pepys, M.B.; Boeing, H.; Muche, R.; Brenner, H.; Koenig, W. Association between C-reactive protein and features of the metabolic syndrome: A population-based study. Diabetes Care 2000, 23, 1835–1839. [Google Scholar] [CrossRef]
- Albert, M.A.; Glynn, R.J.; Ridker, P.M. Plasma concentration of C-reactive protein and the calculated Framingham Coronary Heart Disease Risk Score. Circulation 2003, 108, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Cottone, S.; Mulè, G.; Nardi, E.; Vadalà, A.; Guarneri, M.; Briolotta, C.; Arsena, R.; Palermo, A.; Riccobene, R.; Cerasola, G. Relation of C-reactive protein to oxidative stress and to endothelial activation in essential hypertension. Am. J. Hypertens. 2006, 19, 313–318. [Google Scholar] [CrossRef]
- Kim, M.; Kim, M.; Yoo, H.J.; Lee, S.Y.; Lee, S.H.; Lee, J.H. Age-specific determinants of pulse wave velocity among metabolic syndrome components, inflammatory markers, and oxidative stress. J. Atheroscler. Thromb. 2018, 25, 178–185. [Google Scholar] [CrossRef]
- Verma, S.; Wang, C.H.; Li, S.H.; Dumont, A.S.; Fedak, P.W.; Badiwala, M.V.; Dhillon, B.; Weisel, R.D.; Li, R.K.; Mickle, D.A.; et al. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation 2002, 106, 913–919. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. https://doi.org/10.3390/antiox8030072
Ito F, Sono Y, Ito T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants. 2019; 8(3):72. https://doi.org/10.3390/antiox8030072
Chicago/Turabian StyleIto, Fumiaki, Yoko Sono, and Tomoyuki Ito. 2019. "Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation" Antioxidants 8, no. 3: 72. https://doi.org/10.3390/antiox8030072