The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies




Abstract
:1. Introduction
2. Reactive Oxygen Species—Friend or Foe?
3. Redox Homeostasis
3.1. The NRF2 Pathway in Tumorigenesis
3.2. The Thioredoxin System and Thioredoxin-Domain-Containing Protein Family in Tumorigenesis
3.3. The Glutathione System in Tumorigenesis
4. Modulation of Antioxidant Defense Systems in Anticancer Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Pryczynicz, A.; Dymicka-Piekarska, V.; Kamińska, J.; Koper-Lenkiewicz, O.; Matowicka-Karna, J.; Kędra, B.; Zalewska, A.; et al. Pro-oxidant enzymes, redox balance and oxidative damage to proteins, lipids and DNA in colorectal cancer tissue. Is oxidative stress dependent on tumour budding and inflammatory infiltration? Cancers 2020, 12, 1636. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Cipak, A.; Schaur, R.J.; Zarkovic, N. Pathophysiology of neutrophil-mediated extracellular redox reactions. Front. Biosci. Landmark 2016, 21, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Milkovic, L.; Gegotek, A.; Cindric, M.; Zarkovic, K.; Skrzydlewska, E.; Zarkovic, N. The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Brun, T.; Cnop, M.; Cunha, D.A.; Eizirik, D.L.; Maechler, P. Transient oxidative stress damages mitochondrial machinery inducing persistent β-cell dysfunction. J. Biol. Chem. 2009, 284, 23602–23612. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Tirosh, O.; Cohen, G.; Sasson, S.; Zarkovic, N. Reactive aldehydes-second messengers of free radicals in diabetes mellitus. Free Radic. Res. 2013, 47, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Zivkovic, M.; Poljak-Blazi, M.; Egger, G.; Sunjic, S.B.; Schaur, R.J.; Zarkovic, N. Oxidative burst and anticancer activities of rat neutrophils. BioFactors 2005, 24, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, M.; Poljak-Blazi, M.; Zarkovic, K.; Mihaljevic, D.; Schaur, R.J.; Zarkovic, N. Oxidative burst of neutrophils against melanoma B16-F10. Cancer Lett. 2007, 246, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Poljak-Blazi, M.; Zarkovic, K.; Schaur, R.J.; Zarkovic, N. The involvement of granulocytes in spontaneous regression of Walker 256 carcinoma. Cancer Lett. 2008, 260, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Poljak-Blazi, M.; Schaur, R.J.; Zarkovic, K.; Borovic, S.; Cipak, A.; Cindric, M.; Uchida, K.; Waeg, G.; Zarkovic, N. Elevated neutrophil elastase and acrolein-protein adducts are associated with W256 regression. Clin. Exp. Immunol. 2012, 170, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Poljak-Blazi, M.; Kirac, I.; Borovic, S.; Joerg Schaur, R.; Zarkovic, N. Granulocytes as effective anticancer agent in experimental solid tumor models. Immunobiology 2010, 215, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Matijevic Glavan, T.; Zarkovic, N. The Role of Acrolein and NADPH Oxidase in the Granulocyte-Mediated Growth-Inhibition of Tumor Cells. Cells 2019, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Al-Thani, A.M.; Voss, S.C.; Al-Menhali, A.S.; Barcaru, A.; Horvatovich, P.; Al Jaber, H.; Nikolovski, Z.; Latiff, A.; Georgakopoulos, C.; Merenkov, Z.; et al. Whole blood storage in CPDA1 blood bags alters erythrocyte membrane proteome. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Poljak-Blazi, M.; Jaganjac, M.; Sabol, I.; Mihaljevic, B.; Matovina, M.; Grce, M. Effect of ferric ions on reactive oxygen species formation, cervical cancer cell lines growth and E6/E7 oncogene expression. Toxicol. Vitr. 2011, 25, 160–166. [Google Scholar] [CrossRef]
- Kukulj, S.; Jaganjac, M.; Boranic, M.; Krizanac, S.; Santic, Z.; Poljak-Blazi, M. Altered iron metabolism, inflammation, transferrin receptors, and ferritin expression in non-small-cell lung cancer. Med. Oncol. 2010, 27, 268–277. [Google Scholar] [CrossRef]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Sunjic, S.B.; Zarkovic, N. Utilizing iron for targeted lipid peroxidation as anticancer option of integrative biomedicine: A short review of nanosystems containing iron. Antioxidants 2020, 9, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chen, X.; Li, Z.; Ye, W.; Ding, H.; Li, P.; Aung, L.H.H. Role of RNA oxidation in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5022. [Google Scholar] [CrossRef] [PubMed]
- Schauenstein, E. Autoxidation of polyunsaturated esters in water: Chemical structure and biological activity of the products. J. Lipid Res. 1967, 8, 417–428. [Google Scholar] [PubMed]
- Schaur, R.J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 149–159. [Google Scholar] [CrossRef]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteom. 2013, 92, 239–247. [Google Scholar] [CrossRef]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Jaganjac, M.; Čačev, T.; Čipak, A.; Kapitanović, S.; Trošelj, K.G.; Žarković, N. Even stressed cells are individuals: Second messengers of free radicals in pathophysiology of cancer. Croat. Med. J. 2012, 53, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Al-Menhali, A.S.; Banu, S.; Angelova, P.R.; Barcaru, A.; Horvatovich, P.; Abramov, A.Y.; Jaganjac, M. Lipid peroxidation is involved in calcium dependent upregulation of mitochondrial metabolism in skeletal muscle. Biochim. Biophys. Acta Gen. Subj. 2020, 1864. [Google Scholar] [CrossRef]
- Elrayess, M.A.; Almuraikhy, S.; Kafienah, W.; Al-Menhali, A.; Al-Khelaifi, F.; Bashah, M.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Alsayrafi, M.; et al. 4-hydroxynonenal causes impairment of human subcutaneous adipogenesis and induction of adipocyte insulin resistance. Free Radic. Biol. Med. 2017, 104, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganjac, M.; Almuraikhy, S.; Al-Khelaifi, F.; Al-Jaber, M.; Bashah, M.; Mazloum, N.A.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Kafienah, W.; et al. Combined metformin and insulin treatment reverses metabolically impaired omental adipogenesis and accumulation of 4-hydroxynonenal in obese diabetic patients. Redox Biol. 2017, 12, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrakovcic, L.; Wildburger, R.; Jaganjac, M.; Cindric, M.; Cipak, A.; Sunjic, S.B.; Waeg, G.; Milankovic, A.M.; Zarkovic, N. Lipid peroxidation product 4-hydroxynonenal as factor of oxidative homeostasis supporting bone regeneration with bioactive glasses. Acta Biochim. Pol. 2010, 57, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipak, A.; Mrakovcic, L.; Ciz, M.; Lojek, A.; Mihaylova, B.; Goshev, I.; Jaganjac, M.; Cindric, M.; Sitic, S.; Margaritoni, M.; et al. Growth suppression of human breast carcinoma stem cells by lipid peroxidation product 4-hydroxy-2-nonenal and hydroxyl radical-modified collagen. Acta Biochim. Pol. 2010, 57, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Semlitsch, T.; Tillian, H.M.; Zarkovic, N.; Borovic, S.; Purtscher, M.; Hohenwarter, O.; Schaur, R.J. Differential influence of the lipid peroxidation product 4-hydroxynonenal on the growth of human lymphatic leukaemia cells and human periopherial blood lymphocytes. Anticancer Res. 2002, 22, 1689–1697. [Google Scholar] [PubMed]
- Kreuzer, T.; Zarković, N.; Grube, R.; Schaur, R.J. Inhibition of HeLa cell proliferation by 4-hydroxynonenal is associated with enhanced expression of the c-fos oncogene. Cancer Biother. Radiopharm. 1997, 12, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.H.; Chan, S.H.H. Activation of endogenous antioxidants as a common therapeutic strategy against cancer, neurodegeneration and cardiovascular diseases: A lesson learnt from DJ-1. Pharmacol. Ther. 2015, 156, 69–74. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; De La Vega, M.R.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [Green Version]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. Nrf2 activation in cancer: From DNA to protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Huang, M.T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.N.T. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [Green Version]
- Becks, L.; Prince, M.; Burson, H.; Christophe, C.; Broadway, M.; Itoh, K.; Yamamoto, M.; Mathis, M.; Orchard, E.; Shi, R.; et al. Aggressive mammary carcinoma progression in Nrf2 knockout mice treated with 7,12-dimethylbenz[a]anthracene. BMC Cancer 2010, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Shibata, T.; Takaya, K.; Shiraishi, K.; Kohno, T.; Kunitoh, H.; Tsuta, K.; Furuta, K.; Goto, K.; Hosoda, F.; et al. Regulatory Nexus of Synthesis and Degradation Deciphers Cellular Nrf2 Expression Levels. Mol. Cell. Biol. 2013, 33, 2402–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.C.; Ambrosone, C.B.; Ahn, J.; Choi, J.Y.; McCullough, M.L.; Stevens, V.L.; Rodriguez, C.; Thun, M.J.; Calle, E.E. Genetic variability in iron-related oxidative stress pathways (Nrf2, NQ01, NOS3, and HO-1), iron intake, and risk of postmenopausal breast cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, D.; Ma, Y.; Xu, X.; Zhu, Z.; Wang, X.; Deng, H.; Li, C.; Chen, M.; Tong, J.; et al. Continuous activation of Nrf2 and its target antioxidant enzymes leads to arsenite-induced malignant transformation of human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2015, 289, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Levings, D.C.; Wang, X.; Kohlhase, D.; Bell, D.A.; Slattery, M. A distinct class of antioxidant response elements is consistently activated in tumors with NRF2 mutations. Redox Biol. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, J.A.; Stehr, H.; Das, M.; Padda, S.K.; Ramchandran, K.; Neal, J.W.; Diehn, M.; Wakelee, H.A. Impact of KEAP1/NFE2L2/CUL3 mutations on duration of response to EGFR tyrosine kinase inhibitors in EGFR mutated non-small cell lung cancer. Lung Cancer 2019, 134, 42–45. [Google Scholar] [CrossRef]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence inKRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Qian, Z.; Zhou, T.; Gurguis, C.I.; Xu, X.; Wen, Q.; Lv, J.; Fang, F.; Hecker, L.; Cress, A.E.; Natarajan, V.; et al. Nuclear factor, erythroid 2-like 2-associated molecular signature predicts lung cancer survival. Sci. Rep. 2015, 5, 16889. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Shitara, M.; Yokota, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. MRP3 gene expression correlates with NRF2 mutations in lung squamous cell carcinomas. Mol. Med. Rep. 2012, 6, 705–708. [Google Scholar] [CrossRef]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ma, J.; Lu, Y.; Zhang, S.; Huang, J.; Chen, J.; Bei, J.X.; Yang, K.; Wu, G.; Huang, K.; et al. CDK20 interacts with KEAP1 to activate NRF2 and promotes radiochemoresistance in lung cancer cells. Oncogene 2017, 36, 5321–5330. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. P62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Yang, F.; Li, J.; Deng, H.; Wang, Y.; Lei, C.; Wang, Q.; Xiang, J.; Liang, L.; Xia, J.; Pan, X.; et al. GSTZ 1-1 Deficiency Activates NRF 2/IGF 1R Axis in HCC via Accumulation of Oncometabolite Succinylacetone. EMBO J. 2019, 38, e101964. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, F.P.; Costantini, M.; Copetti, M.; la Torre, A.; Sparaneo, A.; Fontana, A.; Poeta, L.; Gallucci, M.; Sentinelli, S.; Graziano, P.; et al. Keap1/Nrf2 pathway in kidney cancer: Frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 2017, 8, 11187–11198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e18. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Ge, M.K.; Zhang, N.; Xia, L.; Zhang, C.; Dong, S.S.; Li, Z.M.; Ji, Y.; Zheng, M.H.; Sun, J.; Chen, G.Q.; et al. FBXO22 degrades nuclear PTEN to promote tumorigenesis. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, J.; Ning, D.; Liu, Q.; Wang, C.; Zhang, Z.; Chu, L.; Yu, C.; Liang, H.F.; Zhang, B.; et al. FBXO22 promotes the development of hepatocellular carcinoma by regulating the ubiquitination and degradation of p21. J. Exp. Clin. Cancer Res. 2019, 38, 101. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Xie, H.Y.; Qian, J.X.; Huang, Y.N.; Yang, F.; Zhang, F.L.; Shao, Z.M.; Li, D.Q. FBXO22 possesses both protumorigenic and antimetastatic roles in breast cancer progression. Cancer Res. 2018, 78, 5274–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Bowles, K.M.; MacEwan, D.J. High basal nuclear levels of Nrf2 in acute myeloid leukemia reduces sensitivity to proteasome inhibitors. Cancer Res. 2011, 71, 1999–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Ren, Y.; Yan, X.; Luo, Y.; Zhang, H.; Kesarwani, M.; Bu, J.; Zhan, D.; Zhou, Y.; Tang, Y.; et al. The high NRF2 expression confers chemotherapy resistance partly through up-regulated DUSP1 in myelodysplastic syndromes. Haematologica 2019, 104, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.P.; Hayashi, T.; Cottam, H.B.; Jin, G.; Yao, S.; Wu, C.C.N.; Rosenbach, M.D.; Corr, M.; Schwab, R.B.; Carson, D.A. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 7479–7484. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Lopez, E.; Ghia, E.M.; Antonucci, L.; Sharma, N.; Rassenti, L.Z.; Xu, J.; Sun, B.; Kipps, T.J.; Karin, M. NF-κB-p62-NRF2 survival signaling is associated with high ROR1 expression in chronic lymphocytic leukemia. Cell Death Differ. 2020, 27, 2206–2216. [Google Scholar] [CrossRef]
- Yi, X.; Zhao, Y.; Xue, L.; Zhang, J.; Qiao, Y.; Jin, Q.; Li, H. Expression of keap1 and nrf2 in diffuse large b-cell lymphoma and its clinical significance. Exp. Ther. Med. 2018, 16, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Kari, E.; Teppo, H.R.; Haapasaari, K.M.; Kuusisto, M.E.L.; Lemma, A.; Karihtala, P.; Pirinen, R.; Soini, Y.; Jantunen, E.; Turpeenniemi-Hujanen, T.; et al. Nuclear factor erythroid 2-related factors 1 and 2 are able to define the worst prognosis group among high-risk diffuse large B cell lymphomas treated with R-CHOEP. J. Clin. Pathol. 2019, 72, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; Konig, K.; Michels, S.; Fischer, R.; et al. Clinical and pathological characteristics of KEAP1- and NFE2L2-mutated Non–Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Goeman, F.; De Nicola, F.; Scalera, S.; Sperati, F.; Gallo, E.; Ciuffreda, L.; Pallocca, M.; Pizzuti, L.; Krasniqi, E.; Barchiesi, G.; et al. Mutations in the KEAP1-NFE2L2 Pathway Define a Molecular Subset of Rapidly Progressing Lung Adenocarcinoma. J. Thorac. Oncol. 2019, 14, 1924–1934. [Google Scholar] [CrossRef]
- Aljohani, H.M.; Aittaleb, M.; Furgason, J.M.; Amaya, P.; Deeb, A.; Chalmers, J.J.; Bahassi, E.M. Genetic mutations associated with lung cancer metastasis to the brain. Mutagenesis 2018, 33, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, H.L.; Lee, K.B.; Park, J.H.; Chung, W.Y.; Lee, K.S.; Sheen, S.S.; Park, K.J.; Hwang, S.C. Nuclear factor E2-related factor 2 dependent overexpression of sulfiredoxin and peroxiredoxin III in human lung cancer. Korean J. Intern. Med. 2011, 26, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Hikosaka, Y.; Okuda, K.; Kawano, O.; Moriyama, S.; Yano, M.; Fujii, Y. NFE2L2 gene mutation in male japanese squamous cell carcinoma of the lung. J. Thorac. Oncol. 2010, 5, 786–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Cheng, C.; Wang, J.; Wang, J.; Qu, Z.; Ren, H.; Li, Y.; Ning, Q.; Chen, M.; Hu, T. Loss of Beclin1 Expression and Nrf2 Overexpression are Associated with Poor Survival of Patients with Non-Small Cell Lung Cancer. Anticancer Agents Med. Chem. 2018, 18, 1680–1687. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Koh, Y.; Ando, M.; Ito, N.; Takeo, S.; Adachi, H.; Tagawa, T.; Kakegawa, S.; Yamashita, M.; Kataoka, K.; et al. Prospective analysis of oncogenic driver mutations and environmental factors: Japan molecular epidemiology for lung cancer study. J. Clin. Oncol. 2016, 34, 2247–2257. [Google Scholar] [CrossRef]
- Choi, M.; Kadara, H.; Zhang, J.; Parra, E.R.; Rodriguez-Canales, J.; Gaffney, S.G.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Mutation profiles in early-stage lung squamous cell carcinoma with clinical follow-up and correlation with markers of immune function. Ann. Oncol. 2017, 28, 83–89. [Google Scholar] [CrossRef]
- Cescon, D.W.; She, D.; Sakashita, S.; Zhu, C.Q.; Pintilie, M.; Shepherd, F.A.; Tsao, M.S. NRF2 pathway activation and adjuvant chemotherapy benefit in lung squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 2499–2505. [Google Scholar] [CrossRef] [Green Version]
- Cardnell, R.J.G.; Behrens, C.; Diao, L.; Fan, Y.H.; Tang, X.; Tong, P.; Minna, J.D.; Mills, G.B.; Heymach, J.V.; Wistuba, I.I.; et al. An integrated molecular analysis of lung adenocarcinomas identifies potential therapeutic targets among TTF1-negative tumors, including DNA Repair Proteins and Nrf2. Clin. Cancer Res. 2015, 21, 3480–3491. [Google Scholar] [CrossRef] [Green Version]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Gao, Z.; Li, F.; Li, X.; Sun, Y.; Wang, M.; Li, D.; Wang, R.; Li, F.; Fang, R.; et al. Whole exome sequencing identifies frequent somatic mutations in cell-cell adhesion genes in Chinese patients with lung squamous cell carcinoma. Sci. Rep. 2015, 5, 14237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, 1865–1876. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.K.; Singh, A.; Biswal, S.; Askin, F.; Gabrielson, E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J. Hum. Genet. 2011, 56, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Der Yeh, S.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Sasaki, H.; Suzuki, A.; Shitara, M.; Hikosaka, Y.; Okuda, K.; Moriyama, S.; Yano, M.; Fujii, Y. Genotype analysis of the nrf2 gene mutation in lung cancer. Int. J. Mol. Med. 2013, 31, 1135–1138. [Google Scholar] [CrossRef] [Green Version]
- Onodera, Y.; Motohashi, H.; Takagi, K.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Hirakawa, H.; Sasano, H.; Yamamoto, M.; et al. NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr. Relat. Cancer 2014, 21, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Hartikainen, J.M.; Tengström, M.; Winqvist, R.; Jukkola-Vuorinen, A.; Pylkäs, K.; Kosma, V.M.; Soini, Y.; Mannermaa, A. KEAP1 genetic polymorphisms associate with breast cancer risk and survival outcomes. Clin. Cancer Res. 2015, 21, 1591–1601. [Google Scholar] [CrossRef] [Green Version]
- Hartikainen, J.M.; Tengström, M.; Kosma, V.M.; Kinnula, V.L.; Mannermaa, A.; Soini, Y. Genetic polymorphisms and protein expression of NRF2 and sulfiredoxin predict survival outcomes in breast cancer. Cancer Res. 2012, 72, 5537–5546. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B.; Goebel, G.; Hackl, H.; Fiegl, H. Reduced mRNA expression levels of NFE2L2 are associated with poor outcome in breast cancer patients. BMC Cancer 2016, 16, 821. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.; Soares, M.; Ramalhinho, A.C.; Moutinho, J.F.; Breitenfeld, L. Prognosis of hormone-dependent breast cancer seems to be influenced by KEAP1, NRF2 and GSTM1 genetic polymorphisms. Mol. Biol. Rep. 2019, 46, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Seibold, P.; Hall, P.; Schoof, N.; Nevanlinna, H.; Heikkinen, T.; Benner, A.; Liu, J.; Schmezer, P.; Popanda, O.; Flesch-Janys, D.; et al. Polymorphisms in oxidative stress-related genes and mortality in breast cancer patients—Potential differential effects by radiotherapy? Breast 2013, 22, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Roy Chowdhury, S.; Mandal, G.; Purohit, S.; Gupta, A.; Bhattacharyya, A. RelA driven co-expression of CXCL13 and CXCR5 is governed by a multifaceted transcriptional program regulating breast cancer progression. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, E.S.; Koo, J.S. Expression of pentose phosphate pathway-related proteins in breast cancer. Dis. Markers 2018, 2018. [Google Scholar] [CrossRef]
- Hart, P.C.; Ratti, B.A.; Mao, M.; Ansenberger-Fricano, K.; Shajahan-Haq, A.N.; Tyner, A.L.; Minshall, R.D.; Bonini, M.G. Caveolin-1 regulates cancer cell metabolism via scavenging Nrf2 and suppressing MnSOD-driven glycolysis. Oncotarget 2016, 7, 308–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Johnson, A.; Ali, S.M.; Klempner, S.J.; Bekaii-Saab, T.; Vacirca, J.L.; Khaira, D.; Yelensky, R.; Chmielecki, J.; Elvin, J.A.; et al. Comprehensive Genomic Profiling of Advanced Esophageal Squamous Cell Carcinomas and Esophageal Adenocarcinomas Reveals Similarities and Differences. Oncologist 2015, 20, 1132–1139. [Google Scholar] [CrossRef] [Green Version]
- Erkizan, H.V.; Johnson, K.; Ghimbovschi, S.; Karkera, D.; Trachiotis, G.; Adib, H.; Hoffman, E.P.; Wadleigh, R.G. African-American esophageal squamous cell carcinoma expression profile reveals dysregulation of stress response and detox networks. BMC Cancer 2017, 17, 426. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zheng, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic Alterations in Esophageal Tissues from Squamous Dysplasia to Carcinoma. Gastroenterology 2017, 153, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Kitano, Y.; Baba, Y.; Nakagawa, S.; Miyake, K.; Iwatsuki, M.; Ishimoto, T.; Yamashita, Y.-I.Y.I.; Yoshida, N.; Watanabe, M.; Nakao, M.; et al. Nrf2 promotes oesophageal cancer cell proliferation via metabolic reprogramming and detoxification of reactive oxygen species. J. Pathol. 2018, 244, 346–357. [Google Scholar] [CrossRef]
- Hao, J.J.; Lin, D.C.; Dinh, H.Q.; Mayakonda, A.; Jiang, Y.Y.; Chang, C.; Jiang, Y.; Lu, C.C.; Shi, Z.Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507. [Google Scholar] [CrossRef]
- Deng, J.; Chen, H.; Zhou, D.; Zhang, J.; Chen, Y.; Liu, Q.; Ai, D.; Zhu, H.; Chu, L.; Ren, W.; et al. Comparative genomic analysis of esophageal squamous cell carcinoma between Asian and Caucasian patient populations. Nat. Commun. 2017, 8, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 508, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 induces NRF2 stabilization by directly targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, Y.; Ishigami, S.; Arigami, T.; Uenosono, Y.; Yanagita, S.; Uchikado, Y.; Kita, Y.; Nishizono, Y.; Okumura, H.; Nakajo, A.; et al. Clinicopathological significance of nuclear factor (erythroid-2)-related factor 2 (Nrf2) expression in gastric cancer. BMC Cancer 2015, 15, 5. [Google Scholar] [CrossRef] [Green Version]
- Soini, Y.; Eskelinen, M.; Juvonen, P.; Kärjä, V.; Haapasaari, K.M.; Saarela, A.; Karihtala, P. Nuclear Nrf2 expression is related to a poor survival in pancreatic adenocarcinoma. Pathol. Res. Pract. 2014, 210, 35–39. [Google Scholar] [CrossRef]
- Eichenmüller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Häberle, B.; Cairo, S.; Leuschner, I.; Von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [Green Version]
- Ma-on, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Lu, Y.F.; Chen, H.; Shen, Z.Y.; Liu, J. Liver expression of Nrf2-related genes in different liver diseases. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 485–491. [Google Scholar] [CrossRef]
- Chen, J.; Yu, Y.; Ji, T.; Ma, R.; Chen, M.; Li, G.; Li, F.; Ding, Q.; Kang, Q.; Huang, D.; et al. Clinical implication of Keap1 and phosphorylated Nrf2 expression in hepatocellular carcinoma. Cancer Med. 2016, 5, 2678–2687. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Inoue, K.I.; Hachiya, H.; Shibuya, N.; Shimoda, M.; Kubota, K. Frequent alteration of the protein synthesis of enzymes for glucose metabolism in hepatocellular carcinomas. J. Gastroenterol. 2014, 49, 1324–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef]
- Shimokawa, M.; Yoshizumi, T.; Itoh, S.; Iseda, N.; Sakata, K.; Yugawa, K.; Toshima, T.; Harada, N.; Ikegami, T.; Mori, M. Modulation of Nqo1 activity intercepts anoikis resistance and reduces metastatic potential of hepatocellular carcinoma. Cancer Sci. 2020, 111, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Stawiski, E.W.; Durinck, S.; Gowda, H.; Goldstein, L.D.; Barbhuiya, M.A.; Schröder, M.S.; Sreenivasamurthy, S.K.; Kim, S.W.; Phalke, S.; et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat. Commun. 2020, 11, 4225. [Google Scholar] [CrossRef]
- Zhan, M.; Wang, H.; Xu, S.W.; Yang, L.H.; Chen, W.; Zhao, S.X.; Shen, H.; Liu, Q.; Yang, R.M.; Wang, J. Variants in oxidative stress-related genes affect the chemosensitivity through Nrf2-mediated signaling pathway in biliary tract cancer. EBioMedicine 2019, 48, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.C.; Fan, C.W.; Tseng, W.K.; Chen, J.R.; Chein, H.P.; Hwang, C.C.; Hua, C.C. Immunohistochemical study of the Nrf2 pathway in colorectal cancer: Nrf2 expression is closely correlated to keap1 in the tumor and bach1 in the normal tissue. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 511–517. [Google Scholar] [CrossRef]
- Chang, L.-C.; Fan, C.-W.; Tseng, W.-K.; Chein, H.-P.; Hsieh, T.-Y.; Chen, J.-R.; Hwang, C.-C.; Hua, C.-C. The Ratio of Hmox1/Nrf2 mRNA Level in the Tumor Tissue Is a Predictor of Distant Metastasis in Colorectal Cancer. Dis. Markers 2016, 2016, 8143465. [Google Scholar] [CrossRef]
- El-Deek, H.E.M.; Ahmed, A.M.; Mohammed, R.A.A. Aberration of Nrf2-Bach1 pathway in colorectal carcinoma; role in carcinogenesis and tumor progression. Ann. Diagn. Pathol. 2019, 38, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, B.; Kahaly, M.; Mayr, D.; Schmoeckel, E.; Niesler, B.; Kolben, T.; Burges, A.; Mahner, S.; Jeschke, U.; Trillsch, F. Interaction of ERα and NRF2 impacts survival in ovarian cancer patients. Int. J. Mol. Sci. 2019, 20, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinse, G.; Just, P.A.; Rance, B.; Izac, B.; Letourneur, F.; Saidu, N.E.B.; Chouzenoux, S.; Nicco, C.; Goldwasser, F.; Pasmant, E.; et al. The NRF2 transcriptional target NQO1 has low mRNA levels in TP53-mutated endometrial carcinomas. PLoS ONE 2019, 14, e0214416. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Yi, X.; Abushahin, N.; Pang, S.; Zhang, D.; Kong, B.; Zheng, W. Nrf2 expression in endometrial serous carcinomas and its precancers. Int. J. Clin. Exp. Pathol. 2011, 4, 85–96. [Google Scholar]
- Stacy, D.R.; Ely, K.; Massion, P.P.; Yarbrough, W.G.; Hallahan, D.E.; Sekhar, K.R.; Freeman, M.L. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck 2006, 28, 813–818. [Google Scholar] [CrossRef]
- Namani, A.; Matiur Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Hämälaïnen, M.; Teppo, H.R.; Skarp, S.; Haapasaari, K.M.; Porvari, K.; Vuopala, K.; Kietzmann, T.; Karihtala, P. NRF1 and NRF2 mRNA and Protein Expression Decrease Early during Melanoma Carcinogenesis: An Insight into Survival and MicroRNAs. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen Peroxide Sensing and Signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Storr, S.J.; Al-hadyan, K.; Rahman, R.; Smith, S.; Grundy, R.; Paine, S.; Martin, S.G. Thioredoxin System Protein Expression Is Associated with Poor Clinical Outcome in Adult and Paediatric Gliomas and Medulloblastomas. Mol. Neurobiol. 2020, 57, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Tobe, R.; Carlson, B.A.; Tsuji, P.A.; Lee, B.J.; Gladyshev, V.N.; Hatfield, D.L. Differences in redox regulatory systems in human lung and liver tumors suggest different avenues for therapy. Cancers 2015, 7, 2262–2276. [Google Scholar] [CrossRef]
- Ran, D.-M.; Zhang, Q.-W.; Su, H.-L.; Wang, C.; Gao, F.-H. Expression of thioredoxin reductase-1 and its effect in non-small cell lung cancer. Int. J. Clin. Exp. Med. 2016, 9, 7608–7614. [Google Scholar]
- Lee, J.R.; Roh, J.-L.; Lee, S.M.; Park, Y.; Cho, K.-J.; Choi, S.-H.; Nam, S.Y.; Kim, S.Y. Overexpression of glutathione peroxidase 1 predicts poor prognosis in oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2017, 143, 2257–2265. [Google Scholar] [CrossRef]
- Iwasawa, S.; Yamano, Y.; Takiguchi, Y.; Tanzawa, H.; Tatsumi, K.; Uzawa, K. Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncol. Rep. 2011, 25, 637–644. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, C.; Peng, B. Overexpression of thioredoxin system proteins predicts poor prognosis in patients with squamous cell carcinoma of the tongue. Oral Oncol. 2011, 47, 609–614. [Google Scholar] [CrossRef]
- Bu, L.; Li, W.; Ming, Z.; Shi, J.; Fang, P.; Yang, S. Inhibition of TrxR2 suppressed NSCLC cell proliferation, metabolism and induced cell apoptosis through decreasing antioxidant activity. Life Sci. 2017, 178, 35–41. [Google Scholar] [CrossRef]
- Yoon, B.-I.; Kim, Y.H.; Yi, J.-Y.; Kang, M.-S.; Jang, J.-J.; Joo, K.-H.; Kim, Y.; McHugh Law, J.; Kim, D.-Y. Expression of thioredoxin during progression of hamster and human cholangiocarcinoma. Cancer Sci. 2010, 101, 281–288. [Google Scholar] [CrossRef]
- Nagano, M.; Hatakeyama, K.; Kai, M.; Nakamura, H.; Yodoi, J.; Asada, Y.; Chijiiwa, K. Nuclear expression of thioredoxin-1 in the invasion front is associated with outcome in patients with gallbladder carcinoma. HPB 2012, 14, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Hamid, N.M.; Mahmoud, T.K.; Abass, S.A.; El-Shishtawy, M.M. Expression of thioredoxin and glutaredoxin in experimental hepatocellular carcinoma—Relevance for prognostic and diagnostic evaluation. Pathophysiology 2018, 25, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, D.T.; Al-Yatama, F.; Mohammed, F.M.A.; Al-Banaw, A.G.; Al-Bader, M.; Burge, M.; Sinowatz, F.; Singal, P.K. Thioredoxin and thioredoxin reductase expression in thyroid cancer depends on tumour aggressiveness. Anticancer Res. 2010, 30, 767–776. [Google Scholar] [PubMed]
- Gollapalli, K.; Ghantasala, S.; Atak, A.; Rapole, S.; Moiyadi, A.; Epari, S.; Srivastava, S. Tissue Proteome Analysis of Different Grades of Human Gliomas Provides Major Cues for Glioma Pathogenesis. Omics J. Integr. Biol. 2017, 21, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Zhang, P.; Zuo, Z.; Wang, F.; Bi, R.; Shang, W.; Wu, A.; Ye, J.; Li, S.; Sun, X.; et al. Thioredoxin-1 promotes colorectal cancer invasion and metastasis through crosstalk with S100P. Cancer Lett. 2017, 401, 1–10. [Google Scholar] [CrossRef]
- Lim, J.Y.; Yoon, S.O.; Hong, S.W.; Kim, J.W.; Choi, S.H.; Cho, J.Y. Thioredoxin and thioredoxin-interacting protein as prognostic markers for gastric cancer recurrence. World J. Gastroenterol. 2012, 18, 5581–5588. [Google Scholar] [CrossRef]
- Shang, W.; Xie, Z.; Lu, F.; Fang, D.; Tang, T.; Bi, R.; Chen, L.; Jiang, L. Increased Thioredoxin-1 Expression Promotes Cancer Progression and Predicts Poor Prognosis in Patients with Gastric Cancer. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Samaranayake, G.J.; Troccoli, C.I.; Huynh, M.; Lyles, R.D.Z.; Kage, K.; Win, A.; Lakshmanan, V.; Kwon, D.; Ban, Y.; Chen, S.X.; et al. Thioredoxin-1 protects against androgen receptor-induced redox vulnerability in castration-resistant prostate cancer. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Li, J.; Yue, Z.; Xiong, W.; Sun, P.; You, K.; Wang, J. TXNIP overexpression suppresses proliferation and induces apoptosis in SMMC7221 cells through ROS generation and MAPK pathway activation. Oncol. Rep. 2017, 37, 3369–3376. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.H.; Xie, R.; Xie, S.; Wu, Y.; Wang, W.; Li, X.; Xu, Y.; Liu, B.; Zhou, Y.; Wang, T.; et al. Thioredoxin interacting protein (TXNIP) acts as a tumor suppressor in human prostate cancer. Cell Biol. Int. 2020. [Google Scholar] [CrossRef]
- Morrison, J.A.; Pike, L.A.; Sams, S.B.; Sharma, V.; Zhou, Q.; Severson, J.J.; Tan, A.-C.; Wood, W.M.; Haugen, B.R. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noura, M.; Matsuo, H.; Koyama, A.; Adachi, S.; Masutani, H. TXNIP induces growth arrest and enhances ABT263-induced apoptosis in mixed-lineage leukemia-rearranged acute myeloid leukemia cells. FEBS Open Bio 2020, 10, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.; Huang, W.; Zhang, W.; Xu, J.; Song, W.; Wang, Y.; Zhu, A.; Luo, J.; Huang, G.; Wang, Y.; et al. TXNIP interaction with the Her-1/2 pathway contributes to overall survival in breast cancer. Oncotarget 2015, 6, 3003–3012. [Google Scholar] [CrossRef] [PubMed]
- da Motta, L.L.; De Bastiani, M.A.; Stapenhorst, F.; Klamt, F. Oxidative stress associates with aggressiveness in lung large-cell carcinoma. Tumor Biol. 2015, 36, 4681–4688. [Google Scholar] [CrossRef]
- Mo, R.; Peng, J.; Xiao, J.; Ma, J.; Li, W.; Wang, J.; Ruan, Y.; Ma, S.; Hong, Y.; Wang, C.; et al. High TXNDC5 expression predicts poor prognosis in renal cell carcinoma. Tumor Biol. 2016, 37, 9797–9806. [Google Scholar] [CrossRef]
- Tan, F.; Zhu, H.; He, X.; Yu, N.; Zhang, X.; Xu, H.; Pei, H. Role of TXNDC5 in tumorigenesis of colorectal cancer cells: In vivo and in vitro evidence. Int. J. Mol. Med. 2018, 42, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Vincent, E.E.; Elder, D.J.E.; Phillips, L.; Heesom, K.J.; Pawade, J.; Luckett, M.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavaré, J.M. Overexpression of the TXNDC5 protein in non-small cell lung carcinoma. Anticancer Res. 2011, 31, 1577–1582. [Google Scholar]
- Wu, Z.; Zhang, L.; Li, N.; Sha, L.; Zhang, K. An immunohistochemical study of thioredoxin domain-containing 5 expression in gastric adenocarcinoma. Oncol. Lett. 2015, 9, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Wangpu, X.; Han, D.; Feng, H.; Zhao, J.; Ma, J.; Qu, S.; Chen, X.; Liu, B.; Zheng, M. TXNDC9 expression in colorectal cancer cells and its influence on colorectal cancer prognosis. Cancer Investig. 2012, 30, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Zhao, R.; Sun, F.; Lu, Q.; Wang, X.; Hu, J.; Wang, S.; Gao, L.; Zhou, Q.; Xiong, X.; et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 2020, 39, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-F.; Wang, X.-Y.; Fu, Z.-Q.; Peng, Q.-H.; Zhang, J.-Y.; Ye, F.; Fu, Y.-F.; Zhou, C.-Y.; Lu, W.-G.; Cheng, X.-D.; et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 2015, 11, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhani, M.; Taheri, A.-R.; Jafarian, A.-H.; Hashemy, S.I. The activity and tissue distribution of thioredoxin reductase in basal cell carcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 2303–2307. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, A.; Sahaf, B.; Rosén, A. Thioredoxin reductase, a redox-active selenoprotein, is secreted by normal and neoplastic cells: Presence in human plasma. Cancer Res. 2000, 60, 2281–2289. [Google Scholar] [PubMed]
- Shao, L.-E.; Tanaka, T.; Gribi, R.; Yu, J.; Diccianni, M.B.; Yu, A.L.; Pullen, J.D.; Camitta, B.M. Thioredoxin expression in primary T-cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001, 61, 7333–7338. [Google Scholar]
- Cai, Z.; Zhang, C.; Zou, Y.; Lu, C.; Hu, H.; Qian, J.; Jiang, L.; Hu, G. Tissue thioredoxin-interacting protein expression predicted recurrence in patients with meningiomas. Int. J. Clin. Oncol. 2017, 22, 660–666. [Google Scholar] [CrossRef]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Tao, X.; Zhou, L.; Sheng, B.; Zhe, X.; Zhu, X. Expression of thioredoxin 1 and peroxiredoxins in squamous cervical carcinoma and its predictive role in NACT. BMC Cancer 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhao, X.; Li, K.; Luo, G.; Nie, Y.; Shi, Y.; Zhou, Y.; Ren, G.; Feng, B.; Liu, Z.; et al. Thioredoxin-like protein 2 is overexpressed in colon cancer and promotes cancer cell metastasis by interaction with ran. Antioxid. Redox Signal. 2013, 19, 899–911. [Google Scholar] [CrossRef]
- Peng, W.; Zhou, Z.; Zhong, Y.; Sun, Y.; Wang, Y.; Zhu, Z.; Jiao, W.; Bai, M.; Sun, J.; Yin, H.; et al. Plasma activity of Thioredoxin Reductase as a Novel Biomarker in Gastric Cancer. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-L.; Sun, K.-Y.; Tan, Q.; Diao, X.-F.; Liu, T. Expression and prognosis of TrxR in non-small cell lung cancer tissues. Chin. J. Cancer Prev. Treat. 2016, 23, 1224–1228. [Google Scholar]
- Fernandes, A.P.; Capitanio, A.; Selenius, M.; Brodin, O.; Rundlöf, A.-K.; Björnstedt, M. Expression profiles of thioredoxin family proteins in human lung cancer tissue: Correlation with proliferation and differentiation. Histopathology 2009, 55, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylväs, M.; Puistola, U.; Kauppila, S.; Soini, Y.; Karihtala, P. Oxidative stress-induced antioxidant enzyme expression is an early phenomenon in ovarian carcinogenesis. Eur. J. Cancer 2010, 46, 1661–1667. [Google Scholar] [CrossRef]
- Chaiswing, L.; Zhong, W.; Oberley, T.D. Increasing discordant antioxidant protein levels and enzymatic activities contribute to increasing redox imbalance observed during human prostate cancer progression. Free Radic. Biol. Med. 2014, 67, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metere, A.; Frezzotti, F.; Graves, C.E.; Vergine, M.; De Luca, A.; Pietraforte, D.; Giacomelli, L. A possible role for selenoprotein glutathione peroxidase (GPx1) and thioredoxin reductases (TrxR1) in thyroid cancer: Our experience in thyroid surgery. Cancer Cell Int. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, P.; Ramasamy, P.; Ramasamy, P.; Larkin, A.-M.; Larkin, A.-M.; Linge, A.; Tiernan, D.; McAree, F.; Horgan, N.; Moriarty, P.; et al. PRDX3 is associated with metastasis and poor survival in uveal melanoma. J. Clin. Pathol. 2020, 73, 408–412. [Google Scholar] [CrossRef]
- González, R.; Rodríguez-Hernández, M.A.; Negrete, M.; Ranguelova, K.; Rossin, A.; Choya-Foces, C.; Cruz-Ojeda, P.D.L.; Miranda-Vizuete, A.; Martínez-Ruiz, A.; Rius-Pérez, S.; et al. Downregulation of thioredoxin-1-dependent CD95 S-nitrosation by Sorafenib reduces liver cancer. Redox Biol. 2020, 34. [Google Scholar] [CrossRef]
- Shen, X.; Burguillos, M.A.; Osman, A.M.; Frijhoff, J.; Carrillo-Jiménez, A.; Kanatani, S.; Augsten, M.; Saidi, D.; Rodhe, J.; Kavanagh, E.; et al. Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 2016, 17, 1282–1290. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef]
- Zuo, Z.; Zhang, P.; Lin, F.; Shang, W.; Bi, R.; Lu, F.; Wu, J.; Jiang, L. Interplay between Trx-1 and S100P promotes colorectal cancer cell epithelial–mesenchymal transition by up-regulating S100A4 through AKT activation. J. Cell. Mol. Med. 2018, 22, 2430–2441. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Lu, W.; Han, Y.; Yang, J.; Jiang, W.; You, X.; Luo, Y.; Wen, S.; Hu, Y.; et al. Mitochondrial TXNRD3 confers drug resistance via redox-mediated mechanism and is a potential therapeutic target in vivo. Redox Biol. 2020, 36. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Wang, G.; Zhang, J.; Han, Q. Inhibiting TrxR suppresses liver cancer by inducing apoptosis and eliciting potent antitumor immunity. Oncol. Rep. 2018, 40, 3447–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. Cross-talk between two antioxidants, thioredoxin reductase and heme oxygenase-1, and therapeutic implications for multiple myeloma. Redox Biol. 2016, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; Quan, Y.; Zhan, M.; Liao, H.; Li, Y.; Lu, L. MiR-125b-5p inhibits cell proliferation, migration, and invasion in hepatocellular carcinoma via targeting TXNRD1. Cancer Cell Int. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.; Xu, X.; Ma, J.; Xia, J.; Dai, B.; Liu, L.; Ma, Y. MicroRNA-124 regulates the radiosensitivity of non-small cell lung cancer cells by targeting TXNRD1. Oncol. Lett. 2017, 13, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhang, Y.; Ding, J.; Hu, W.; Tan, C.; Wang, M.; Tang, J.; Xu, Y. miR-17-3p Downregulates Mitochondrial Antioxidant Enzymes and Enhances the Radiosensitivity of Prostate Cancer Cells. Mol. Ther. Nucleic Acids 2018, 13, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Engelman, R.; Ziv, T.; Arnér, E.S.J.; Benhar, M. Inhibitory nitrosylation of mammalian thioredoxin reductase 1: Molecular characterization and evidence for its functional role in cellular nitroso-redox imbalance. Free Radic. Biol. Med. 2016, 97, 375–385. [Google Scholar] [CrossRef]
- Wright, D.E.; Altaany, Z.; Bi, Y.; Alperstein, Z.; O’Donoghue, P. Acetylation regulates thioredoxin reductase oligomerization and activity. Antioxid. Redox Signal. 2018, 29, 377–388. [Google Scholar] [CrossRef]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Miao, L.-Y.; Xiao, Y.-L.; Huang, M.; Yu, M.; Meng, K.; Cai, H.-R. Hypoxia induced high expression of thioredoxin interacting protein (TXNIP) in non-small cell lung cancer and its prognostic effect. Asian Pac. J. Cancer Prev. 2015, 16, 2953–2958. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, Z.; Ma, Q.; Liu, J.; Xu, Q.; Han, L.; Duan, W.; Lv, Y.; Wang, F.; Reindl, K.M.; et al. Hyperglycemia regulates TXNIP/TRX/ROS axis via p38 MAPK and ERK pathways in pancreatic cancer. Curr. Cancer Drug Targets 2014, 14, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Ayer, D.E. Ras suppresses TXNIP expression by restricting ribosome translocation. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuljaca, S.; Liu, T.; Dwarte, T.; Kavallaris, M.; Haber, M.; Norris, M.D.; Martin-Caballero, J.; Marshall, G.M. The cyclin-dependent kinase inhibitor, p21WAF1, promotes angiogenesis by repressing gene transcription of thioredoxin-binding protein 2 in cancer cells. Carcinogenesis 2009, 30, 1865–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Zhou, L.; Liu, H.; Shan, Y.; Zhang, X. MicroRNA-224 promotes pancreatic cancer cell proliferation and migration by targeting the TXNIP-mediated HIF1α pathway. Cell. Physiol. Biochem. 2018, 48, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Dang, B.-L.; Huang, J.; Chen, M.; Wu, D.; Xu, M.-L.; Li, R.; Yan, G.-R. MiR-373 drives the epithelial-to-mesenchymal transition and metastasis via the miR-373-TXNIP-HIF1a-TWIST signaling axis in breast cancer. Oncotarget 2015, 6, 32701–32712. [Google Scholar] [CrossRef] [Green Version]
- Masaki, S.; Masutani, H.; Yoshihara, E.; Yodoi, J. Deficiency of Thioredoxin binding protein-2 (TBP-2) enhances TGF-β signaling and promotes epithelial to mesenchymal transition. PLoS ONE 2012, 7, e39900. [Google Scholar] [CrossRef]
- Zhang, B.; Lyu, J.; Yang, E.J.; Liu, Y.; Wu, C.; Pardeshi, L.; Tan, K.; Chen, Q.; Xu, X.; Deng, C.-X.; et al. Class I histone deacetylase inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells. Acta Pharm. Sin. B 2020, 10, 615–627. [Google Scholar] [CrossRef]
- Lee, J.-H.; Jeong, E.-G.; Choi, M.-C.; Kim, S.-H.; Park, J.-H.; Song, S.-H.; Park, J.; Bang, Y.-J.; Kim, T.-Y. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol. Cells 2010, 30, 107–112. [Google Scholar] [CrossRef]
- Zhang, B.; Lyu, J.; Liu, Y.; Wu, C.; Yang, E.J.; Pardeshi, L.; Tan, K.; Wong, K.H.; Chen, Q.; Xu, X.; et al. BRCA1 deficiency sensitizes breast cancer cells to bromodomain and extra-terminal domain (BET) inhibition. Oncogene 2018, 37, 6341–6356. [Google Scholar] [CrossRef]
- Hong, S.Y.; Yu, F.-X.; Luo, Y.; Hagen, T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell. Signal. 2016, 28, 377–383. [Google Scholar] [CrossRef]
- Ho, B.; Huang, G.; Golubovskaya, V.M. Focal adhesion kinase regulates expression of thioredoxin-interacting protein (TXNIP) in cancer cells. Anticancer Agents Med. Chem. 2014, 14, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riahi, Y.; Kaiser, N.; Cohen, G.; Abd-Elrahman, I.; Blum, G.; Shapira, O.M.; Koler, T.; Simionescu, M.; Sima, A.V.; Zarkovic, N.; et al. Foam cell-derived 4-hydroxynonenal induces endothelial cell senescence in a TXNIP-dependent manner. J. Cell. Mol. Med. 2015, 19, 1887–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, G.; Martinotti, S.; Fazio, V.; Manzari, V.; Paradisi, L.; Parola, M.; Frati, L.; Dianzani, M.U. Effect of 4-Hydroxynonenal on c-myc Expression. Toxicol. Pathol. 1987, 15, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Mohankumar, K.; Lacey, A.; Safe, S. Inhibition of NR4A1 promotes ROS accumulation and IL24-dependent growth arrest in rhabdomyosarcoma. Mol. Cancer Res. 2019, 17, 2221–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, E.; Lee, S.-O.; Kim, G.; Abdelrahim, M.; Jin, U.-H.; Safe, S.; Abudayyeh, A. Nuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinoma. PLoS ONE 2015, 10, e0128308. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Lee, S.-O.; Doddapaneni, R.; Singh, M.; Safe, S. Nuclear receptor 4A1 as a drug target for breast cancer chemotherapy. Endocr. Relat. Cancer 2015, 22, 831–840. [Google Scholar] [CrossRef]
- Xu, B.; Li, J.; Liu, X.; Li, C.; Chang, X. TXNDC5 is a cervical tumor susceptibility gene that stimulates cell migration, vasculogenic mimicry and angiogenesis by downregulating SERPINF1 and TRAF1 expression. Oncotarget 2017, 8, 91009–91024. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Song, G.; Chang, X.; Tan, W.; Pan, J.; Zhu, X.; Liu, Z.; Qi, M.; Yu, J.; Han, B. The role of TXNDC5 in castration-resistant prostate cancer—Involvement of androgen receptor signaling pathway. Oncogene 2015, 34, 4735–4745. [Google Scholar] [CrossRef] [Green Version]
- Gurjar, S.A.; Wheeler, J.X.; Wadhwa, M.; Thorpe, R.; Kimber, I.; Derrick, J.P.; Dearman, R.J.; Metcalfe, C. The impact of thioredoxin reduction of allosteric disulfide bonds on the therapeutic potential of monoclonal antibodies. J. Biol. Chem. 2019, 294, 19616–19634. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.-L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid. Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef]
- Chen, X.; Tang, W.; Liu, S.; Yu, L.; Chen, Z. Thioredoxin-1 phosphorylated at T100 is needed for its anti-apoptotic activity in HepG2 cancer cells. Life Sci. 2010, 87, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, M.; Kwon, Y.-W.; Yodoi, J.; Masutani, H. Thioredoxin regulates cell cycle via the ERK1/2-cyclin d1 pathway. Antioxid. Redox Signal. 2009, 11, 2957–2971. [Google Scholar] [CrossRef] [PubMed]
- Reynoso, E.; Liu, H.; Li, L.; Yuan, A.L.; Chen, S.; Wang, Z. Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J. Biol. Chem. 2017, 292, 17514–17524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.R.; Cappabianca, L.; Desantis, G.; Ianni, N.D.; Ruggeri, P.; Ragone, M.; Merolle, S.; Tonissen, K.F.; Gulino, A.; MacKay, A.R. Thioredoxin stimulates MMP-9 expression, de-regulates the MMP-9/TIMP-1 equilibrium and promotes MMP-9 dependent invasion in human MDA-MB-231 breast cancer cells. FEBS Lett. 2011, 585, 3328–3336. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kan, G.; Mao, Y.; Wu, Z.; Tan, X.; He, H.; Lee, C. UHRF1 downmodulation enhances antitumor effects of histone deacetylase inhibitors in retinoblastoma by augmenting oxidative stress-mediated apoptosis. Mol. Oncol. 2020, 14, 329–346. [Google Scholar] [CrossRef] [Green Version]
- Chepikova, O.E.; Malin, D.; Strekalova, E.; Lukasheva, E.V.; Zamyatnin, A.A.; Cryns, V.L. Lysine oxidase exposes a dependency on the thioredoxin antioxidant pathway in triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020. [Google Scholar] [CrossRef]
- Shin, B.; Feser, R.; Nault, B.; Hunter, S.; Maiti, S.; Ugwuagbo, K.C.; Majumder, M. miR526b and miR655 induce oxidative stress in breast cancer. Int. J. Mol. Sci. 2019, 20, 4039. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Yang, M.; Zhou, B.; Luo, J.; Zhang, Z.; Zhang, W.; Yan, Z. CircRNA-104718 acts as competing endogenous RNA and promotes hepatocellular carcinoma progression through microRNA-218-5p/TXNDC5 signaling pathway. Clin. Sci. 2019, 133, 1487–1503. [Google Scholar] [CrossRef]
- Chawsheen, H.A.; Jiang, H.; Ying, Q.; Ding, N.; Thapa, P.; Wei, Q. The redox regulator sulfiredoxin forms a complex with thioredoxin domain–containing 5 protein in response to ER stress in lung cancer cells. J. Biol. Chem. 2019, 294, 8991–9006. [Google Scholar] [CrossRef]
- Kamitori, K.; Yamaguchi, F.; Dong, Y.; Hossain, A.; Katagi, A.; Noguchi, C.; Hirata, Y.; Tsukamoto, I.; Hatano, N.; Tokuda, M. Both Ser361 phosphorylation and the C-arrestin domain of thioredoxin interacting protein are important for cell cycle blockade at the G1/S checkpoint. FEBS Open Bio 2018, 8, 1804–1819. [Google Scholar] [CrossRef]
- Qu, X.; Sun, J.; Zhang, Y.; Li, J.; Hu, J.; Li, K.; Gao, L.; Shen, L. c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 415–421. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukthapura, A.; Shimogga, A.; Sudha, K.V.; Shetty, B.; Rao, G.M. Oxidative products of proteins and antioxidant potential of thiols in gastric carcinoma patients. J. Med. Biochem. 2010, 29, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.U.; Mahjabeen, I.; Malik, F.A.; Kayani, M.A. Prognostic significance of altered blood and tissue glutathione levels in head and neck squamous cell carcinoma cases. Asian Pac. J. Cancer Prev. 2014, 15, 7603–7609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murawaki, Y.; Tsuchiya, H.; Kanbe, T.; Harada, K.; Yashima, K.; Nozaka, K.; Tanida, O.; Kohno, M.; Mukoyama, T.; Nishimuki, E.; et al. Aberrant expression of selenoproteins in the progression of colorectal cancer. Cancer Lett. 2008, 259, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Patel, A.K.; Kumari, R.; Chugh, S.; Shrivastav, C.; Mehra, S.; Sharma, A.N. Interactions between oxidative stress, lipid profile and antioxidants in breast cancer: A case control study. Asian Pac. J. Cancer Prev. 2012, 13, 6295–6298. [Google Scholar] [CrossRef] [Green Version]
- Himmetoglu, S.; Dincer, Y.; Ersoy, Y.E.; Bayraktar, B.; Celik, V.; Akcay, T. DNA oxidation and antioxidant status in breast cancer. J. Investig. Med. 2009, 57, 720–723. [Google Scholar] [CrossRef]
- Sehitogullari, A.; Aslan, M.; Sayir, F.; Kahraman, A.; Demir, H. Serum paraoxonase-1 enzyme activities and oxidative stress levels in patients with esophageal squamous cell carcinoma. Redox Rep. 2014, 19, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Tripathi, M.; Satyam, A.; Kumar, L. Study of antioxidant levels in patients with multiple myeloma. Leuk. Lymphoma 2009, 50, 809–815. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sabet, S.; Peng, D.-F.; Nouh, M.A.; El-Shinawi, M.; El-Rifai, W. Promoter hypermethylation and suppression of glutathione peroxidase 3 are associated with inflammatory breast carcinogenesis. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, T.; Zhu, S.; Mukaisho, K.; El-Rifai, W.; Peng, D.-F. Glutathione peroxidase 7 suppresses cancer cell growth and is hypermethylated in gastric cancer. Oncotarget 2017, 8, 54345–54356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Bai, W.; Huang, F.; Tang, J.; Lin, X. Downregulation of microRNA-196a inhibits stem cell self-renewal ability and stemness in non-small-cell lung cancer through upregulating GPX3 expression. Int. J. Biochem. Cell Biol. 2019, 115. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Yu, X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/snail signaling. Oncogene 2018, 37, 5843–5857. [Google Scholar] [CrossRef] [PubMed]
- Naiki, T.; Naiki-Ito, A.; Iida, K.; Etani, T.; Kato, H.; Suzuki, S.; Yamashita, Y.; Kawai, N.; Yasui, T.; Takahashi, S. GPX2 promotes development of bladder cancer with squamous cell differentiation through the control of apoptosis. Oncotarget 2018, 9, 15847–15859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cao, P.; Alshwmi, M.; Jiang, N.; Xiao, Z.; Jiang, F.; Gu, J.; Wang, X.; Sun, X.; Li, S. GPX2 suppression of H2O2 stress regulates cervical cancer metastasis and apoptosis via activation of the β-catenin-WNT pathway. Onco Targets. Ther. 2019, 12, 6639–6651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Sun, L.; Tong, J.; Chen, X.; Li, H.; Zhang, Q. Prognostic significance of glutathione peroxidase 2 in gastric carcinoma. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Kinowaki, Y.; Kurata, M.; Ishibashi, S.; Ikeda, M.; Tatsuzawa, A.; Yamamoto, M.; Miura, O.; Kitagawa, M.; Yamamoto, K. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab. Investig. 2018, 98, 609–619. [Google Scholar] [CrossRef]
- Liu, X.; Sui, X.; Zhang, C.; Wei, K.; Bao, Y.; Xiong, J.; Zhou, Z.; Chen, Z.; Wang, C.; Zhu, H.; et al. Glutathione S-transferase A1 suppresses tumor progression and indicates better prognosis of human primary hepatocellular carcinoma. J. Cancer 2020, 11, 83–91. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, J.; Zhang, J.; Wang, Z.; Yu, Y.; Miao, M.; Yao, Q. Dual roles of glutathione S-transferase mu 1 in the development and metastasis of hepatocellular carcinoma. Biomed. Pharmacother. 2019, 120. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Yang, Y.; Lei, C.; Yang, F.; Liang, L.; Chen, C.; Xia, J.; Wang, K.; Tang, N. GSTZ1 deficiency promotes hepatocellular carcinoma proliferation via activation of the KEAP1/NRF2 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 438. [Google Scholar] [CrossRef]
- Gómez-Martín, A.; Martinez-Gonzalez, L.J.; Puche-Sanz, I.; Cozar, J.M.; Lorente, J.A.; Hernández, A.F.; Alvarez-Cubero, M.J. GSTM1 gene expression and copy number variation in prostate cancer patients—Effect of chemical exposures and physical activity. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 290.e9–290.e15. [Google Scholar] [CrossRef] [PubMed]
- Djukic, T.; Simic, T.; Pljesa-Ercegovac, M.; Matic, M.; Suvakov, S.; Coric, V.; Dragicevic, D.; Savic-Radojevic, A. Upregulated glutathione transferase omega-1 correlates with progression of urinary bladder carcinoma. Redox Rep. 2017, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Bulus, H.; Oguztuzun, S.; Güler Simsek, G.; Kilic, M.; Ada, A.O.; Göl, S.; Kocdogan, A.K.; Kaygın, P.; Bozer, B.; Iscan, M. Expression of CYP and GST in human normal and colon tumor tissues. Biotech. Histochem. 2019, 94, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tan, N.; Liao, H.; Pan, G.; Xu, Q.; Zhu, R.; Zou, L.; He, S.; Zhu, H. High GSTP1 inhibits cell proliferation by reducing Akt phosphorylation and is associated with a better prognosis in hepatocellular carcinoma. Oncotarget 2018, 9, 8957–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, K.; Zheng, Z.; Huang, Y. Long intergenic noncoding RNA 00844 promotes apoptosis and represses proliferation of prostate cancer cells through upregulating GSTP1 by recruiting EBF1. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hafez, M.M.; Al-Shabanah, O.A.; Al-Rejaie, S.S.; Al-Harbi, N.O.; Hassan, Z.K.; Alsheikh, A.; Al Theyab, A.I.; Aldelemy, M.L.; Sayed-Ahmed, M.M. Increased Hypermethylation of Glutathione S-transferase P1, DNA-binding protein inhibitor, death associated protein kinase and paired box protein-5 genes in triple-negative breast cancer Saudi females. Asian Pac. J. Cancer Prev. 2015, 16, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richiardi, L.; Fiano, V.; Grasso, C.; Zugna, D.; Delsedime, L.; Gillio-Tos, A.; Merletti, F. Methylation of APC and GSTP1 in Non-Neoplastic Tissue Adjacent to Prostate Tumour and Mortality from Prostate Cancer. PLoS ONE 2013, 8, e68162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.-Y.; Kim, Y.-W.; Kang, H.-W.; Kim, W.T.; Yun, S.-J.; Lee, S.-C.; Kim, W.-J.; Kim, Y.-J. DNA methylation of GSTP1 in human prostate tissues: Pyrosequencing analysis. Korean J. Urol. 2012, 53, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Dumache, R.; Puiu, M.; Motoc, M.; Vernic, C.; Dumitrascu, V. Prostate cancer molecular detection in plasma samples by glutathione S-transferase P1 (GSTP1) methylation analysis. Clin. Lab. 2014, 60, 847–852. [Google Scholar] [CrossRef]
- Mahon, K.L.; Qu, W.; Lin, H.-M.; Spielman, C.; Cain, D.; Jacobs, C.; Stockler, M.R.; Higano, C.S.; de Bono, J.S.; Chi, K.N.; et al. Serum Free Methylated Glutathione S-transferase 1 DNA Levels, Survival, and Response to Docetaxel in Metastatic, Castration-resistant Prostate Cancer: Post Hoc Analyses of Data from a Phase 3 Trial. Eur. Urol. 2019, 76, 306–312. [Google Scholar] [CrossRef]
- Hopkins, J.; Tudhope, G.R. Glutathione Peroxidase in Human Red Cells in Health and Disease. Br. J. Haematol. 1973, 25, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Devi, G.S.; Prasad, M.H.; Saraswathi, I.; Raghu, D.; Rao, D.N.; Reddy, P.P. Free radicals antioxidant enzymes and lipid peroxidation in different types of leukemias. Clin. Chim. Acta 2000, 293, 53–62. [Google Scholar] [CrossRef]
- Oltra, A.M.; Carbonell, F.; Tormos, C.; Iradi, A.; Sáez, G.T. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic. Biol. Med. 2001, 30, 1286–1292. [Google Scholar] [CrossRef]
- Moscow, J.A.; Fairchild, C.R.; Madden, M.J.; Ransom, D.T.; Wieand, H.S.; O’Brien, E.E.; Poplack, D.G.; Cossman, J.; Myers, C.E.; Cowan, K.H. Expression of Anionic Glutathione-S-transferase and P-Glycoprotein Genes in Human Tissues and Tumors. Cancer Res. 1989, 49, 1422–1428. [Google Scholar]
- Tome, M.E.; Johnson, D.B.F.; Rimsza, L.M.; Roberts, R.A.; Grogan, T.M.; Miller, T.P.; Oberley, L.W.; Briehl, M.M. A redox signature score identifies diffuse large B-cell lymphoma patients with a poor prognosis. Blood 2005, 106, 3594–3601. [Google Scholar] [CrossRef] [Green Version]
- Kangari, P.; Farahany, T.Z.; Golchin, A.; Ebadollahzadeh, S.; Salmaninejad, A.; Mahboob, S.A.; Nourazarian, A. Enzymatic antioxidant and lipid peroxidation evaluation in the newly diagnosed breast cancer patients in Iran. Asian Pac. J. Cancer Prev. 2018, 19, 3511–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diakowska, D.; Nienartowicz, M.; Grabowski, K. Activities of antioxidant enzymes in erythrocytes and tumor tissue in colorectal cancer patients. Gastroenterol. Pol. 2013, 20, 10–14. [Google Scholar]
- Zhu, X.; Wang, J.; Li, L.; Deng, L.; Wang, J.; Liu, L.; Zeng, R.; Wang, Q.; Zheng, Y. GPX3 suppresses tumor migration and invasion via the FAK/AKT pathway in esophageal squamous cell carcinoma. Am. J. Transl. Res. 2018, 10, 1908–1920. [Google Scholar]
- Handayani, E.; Edianto, D.; Sahil, M.F.; Barus, R.P.; Tobing, C.L.; Ardiansyah, E.; Yaznil, M.R. Glutathione peroxidase in ovarian cancer patients in Indonesia. S. Afr. J. Obstet. Gynaecol. 2019, 25. [Google Scholar] [CrossRef]
- Agnani, D.; Camacho-Vanegas, O.; Camacho, C.; Lele, S.; Odunsi, K.; Cohen, S.; Dottino, P.; Martignetti, J.A. Decreased levels of serum glutathione peroxidase 3 are associated with papillary serous ovarian cancer and disease progression. J. Ovarian Res. 2011, 4. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.-D.; Zhang, Y.-N.; Wang, L.-F. GPX7 promotes the growth of human papillary thyroid carcinoma via enhancement of cell proliferation and inhibition of cell apoptosis. Transl. Cancer Res. 2019, 8, 2570–2580. [Google Scholar] [CrossRef]
- Chew, S.H.; Okazaki, Y.; Akatsuka, S.; Wang, S.; Jiang, L.; Ohara, Y.; Ito, F.; Saya, H.; Sekido, Y.; Toyokuni, S. Rheostatic CD44 isoform expression and its association with oxidative stress in human malignant mesothelioma. Free Radic. Biol. Med. 2017, 106, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019, 29, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Kulak, M.V.; Cyr, A.R.; Woodfield, G.W.; Bogachek, M.; Spanheimer, P.M.; Li, T.; Price, D.H.; Domann, F.E.; Weigel, R.J. Transcriptional regulation of the GPX1 gene by TFAP2C and aberrant CpG methylation in human breast cancer. Oncogene 2013, 32, 4043–4051. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Luo, K.; Tan, L.-Z.; Ren, B.-G.; Gu, L.-Q.; Michalopoulos, G.; Luo, J.-H.; Yu, Y.P. p53-induced gene 3 mediates cell death induced by glutathione peroxidase 3. J. Biol. Chem. 2012, 287, 16890–16902. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Xie, L.; Lu, Y.; Hu, Z.; Chang, J. MiR-133b reverses cisplatin resistance by targeting GSTP1 in cisplatin-resistant lung cancer cells. Int. J. Mol. Med. 2018, 41, 2050–2058. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Meng, X.; Pan, C.; Qu, F.; Gan, W.; Xiang, Z.; Han, X.; Li, D. piR-31470 epigenetically suppresses the expression of glutathione S-transferase pi 1 in prostate cancer via DNA methylation. Cell. Signal. 2020, 67. [Google Scholar] [CrossRef]
- Peng, G.; Tang, Z.; Xiang, Y.; Chen, W. Glutathione peroxidase 4 maintains a stemness phenotype, oxidative homeostasis and regulates biological processes in Panc-1 cancer stem-like cells. Oncol. Rep. 2019, 41, 1264–1274. [Google Scholar] [CrossRef] [Green Version]
- Shireman, L.M.; Kripps, K.A.; Balogh, L.M.; Conner, K.P.; Whittington, D.; Atkins, W.M. Glutathione transferase A4-4 resists adduction by 4-hydroxynonenal. Arch. Biochem. Biophys. 2010, 504, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, L.; Jiang, S.; Yang, T.; Lan, J.; Lei, Y.; Tan, H.; Pan, K. HMGB1 mediates lipopolysaccharide-induced inflammation via interacting with GPX4 in colon cancer cells. Cancer Cell Int. 2020, 20. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.; Zhang, X.; Chen, W. Glutathione reductase-mediated thiol oxidative stress suppresses metastasis of murine melanoma cells. Free Radic. Biol. Med. 2018, 129, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Dong, J.-L.; Chen, Y.-L.; Liu, Y.; Huang, S.-S.; Zhong, X.-L.; Cheng, Y.-H.; Wang, Z.-G. Nrf2 mediates the protective effects of homocysteine by increasing the levels of GSH content in HepG2 cells. Mol. Med. Rep. 2017, 16, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Ye, J.; You, J.; Shi, X.; Kang, W.; Wang, T. Role of the cystathionine β-synthase/H2S system in liver cancer cells and the inhibitory effect of quinolone-indolone conjugate QIC2 on the system. Oncol. Rep. 2017, 37, 3001–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. CMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-L.; Lin, Z.-X.; Qin, Y.-S.; She, Y.-Q.; Chen, Y.; Chen, C.; Qiu, G.-D.; Zheng, J.-T.; Chen, Z.-L.; Zhang, S.-Y. Overexpression of long noncoding RNA LINC01419 in esophageal squamous cell carcinoma and its relation to the sensitivity to 5-fluorouracil by mediating GSTP1 methylation. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Yang, J.H.; Huang, F.Z.; Nie, W.P.; Liu, C.P.; Mao, X.H.; Yin, X.M.; Shen, X.B.; Peng, C.; Chen, M.F.; et al. Glutathione-s-transferase A 4 (GSTA4) suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting AKT pathway. Am. J. Transl. Res. 2017, 9, 301–315. [Google Scholar]
- Gaziano, J.M.; Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.; Glynn, R.J.; Buring, J.E. Multivitamins in the prevention of cancer in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2012, 308, 1871–1880. [Google Scholar] [CrossRef]
- Qiao, Y.-L.; Dawsey, S.M.; Kamangar, F.; Fan, J.-H.; Abnet, C.C.; Sun, X.-D.; Johnson, L.L.; Gail, M.H.; Dong, Z.-W.; Yu, B.; et al. Total and cancer mortality after supplementation with vitamins and minerals: Follow-up of the Linxian General Population Nutrition Intervention Trial. J. Natl. Cancer Inst. 2009, 101, 507–518. [Google Scholar] [CrossRef]
- Myung, S.K.; Kim, Y.; Ju, W.; Choi, H.J.; Bae, W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol. 2009, 21, 166–179. [Google Scholar] [CrossRef]
- The Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin e and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Haider, C.; Ferk, F.; Bojaxhi, E.; Martano, G.; Stutz, H.; Bresgen, N.; Knasmüller, S.; Alija, A.; Eckl, P.M. Effects of β-carotene and its cleavage products in primary pneumocyte type II cells. Antioxidants 2017, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanes, D.; Heinonen, O.P.; Taylor, P.R.; Virtamo, J.; Edwards, B.K.; Rautalahti, M.; Hartman, A.M.; Palmgren, J.; Freedman, L.S.; Haapakoski, J.; et al. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: Effects of base-line characteristics and study compliance. J. Natl. Cancer Inst. 1996, 88, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Virtamo, J.; Taylor, P.R.; Kontto, J.; Männistö, S.; Utriainen, M.; Weinstein, S.J.; Huttunen, J.; Albanes, D. Effects of α-tocopherol and β-carotene supplementation on cancer incidence and mortality: 18-Year postintervention follow-up of the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Int. J. Cancer 2014, 135, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkovic, L.; Siems, W.; Siems, R.; Zarkovic, N. Oxidative stress and antioxidants in carcinogenesis and integrative therapy of cancer. Curr. Pharm. Des. 2014, 20, 6529–6542. [Google Scholar] [CrossRef] [PubMed]
- Mut-Salud, N.; Álvarez, P.J.; Garrido, J.M.; Carrasco, E.; Aránega, A.; Rodríguez-Serrano, F. Antioxidant Intake and Antitumor Therapy: Toward Nutritional Recommendations for Optimal Results. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, A.Y.; Cai, X.; Thoene, K.; Obi, N.; Jaskulski, S.; Behrens, S.; Flesch-Janys, D.; Chang-Claude, J. Antioxidant supplementation and breast cancer prognosis in postmenopausal women undergoing chemotherapy and radiation therapy. Am. J. Clin. Nutr. 2019, 109, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.-C.; Ji, J.-A.; Jiang, Z.-Y.; You, Q.-D. The Keap1-Nrf2-ARE Pathway as a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A.; et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investig. New Drugs 2015, 33, 480–489. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Kujundžić, R.N.; Stepanić, V.; Milković, L.; Gašparović, A.Č.; Tomljanović, M.; Trošelj, K.G. Curcumin and its potential for systemic targeting of inflamm-aging and metabolic reprogramming in cancer. Int. J. Mol. Sci. 2019, 20, 1180. [Google Scholar] [CrossRef] [Green Version]
- Ferri, C.; West, K.; Otero, K.; Kim, Y.H. Effectiveness of Curcumin for Treating Cancer during Chemotherapy. Altern. Complementary Ther. 2018, 24, 13–18. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [Green Version]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 2020, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Jung, B.J.; Lee, S.H.; Yoo, H.S.; Shin, E.A.; Ko, H.J.; Chang, S.; Kim, S.Y.; Jeon, S.M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Gao, A.; Ke, Z.; Wang, J.; Yang, J.; Chen, S.-Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, D.L.; Powis, G. Clinically Evaluated Cancer Drugs Inhibiting Redox Signaling. Antioxid. Redox Signal. 2017, 26, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. (Tokyo) 2019, 67, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Brenneisen, P.; Reichert, A.S. Nanotherapy and Reactive Oxygen Species (ROS) in Cancer: A Novel Perspective. Antioxidants 2018, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Yoo, S.Y.; Bartholomeusz, G.; Graham, R.A.; Majidi, M.; Yan, S.; Meng, J.; Ji, L.; Coombes, K.; Minna, J.D.; et al. KEAP1-dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013, 73, 5532–5543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef]
- Xu, X.; Yang, Y.; Liu, X.; Cao, N.; Zhang, P.; Zhao, S.; Chen, D.; Li, L.; He, Y.; Dong, X.; et al. NFE2L2/KEAP1 Mutations Correlate with Higher Tumor Mutational Burden Value/PD-L1 Expression and Potentiate Improved Clinical Outcome with Immunotherapy. Oncologist 2020, 25, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonenn, A.L.; Levonen, A. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, H.M.; Ruotsalainen, A.K.; Määttä, A.M.; Laitinen, H.M.; Kuosmanen, S.M.; Kansanen, E.; Pikkarainen, J.T.; Lappalainen, J.P.; Samaranayake, H.; Lesch, H.P.; et al. Oxidative stress-regulated lentiviral TK/GCV gene therapy for lung cancer treatment. Cancer Res. 2012, 72, 6227–6235. [Google Scholar] [CrossRef] [Green Version]
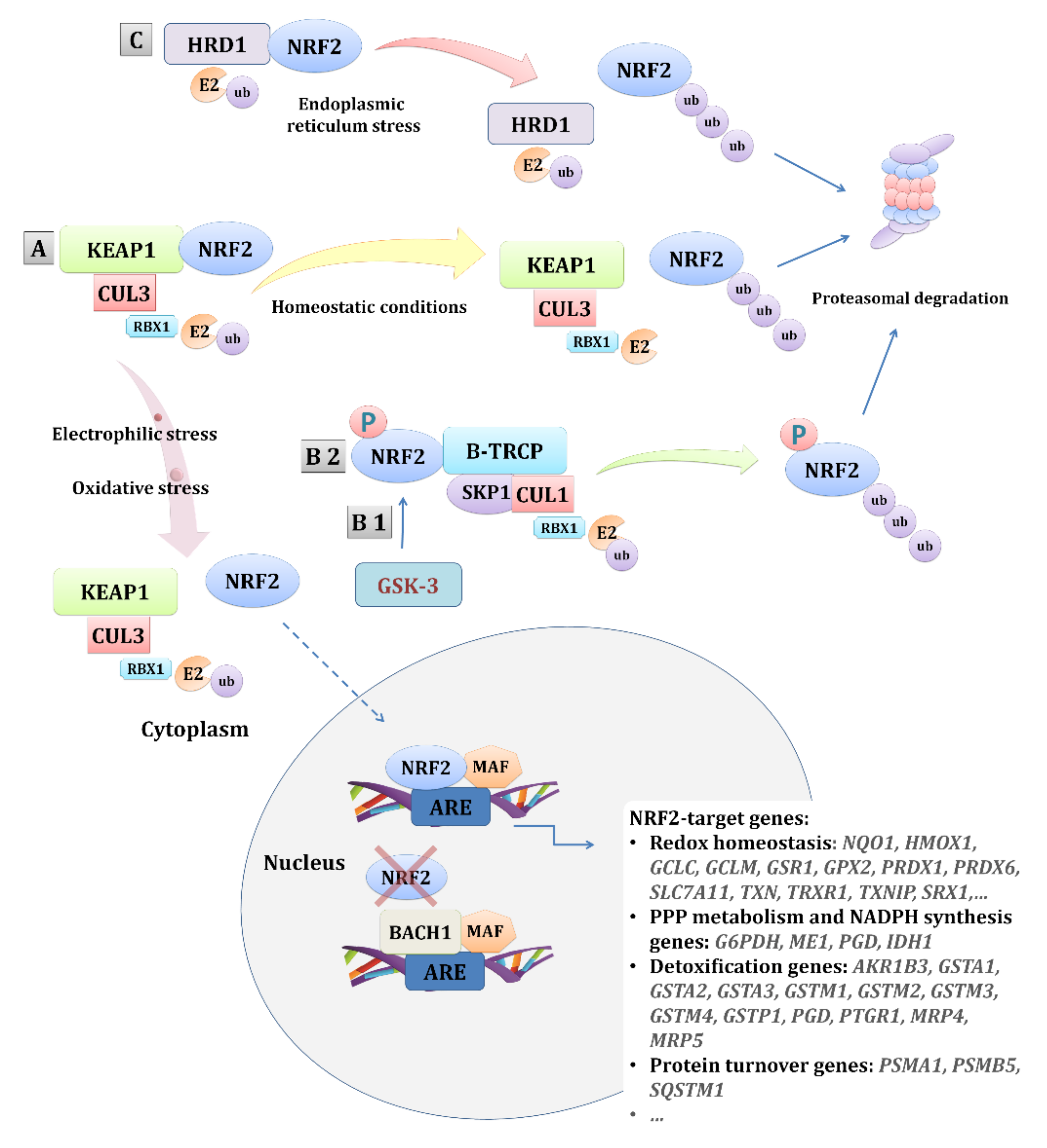


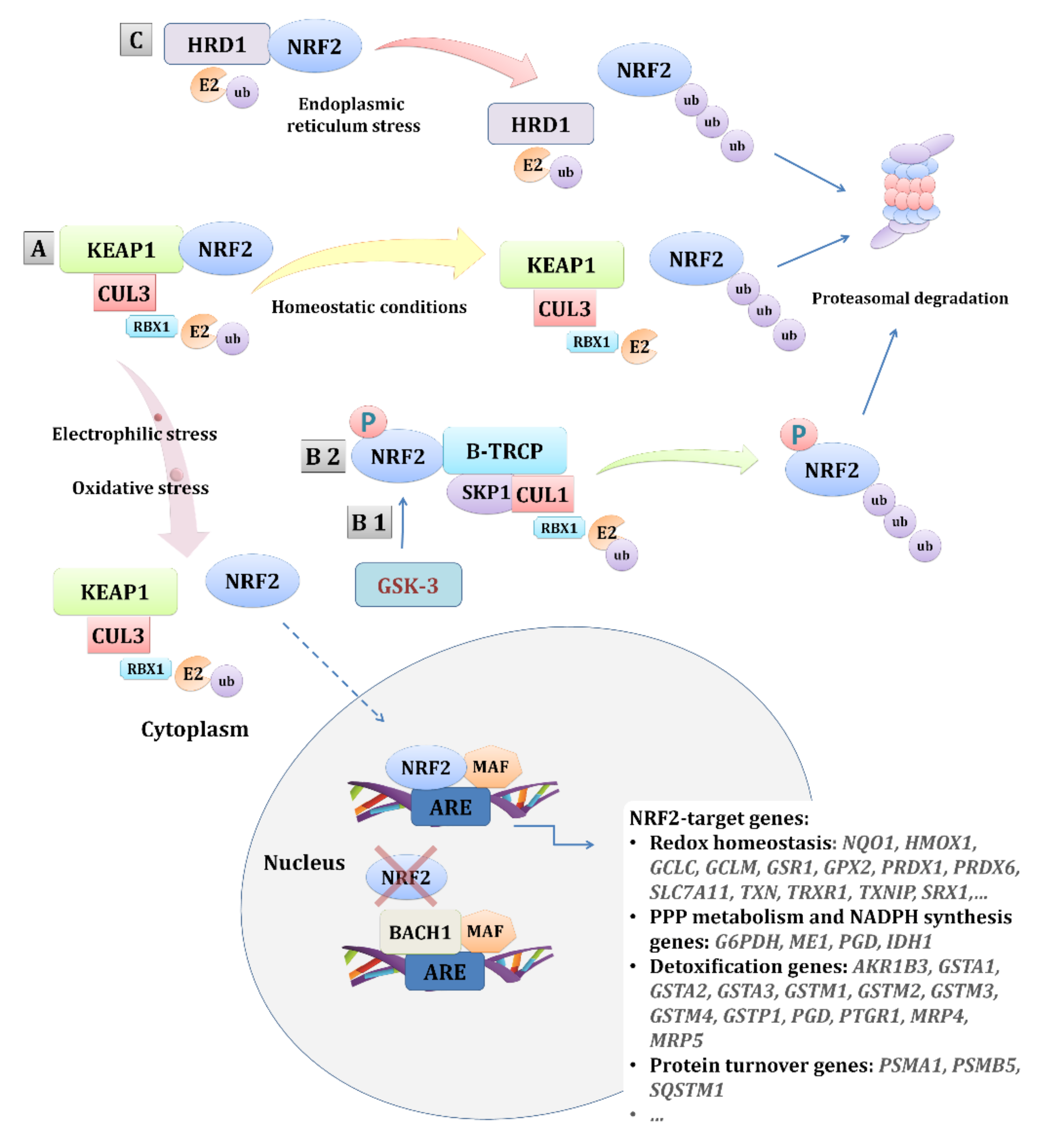{kind=link}
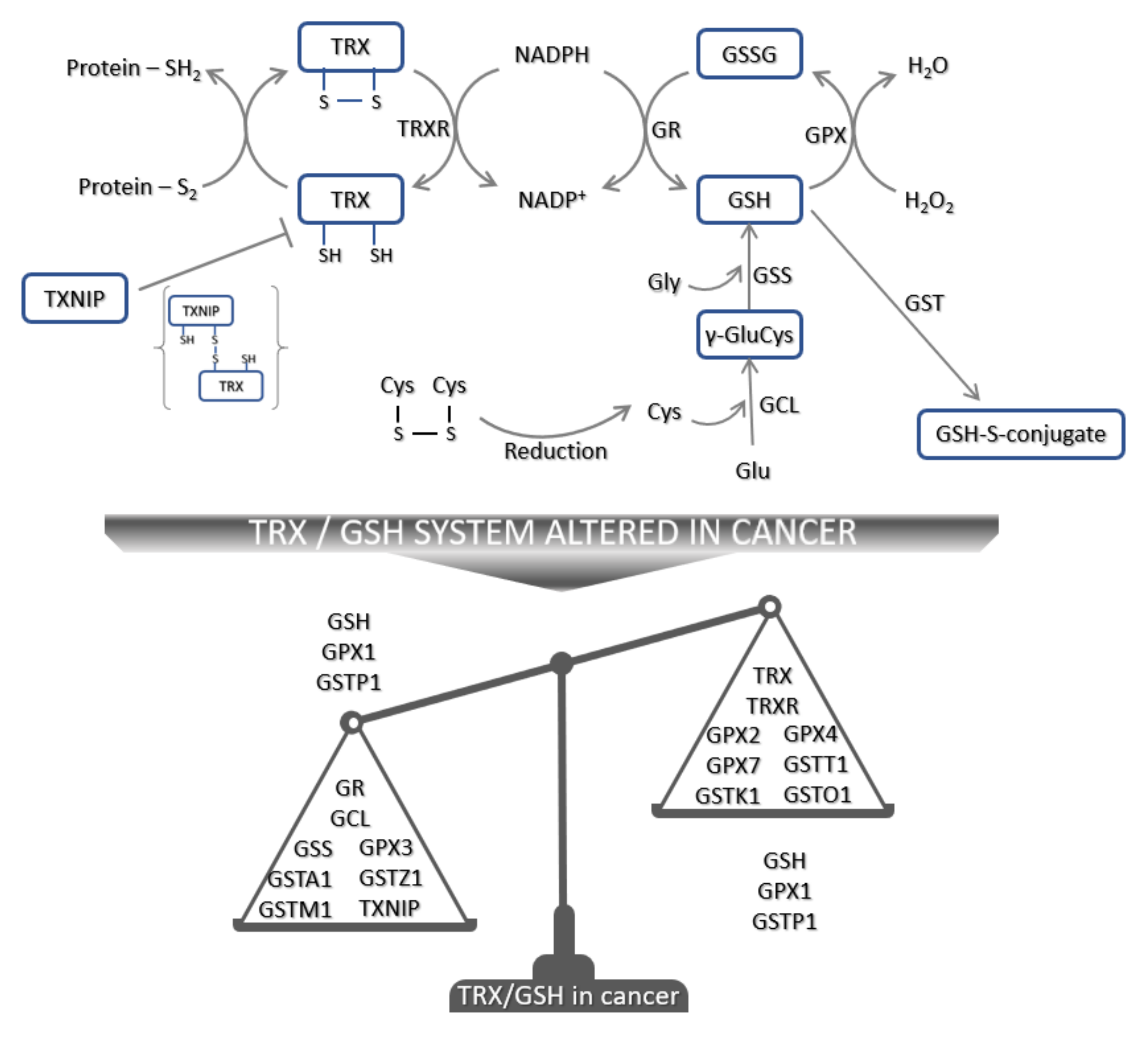{kind=link}
| Cancer Type | The NRF2 Pathway Activities | Ref. |
|---|---|---|
| Blood cancers | High expression of NRF2 in AML patients is governed by NF-κB and leads to chemoresistance. | [72] |
| Nuclear accumulation of NRF2; lower nuclear levels of BACH1; and a higher expression of HMOX-1, NQO1, GCLM, and GSR were found to be protective mechanisms of bortezomib-resistant AML patients. | [73] | |
| IHC expression of NRF2 in bone marrow correlates with a risk of myelodysplastic syndromes and with the worse overall survival of high-risk patients. | [74] | |
| Higher NRF2 and HMOX-1 levels are found in the peripheral blood mononuclear cells of CLL patients vs. healthy donors. | [75] | |
| NF-κB-dependent activation of P62 activates the NRF2 pathway, ensuring resistance to ROS-inducible therapy in ROR1-high CLL patients. | [76] | |
| IHC expression of NRF2 and KEAP1 was higher in patients with diffuse large B-cell lymphoma than with reactive lymph node hyperplasia and rises with the tumor stage. | [77] | |
| Combination of NRF2, NRF1, and KEAP1 localized expression (high nuclear NRF2, high cytoplasmic and low nuclear NRF1, and low cytoplasmic KEAP1) is linked to worse overall survival in patients with diffuse large B cell lymphoma. | [78] | |
| Lung cancer | Heterogenic distribution of KEAP1 and NFE2L2 mutations among NSCLC patients (with a frequency of 11.3% and NFE2L2 3.5%, respectively) is linked to chemotherapy resistance.In 80% of patients, co-occurrence with other cancer-related mutations was observed. | [79] |
| KEAP1/NFE2L2 mutations in metastatic lung adenocarcinoma are linked with a chemotherapy-resistant subtype and more progressive disease. | [80] | |
| NSCLC patients with brain metastasis have mutations in the KEAP1-NRF2-ARE pathway that provide a survival advantage and dissemination of circulating tumor cells. | [81] | |
| Higher protein expression of NRF2, peroxiredoxin, TRX, and sulfiredoxin in lung cancer tissues in comparison to the paired normal lung tissue implies their protective role against oxidative injury and compensation due to the high mitochondrial metabolism. | [82] | |
| In Japanese patients with lung cancer, NFE2L2 mutations were mainly found in males with advanced stages of squamous cell carcinoma and worse overall survival. | [83] | |
| The most frequent co-mutations found within the NSCLC patients with the KRAS mutations include KEAP1/NFE2L2 (27%). These co-mutations are a negative prognostic factor, predicting shorter survival and response to therapy. | [84] | |
| KEAP1/ NFE2L2 mutation status predicts the risk of local recurrence after radiotherapy in NSCLC patients. | [85] | |
| NRF2 overexpression and lower expression of Beclin 1 are associated with worse prognosis in NSCLC patients. Higher expression of NRF2 was linked to a larger tumor, more advanced TNM stage, lymph node, and distant metastasis. | [86] | |
| NFE2L2 mutations were observed more frequently in the advanced stages of NSCLC, particularly squamous cell carcinoma in the Japanese cohort. | [87] | |
| Early-stage squamous cell carcinoma is enriched with several somatic mutations, including mutually exclusive NFE2L2 and KEAP1. Patients with NFE2L2 mutations, especially co-occurring with TP53 mutations, were linked with worse recurrence-free survival, while KEAP1 and TP53 co-mutants were associated with a poor response to adjuvant therapy. | [88] | |
| Somatic alterations of NFE2L2, KEAP1, or CUL3 upregulate a specific set of 28 genes used to discriminate SCC in subgroups with an active NRF2 pathway and WT. SCC patients with the expression signature of an active NFR2 pathway have shown reduced benefit from adjuvant cisplatin/vinorelbine therapy. | [89] | |
| NAMS comprised of 50 tumor-associated genes can be used as an independent prognostic marker of recurrence-free survival and overall survival, with NAMS+ patients having a worse prognosis. | [57] | |
| Enrichment with KEAP1 mutations and NRF2 overexpression is observed in more than 50% of TTF1-negative lung adenocarcinoma patients, who are known to have shorter survival. | [90] | |
| Nuclear expression of NRF2 was observed in 26% of NSCLC patients and more commonly seen in SCC than adenocarcinoma, while low or absent KEAP1 expression was detected in 56% of NSCLC and more commonly in adenocarcinoma. While nuclear NRF2 expression was associated with a worse overall survival in NSCLC and worse recurrence-free survival in SCC patients who underwent platinum-based adjuvant treatment, low or absent KEAP1 was linked with worse overall survival in SCC. | [91] | |
| Somatic mutations of the genes involved in oxidative stress response (NFE2L2, KEAP1) present in 21.1% of Chinese SCC patients. Frequent NFE2L2, MAGEC1, NLRP3, and FAM5C mutations were detected only in smokers. | [92] | |
| In 34% of SCC patients, there is the activation of the NRF2/KEAP1 pathway due to gene alterations in NFE2L2, KEAP1, CUL. | [93] | |
| Biallelic inactivation of KEAP1 and activation of the NRF2 pathway (high nuclear and cytoplasmic staining of NRF2) is found in 41% of NSCLC tumors. | [94] | |
| KEAP1 alterations and the overexpression of nuclear NRF2 were observed in 60% of true papillary adenocarcinoma. | [95] | |
| Higher NRF2, BCL-2, and BCL-XL mRNA levels observed in TP53-mutant NSCLC patients were linked with cisplatin-based chemotherapy resistance. | [96] | |
| NFE2L2 mutations were observed predominantly in male smokers with SCC. | [97] | |
| Breast cancer | Nuclear NRF2 predominantly in breast carcinoma cells observed in 44% of breast cancer patients was associated with worse recurrence and disease-free survival. | [98] |
| Certain genetic polymorphisms in KEAP1 can increase the risk of breast cancer and worsen patients’ survival, particularly when receiving adjuvant therapy. | [99] | |
| NRF2 and SRXN1 genetic polymorphisms could predict breast cancer risk and a survival outcome. For example, the NRF2 rs2886162 AA genotype was associated with a worse survival, while the NRF2 rs2706110 AA genotype was linked with an increased risk and the SRXN1 rs6053666 C allele with a decreased risk of breast cancer. | [100] | |
| Low NRF2 mRNA expression levels were associated with worse disease-specific survival and overall survival, while higher levels of NRF2 mRNA in ER-positive tumors predict a better outcome. Comparison of the mRNA NRF2 expression levels in tumor vs. normal breast tissues revealed lower levels in tumors. | [101] | |
| GSTM1 * 1/0 genotype and genetic alterations in KEAP1 and/or NFE2L2 are linked with a worse prognosis. | [102] | |
| Out of 109 investigated SNPs related to oxidative stress genes, SNPs located in NFE2L2, metallothionein, NQO1, and peroxiredoxin 1 and 6 were associated with overall mortality. | [103] | |
| CXCL13-CXCR5 co-expression within breast tumors governed by high RelA conditions, low NRF2, and a lack of cxcr5 promoter DNA-methylation drive tumor progression and metastasis. NRF2 negatively regulates the transcription of CXCL13. | [104] | |
| NRF2 level decreased in the tumor in comparison to normal breast tissue. Lower NRF2 in the luminal B subtype is associated with a longer overall survival. | [105] | |
| The aggressive phenotype of breast cancer showing inverse expression of Caveolin-1 (low) and Mn-SOD (high) in tumor vs. normal tissue is associated with the activation of the NRF2 pathway, upregulation of Mn-SOD that leads to ROS production, and AMPK activation inducing glycolytic shift. | [106] | |
| Esophageal cancer | Genetic alterations of NFE2L2 are more common in ESCC (24%) vs. esophageal adenocarcinomas (1%). | [107] |
| Enrichment of the NRF2-mediated oxidative stress pathway was suggested as a potential distinctive molecular mechanism of ESCC in African Americans. | [108] | |
| Genetic alterations of NFE2L2 were one of the trunk mutations found in both precancerous lesions and ESCC, suggesting them to be early CNA events. | [109] | |
| High IHC expression of NRF2 was linked with metabolic reprogramming to glutathione synthesis and ROS detoxification and was associated with poor recurrence-free and overall survival in esophageal cancer patients. | [110] | |
| Evaluation of spatial intratumoral heterogeneity revealed NFE2L2 and KEAP1 mutations on branches, thus suggesting them as late events in ESCC. | [111] | |
| Comparison of ESCC in Asian and Caucasian patients identified NFE2L2 as a race-biased gene, with a higher mutational rate in Asian patients. | [112] | |
| NFE2L2 gain-of-function mutation occurred in 22% advanced ESCC and was linked with tumor recurrence and poor prognosis. Additionally, a molecular signature associated with NFE2L2 mutation was linked with poor response to therapy and suggested as a potential prognostic marker to therapy. | [113] | |
| Somatic gene alterations of NFE2L2 was found in 10% of ESCC. In addition, NFE2L2, KEAP1, and CUL3 mutations were shared among squamous cell carcinomas that originated from different parts of the body. | [114] | |
| NFE2L2 gene is significantly mutated in ESCC. | [115] | |
| Overexpression of miR-432-3p and negative relation with KEAP1 was observed in primary ESCC. Experimentally, miR-432-3p directly binds to the coding region of KEAP1, thus downregulating it and inducing the stabilization of NRF2. | [116] | |
| Gastric cancer | NRF2 nuclear positivity was mostly present in cancer cells and associated with more aggressive tumors, worse overall survival, and resistance to 5FU-based adjuvant chemotherapy. | [117] |
| Pancreatic cancer | Nuclear NRF2 expression is associated with the expression of sulfiredoxin and predicts a worse survival in pancreatic adenocarcinoma. | [118] |
| Liver cancer | NFE2L2 mutations were detected in 9.8% of hepatoblastoma, mainly in regions that are essential for binding with the KEAP1/CUL3 complex. Overexpression of NFE2L2 target gene NQO1 was the highest in NFE2L2-mutated tumors and was associates=d with metastasis, vascular invasion, and a worse outcome. | [119] |
| Higher nuclear expression of NRF2 was observed in bigger tumors with poor differentiation and metastasis and was associated with a worse survival in HCC patients. | [120] | |
| Higher levels of NRF2 and 8-OHdG were observed in HCC cells. High 8-OHdG was associated with short survival. Experimentally, oxidative stress was suggested as a driver of HCC progression. | [121] | |
| mRNA expression of NRF2 and NRF2-related genes differs between HCC, adjacent tissue, normal liver, and liver diseases. Expression of NRF2 was the lowest in HCC and increased in cirrhosis and end-stage liver disease, while KEAP1 was higher in HCC vs. normal liver and increased in cirrhosis and end-stage disease. The expression of NQO1 was the highest in HCC and suggested as a possible biomarker of HCC. | [122] | |
| Out of 107 HCC samples, a high nuclear expression of NRF2 was observed in 75 samples. Expression of nuclear NRF2 and KEAP1 was inversely related and patients with high NRF2 and reduced KEAP1 had worse overall and disease-free survival. HCC patients with high NRF2 had a higher mRNA expression of AKR1B10, NQO1, and GCLM in tumor tissue. | [123] | |
| In HCC, the higher nuclear NRF2 observed in tumors vs. matched controls is linked with the increased production of PPP enzymes and the loss of aldolase A. | [124] | |
| KEAP1 mutations were observed in 8% of HCC patients and linked with shorter disease-free survival. | [125] | |
| Overexpression of NRF2 and NQO1 was linked with tumor size, multiple intrahepatic recurrences, and poor prognosis. | [126] | |
| The upregulation of TRIM25 is correlated with a high NRF2 expression and low KEAP1 expression and predicts a poor prognosis in HCC patients. | [66] | |
| Biliary tract cancer | NFE2L2 is one of the significantly mutated gene in gallbladder carcinoma. Additionally, KEAP1 and NFE2L2 (exon 2 deletion) splice variants were also observed. KEAP1/NFE2L2 pathway activation was suggested as a significant prognostic predictor of survival. | [127] |
| Higher NRF2 expression is associated with a worse overall survival in BTC patients receiving chemotherapy. SNPs located in GPX4, CAT, and GSR might modify chemotherapy effects on overall survival. Experimentally, the knockdown of GPX4, CAT, or GSR induced chemoresistance by increasing the ROS level and activating the NRF2-ABCG2 pathway. | [128] | |
| Colorectal cancer | The expression of the proteins in the NRF2 pathway differs between cancer and normal tissue. Mean IHC density of KEAP1 and prohibitin was higher in tumor vs. normal tissue, with lower levels of NRF2, P62, and PARK7 than the distant normal tissue. The lowest level of KEAP1 and p21 was found in the adjacent normal tissue. NRF2 levels correlated with KEAP1 in the tumor and BACH1 in the normal tissue. | [129] |
| A lower ratio of HMOX1/NRF2 mRNA level found in the tumor tissue of patients with distant metastasis might be used as a predictor of distant metastasis in CRC. | [130] | |
| Distinctive expression patterns of NRF2 and BACH1 were observed in CRC. While the increase in the NRF2 expression with the grade of malignancy did not contribute to the tumor invasiveness, the expression of BACH1 (the highest in normal mucosa, lower in adenoma, and again high in carcinoma) was associated with tumor invasiveness and metastasis. | [131] | |
| Ovarian cancer | High cytoplasmic NRF2 was associated with low-grade histology and, together with high ERα expression, was associated with a better overall survival in patients with a serous cancer subtype. | [132] |
| Endometrial cancer | High nuclear NRF2 staining in 24.7% of EC mainly in TP53/CNH-like tumors (tumors with a mutation within the TP53 coding sequence) and no nuclear staining in normal epithelial and stromal cells. No correlation between the nuclear NRF2 and mRNA levels of its target genes: NQO1, GCLC, AKR1C3. A subset of TP53/CNH-like tumors with a low mRNA NQO1 was associated with NRF2/TP53 cooperation that drives a more aggressive phenotype but initial better sensitivity to chemotherapy. | [133] |
| NRF2 overexpression observed in ESC and its precancers might contribute to the worse overall prognosis in patients with ESC. | [134] | |
| Head and neck cancer | Increased expression of NRF2 and to some extent thioredoxin was observed in head and neck squamous cell carcinomas, while KEAP1 overexpression was anatomic site-dependent and not negatively correlated with NRF2. | [135] |
| Genetic alteration of the KEAP1-NFE2L2-CUL3 axis in HNSCC induces the expression of genes, of which 17 selected are related to poor survival. They include genes associated with drug resistance, glutathione metabolism, oxidation-reduction processes, etc. | [136] | |
| Skin cancer | mRNA and protein levels of NRF2 and NRF1 were the highest in benign naevi and decreased during melanoma carcinogenesis. High nuclear NRF2 or NRF1 expression in pigment cells was associated with a worse survival in patients without distant metastasis or without nodal metastasis, respectively. | [137] |
| NFE2L2 mutations were observed in 6.3% of skin SCC. | [138] |
| Tumor Type | Involvement of TRX System | Ref. |
|---|---|---|
| Basal cell carcinoma | TRXR activity is higher in tumor tissues compared to adjacent healthy tissue. | [172] |
| Blood cancer | Poor survival is correlated with a lower expression of TXNIP in acute myeloid leukemia. | [162] |
| The human histiocytic/monocytic leukemia cells have several-fold higher TRXR expression compared to non-transformed cells. Both normal and transformed cells were found to secrete TRXR. | [173] | |
| TRX is overexpressed in T-Cell acute lymphoblastic leukemia cells. | [174] | |
| Brain cancer | Excessive cytoplasmic TRXR is correlated with a worse prognosis of brain cancer patients. | [142] |
| TRX expression is positively correlated with increasing grades of glioma. | [154] | |
| TXNIP high expression is associated with a lower pathological grade of meningioma. | [175] | |
| Breast cancer | TRX1 and TRXR1 are overexpressed in tumor tissue and are correlated with poor survival. | [143] |
| TXNIP overexpression is correlated with better survival. | [163] | |
| TRXR1 overexpression is associated with the occurrence of metastasis, while TXNIP overexpression correlated with a better prognosis. | [176] | |
| Poor survival of triple-negative breast cancer patients correlates with high c-MYC and low TXNIP expression. | [177] | |
| Cervical squamous cell carcinoma | High expression of TRX1 is associated with poor response to cisplatin-based neoadjuvant chemotherapy. | [178] |
| Cholangiocarcinoma | TRX is overexpressed in tumor tissue and in dysplastic bile ducts with highly abnormal growth patterns. | [150] |
| Clear cell renal cell carcinoma | TXNDC5 is overexpressed in tumor tissues compared to adjacent normal tissues. | [165] |
| Colorectal cancer | Thioredoxin-like protein 2 expression is increased in tumor tissues and correlates with its histological grade and prognosis. | [179] |
| TRX1 is overexpressed in tumor tissues and associated with clinicopathological features and poor survival. | [155] | |
| TXNDC5 is overexpressed in tumor tissues. | [166] | |
| TXNDC9 expression is associated with tumor histological grade and survival. | [169] | |
| Gallbladder carcinoma | TRX1 expression is higher in gallbladder carcinoma. | [151] |
| Gastric cancer | High TXNDC5 expression correlates with poor prognosis. | [168] |
| High TXNIP and low TRX correlates with better prognosis, while low TXNIP and high TRX correlates with a poor prognosis. | [156] | |
| High TRX1 expression in gastric cancer tissues is associated with poor survival. | [157] | |
| TRXR activity is significantly higher in the plasma of gastric cancer patients compared to healthy controls. | [180] | |
| Hepatocellular carcinoma | TRX expression is overexpressed in HCC compared to the control group. | [152] |
| TRXR1 and TRX are upregulated in the tumor. | [144] | |
| TXNIP expression is significantly decreased in tumor tissues. | [159] | |
| Lung cancer | High TRXR expression is associated with the poor prognosis of NSCLC patients. | [181] |
| TRXR1 mRNA and protein are overexpressed in NSCLC. | [145] | |
| TRXR2 is upregulated in NSCLC tumor tissues. | [149] | |
| TXNDC5 is upregulated in NSCLC tumor tissue. | [167] | |
| TRX1 expression correlated with the degree of NSCLC tumor differentiation. | [182] | |
| TXNIP is correlated with a good prognosis of lung large-cell carcinoma patients. | [164] | |
| TRXR1 and TRX are upregulated in lung adenocarcinoma. | [144] | |
| Oral squamous cell carcinoma | TRXR1 is overexpressed in oral carcinoma patients. | [146] |
| TRXR1 is overexpressed in tumors and correlates with the clinical stage and metastasis. | [147] | |
| Ovarian cancer | Nuclear TRX expression was lower in borderline tumors compared to benign ovarian epithelial tumors. | [183] |
| TXNDC17 is overexpressed in tumor tissue and correlates with poor prognosis and shorter survival of patients. | [171] | |
| Prostate cancer | Levels of TRX1 increase with cancer progression in androgen-deprived castration-resistant prostate cancer cells. | [158] |
| TRX1 protein is overexpressed, but its activity unchanged, in high-grade prostate cancer compared with adjacent normal tissue. | [184] | |
| Tumors have increased TXNDC9, and it correlates with advanced clinical stages. | [170] | |
| TXNIP expression is decreased in prostate cancer. | [160] | |
| Thyroid cancer | TRXR1 expression is decreased in thyroid cancer cells compared to healthy cells. | [185] |
| TXNIP is highly expressed in differentiated thyroid cancer, while its expression is low in anaplastic thyroid cancer. | [161] | |
| TRX and TRXR are overexpressed in the cytoplasm and nuclei of tumor cells compared to normal tissue. | [153] | |
| Tongue squamous cell carcinoma | TRX and TRXR1 are highly expressed in tumor tissue. | [148] |
| Uveal melanoma | Poor survival and metastasis are associated with the high uveal melanoma tissue expression of peroxiredoxin-3. | [186] |
| TRX System/TXNDC | Mechanism | Ref. |
|---|---|---|
| TRX1/2 | TRX alters the function of therapeutic monoclonal antibodies by reducing the antibodies’ interchain disulfide bonds. | [219] |
| Joint inhibition of TRX, GSH, and NRF2 promotes intracellular ROS and suppresses the growth of head and neck cancer cells. | [220] | |
| TRX phosphorylation at T100 attributes to its anti-apoptotic effects in tumor cells. | [221] | |
| TRX knockdown induces G1 phase cell-cycle arrest through the ERK1/2-cyclin D1 pathway. | [222] | |
| Nitric oxide synthase type 3 and S-nitrosation of the CD95 receptor is induced by TRX1. This results in the increased activity of caspase-8, while the activity of caspase-3 is decreased promoting the proliferation of liver cancer cells. | [187] | |
| TRX1 overexpression decreases PTEN; increases the amount of phosphorylated AKT; and promotes the growth, migration, and invasion of gastric cancer cells. Contrary, TRX1 silencing has the opposite effect. | [157] | |
| TRX1 promotes epithelial to mesenchymal transition of colorectal cancer cells through the phosphorylation of AKT, leading to the upregulation of S100A4. | [190] | |
| TRX1 inhibition induces intracellular ROS, elevates TP53 and androgen receptor levels, and promotes cell death. Additionally, the androgen receptor levels under androgen deprivation are increased in castration-resistant prostate cancer cells when TRX1 is inhibited. | [158] | |
| TRX1 plays a role in keeping mixed-lineage kinase domain-like protein, necessary for necroptosis activation, in a reduced inactive state. | [223] | |
| TRX1 activates the transcription of S100P, which in turn downregulates TXNIP and upregulates p-ERK1/2, thus promoting TRX1 expression in colorectal cancer cells. | [155] | |
| Upregulation of TRX1 induces matrix metalloproteinase 9 expression, promoting the invasion of breast cancer cells. | [224] | |
| Depletion of ubiquitin-like with PHD and RING finger domains 1 reduces TRX2 and increases intracellular ROS in retinoblastoma cells. | [225] | |
| Glioma nitric oxide synthase 2 induces the S-nitrosylation of TRX2 and mitochondrial caspase 3 in microglial cells, reducing their activity and promoting tumorigenesis. | [188] | |
| TRXR1/2/3 | Mitochondrial TRXR3 reduces TRX2 and stabilizes mitochondrial-associated survival molecules, thus promoting cell survival. | [191] |
| TRXR inhibition alters the mitochondrial membrane and induces the apoptosis of liver cancer cells. | [192] | |
| TRXR inhibition promotes heme oxygenase-1 overexpression, allowing tumor cells to survive, while the simultaneous inhibition of both induces the apoptosis of myeloma cells. | [193] | |
| Lysine oxidase induces ROS, activates caspase-independent cell death, and promotes TRXR1 via NRF2 in triple-negative breast cancer cells. | [226] | |
| ROS promotes miR-526b/miR-655 expression, consequently leading to the upregulation of TRXR1 in cancer cells. | [227] | |
| miR-125b-5p inhibits TRXR1 in HCC cells. | [194] | |
| Acetylation of TRXR1 multimers promotes the formation of more active TRXR1 dimers. Additionally, acetylation of TRXR1 at Lys307 results in a2.7-fold increased catalytic activity. | [198] | |
| Overexpression of miR-124 binds to 3’-UTR of TRXR1 and reduces its expression. | [195] | |
| TRXR1 is susceptible to nitrosylation, resulting in TRXR1 inactivation. | [197] | |
| Upregulation of mature miR-17-3p inhibits TRXR2 and suppresses mitochondrial respiration, rendering prostate cancer cells more sensitive to ionizing radiation. | [196] | |
| TRXR2 inhibition promotes ROS formation; decreases the activity of SOD, CAT, and GPX1, and reduces growth; and induces the apoptosis of NSCLC cells. | [149] | |
| TXNDC | Circular RNA, circRNA-104718, competes with TXNDC5 mRNA for miR-218-5p, and its overexpression promotes tumor growth and metastasis. | [228] |
| ER stress induces the association of sulfiredoxin with TXNDC5, and, depending on the levels of each, they have a different impact on cancer patient survival. | [229] | |
| Inactivation of NR4A1 downregulates TXNDC5, thus promoting intracellular ROS and IL24 expression. This in turn inhibits the growth and induces apoptosis of rhabdomyosarcoma. | [214] | |
| TXNDC5 expression might be induced under hypoxic conditions by upregulating HIF1α and thus supporting the tumorigenesis of colorectal cancer cells. | [166] | |
| Inhibition of TXNDC5 promotes the expression of serpin peptidase inhibitor, clade F, and TNF receptor-associated factor 1, inducing apoptosis and inhibiting angiogenesis in cervical cancer. | [217] | |
| Androgen deprivation induces the hypoxia of prostate cancer cells by downregulating miR-200b, promoting HIF1α, and increasing TXNDC5, which directly interacts with the androgen receptor, promoting its stability during cancer progression. | [218] | |
| Inactivation of NR4A1 downregulates TXNDC5, isocitrate dehydrogenase 1, and the mTOR pathway, promoting intracellular ROS, inducing apoptosis, and inhibiting the growth of kidney cancer cells. | [215] | |
| Downregulation of NR4A1 downregulates TXNDC5 and isocitrate dehydrogenase 1, activates oxidative and ER stress, and inhibits the mTOR pathway in breast cancer cells. | [216] | |
| TXNDC9 interacts with peroxiredoxin-1 and MDM2 in prostate cancer cells. Elevated ROS induce TXNDC9 overexpression, triggering the dissociation of peroxiredoxin-1 and the degradation of MDM2, thus promoting the androgen receptor signaling, growth, and progression of prostate cancer cells. | [170] | |
| TXNIP | Inhibition of class I histone deacetylases promotes TXNIP expression, promoting the ROS-induced DNA damage and apoptosis of BRCA1-deficient breast cancer cells. | [207] |
| Overexpression of TXNIP promotes the apoptosis of prostate cancer cells and induces G0/G1 cell cycle arrest. | [160] | |
| Inhibition of bromodomain and extra-terminal domain downregulates MYC, leading to the upregulation of TXNIP, excessive intracellular ROS, and promoting the apoptosis of BRCA1-deficient breast cancer cell death. | [209] | |
| p38 mitogen-activated protein kinase phosphorylates TXNIP, predominantly at Ser361, promoting its association with JAB1 and inducing G1/S cell cycle arrest. | [230] | |
| c-MYC-driven glycolysis in prostate cancer cells is accomplished through the activation of glutaminolysis via glutaminase, inducing the blockage of MondoA activity and yielding the suppression of TXNIP. | [231] | |
| Oncogenic Ras targets the N-terminus of TXNIP, suppressing its synthesis via altered translation rate by ribosomes. | [202] | |
| TXNIP forms a complex with hypoxia-inducible factor 1α and mediates its nuclear export and degradation. miR-224 binds to the 3’-UTR of TXNIP, altering the nuclear export of hypoxia-inducible factor 1α and promoting the proliferation and migration of pancreatic cancer cells. | [204] | |
| Metabolic/oxidative stress induces TXNIP expression, while insulin-like growth factor 1 inhibits TXNIP. | [199] | |
| TXNIP overexpression induces ROS generation by mitochondria, activates the MAPK pathway, promotes apoptosis, and decreases the growth of HCC cells. | [159] | |
| Inhibition of the PI3K/AKT pathway promotes TXNIP expression, which inhibits the plasma membrane localization of glucose transporter 1 in NSCLC cells. | [210] | |
| c-MYC binds to an E-box-containing region of TXNIP promoter, downregulating TXNIP and leading to elevated glucose uptake in triple-negative breast cancer cells. | [177] | |
| Downregulation of the HER-1/2 pathway induces TXNIP expression, which further promotes the p27 expression, apoptosis, and G1 cell cycle arrest of breast cancer cells. | [163] | |
| TWIST acts as a transcription factor that, by binding to the miR-371-373 gene cluster promoter, upregulates miR-373 expression. MiR-373 targets 3’-UTR of TXNIP, suppressing it, which in turn induces hypoxia-inducible-factor 1α and TWIST, promoting the epithelial-to-mesenchymal transition and metastasis of breast cancer cells. | [205] | |
| Hypoxia induces TXNIP expression in NSCLC cells. | [200] | |
| Hyperglycemia induces TXNIP overexpression in pancreatic cancer cells. | [201] | |
| Focal adhesion kinase overexpression inhibits TXNIP expression, while its downregulation upregulates TXNIP in cancer cells. | [211] | |
| TXNIP inhibition upregulates the transforming growth factor-β pathway and promotes epithelial to mesenchymal transition in lung cancer cells. | [206] | |
| Downregulation of histone deacetylase 10 induces TXNIP expression in gastric cancer cells. | [208] | |
| p21WAF1 binds to the TXNIP promoter, suppressing its expression and inducing TRX and angiogenesis in breast, lung, and prostate cancer cells. | [203] |
| Tumor Type | Involvement of the GSH System | Ref. |
|---|---|---|
| Bladder cancer | GPX2 is overexpressed in papillary urothelial carcinoma. | [244] |
| GSTO1 expression correlates with tumor grade and stage of urinary bladder carcinoma. | [252] | |
| Blood cancer | GPX is increased in acute myeloblastic leukemia. | [261] |
| GPX4 expression correlates with the poor survival of patients with large B-cell lymphoma. | [247] | |
| Blood GPX level is decreased in multiple myeloma patients. | [239] | |
| Leukemia patients have excessive leukocyte superoxide anion generation and elevated red cell GPX and SOD activity. | [262] | |
| Lymphocytes of chronic lymphocytic leukemia patients have increased GPX, GSH, 8-OHdG, and lipid peroxidation, while SOD and CAT are decreased. | [263] | |
| GSTP1 is decreased in lymphoma. | [264] | |
| Downregulation of CAT, GPX, SOD, and TRX inhibitor is associated with the poor prognosis of diffuse large B-cell lymphoma patients. | [265] | |
| Breast cancer | Breast cancer patients have a lower GPX activity in serum. | [266] |
| GPX3 promoter is hypermethylated and GPX3 expression downregulated in inflammatory breast cancer tissues. | [240] | |
| GSTP1 hypermethylation correlates with the increased tumor grade of triple-negative breast cancer patients. | [256] | |
| Serum GPX activity is decreased in cancer patients compared to healthy control. | [236] | |
| Serum GPX activity is decreased in cancer patients. | [237] | |
| Cervical cancer | GPX2 expression is upregulated in tumor tissue. | [245] |
| Colorectal cancer | GPX activity is increased in tumor tissue compared to normal tissue. | [267] |
| GSH level and expression of GPX1 and GPX3 are lower in tumor tissue compared to normal tissue. On the contrary, GPX2 expression is increased. | [235] | |
| GSTP1, GSTT1, GSTO1, and GSTK1 expression is upregulated in tumor tissue compared to adjacent normal tissue. | [253] | |
| Esophageal carcinoma | Serum GPX and GR activities are decreased in esophageal squamous cell carcinoma cancer patients. | [238] |
| The tumor has higher GPX3 methylation and lower GPX3 activity compared to paired normal tissue. | [268] | |
| Gastric cancer | Blood GSH is decreased in cancer patients. | [233] |
| GPX2 expression is upregulated in tumor tissue and lymph node metastases. | [246] | |
| GPX7 is downregulated in almost 50% of gastric cancer samples. | [241] | |
| Head and neck carcinoma | Blood GSH is decreased in cancer patients, while it is increased in tumor tissue compared to adjacent normal tissue. | [234] |
| Hepatocellular carcinoma | GPX4 and gamma-glutamyltransferase 1 expression is increased, while GCL, GR, GPX1, and GSS are decreased in liver tumor tissue compared to the surrounding normal tissue. | [144] |
| GSTA1 expression is downregulated in HCC and correlates with poor prognosis. | [248] | |
| GSTM1 expression is downregulated in HCC. | [249] | |
| GSTZ1 expression is downregulated in HCC. | [63] | |
| GSTZ1 expression is downregulated in tumor tissue compared to adjacent normal tissue and correlates with poor prognosis. | [250] | |
| High GSTP1 expression correlates with better survival and smaller tumor size. | [254] | |
| Lung cancer | GPX3 expression is decreased in NSCLC tissues. | [242] |
| GSTP1 expression is increased while GCL and gamma-glutamyltransferase 1 are decreased in tumor tissue compared to the surrounding normal tissue. | [144] | |
| Oral squamous cell carcinoma | GPX1 and GPX4 are overexpressed in oral carcinoma correlates with grade and stage and with poor survival. | [146] |
| Ovarian cancer | GPX levels are decreased in cancer patients. | [269] |
| Serum GPX3 is decreased in cancer patients and correlates with the stage of the disease. | [270] | |
| Pancreatic cancer | GPX1 expression is lower in tumor tissues compared to adjacent normal tissue and correlates with poorer prognosis. | [243] |
| Prostate cancer | GSTM1 expression is downregulated in prostate cancer. | [251] |
| GSTP1 methylation was detected in more than 80% of tumor tissues and approximately 40% of adjacent non-neoplastic tissue. | [257] | |
| The incidence of GSTP1 methylation is higher in malign than in benign tissue samples. | [258] | |
| Plasma GSTP1 is hypermethylated in cancer patients. | [259] | |
| Tumor tissues have low GSTP1 expression. | [255] | |
| Undetectable methylated GSTP1 DNA in serum correlates with a better prognosis. | [260] | |
| Thyroid cancer | Papillary thyroid carcinoma tissue has a higher expression of GPX7 compared to nodular goiter. | [271] |
| GSH System | Mechanism | Ref. |
|---|---|---|
| GCL | NRF2 overexpression promotes the expression of GCL, elevating GSH and supporting tumorigenesis, while its downregulation elevates ROS and induces G1 cell cycle arrest and apoptosis. | [110] |
| NRF2/AP-1 induces the upregulation of the GCL subunit, leading to increased GSH. | [272] | |
| GPX | GPX4 activity is negatively regulated by acetylated high-mobility group box-1, consequently promoting inflammation. | [280] |
| MiR-196a targets GPX3, downregulating its expression and promoting the tumorigenicity of NSCLC cells. | [242] | |
| GPX4 deficiency induces G1/G0 cell cycle arrest and inhibits tumorigenesis in pancreatic cancer stem-like cells. | [278] | |
| GPX2 overexpression correlates with the activation of epithelial to mesenchymal transition, the activation of β-catenin-WNT signaling, and the increased proliferation and metastasis of cervical cancer cells. | [245] | |
| GPX1 downregulation activates the AKT/GSK-3β/SNAIL pathway, promoting the epithelial to mesenchymal transition of pancreatic ductal adenocarcinoma cells. | [243] | |
| TFAP2C targets GPX1 promoter inducing GPX1 expression, while the CpG island methylation of GPX1 promoter downregulates its transcription in breast cancer. | [274] | |
| GPX3 interacts with TP53-induced gene 3, enhancing ROS production in prostate cancer cells. When this interaction is affected, the GPX3-mediated cell death is decreased. | [275] | |
| Upregulation of mature miR-17-3p inhibits GPX2 and suppresses mitochondrial respiration, rendering prostate cancer cells more sensitive to ionizing radiation. | [196] | |
| GR | GR inhibition reduces vimentin, ERK1/2, and SNAIL transcription, while it increases the E-cadherin expression, altering the epithelial to mesenchymal transition of melanoma cells. | [281] |
| GSH | When GSH is depleted, protein homeostasis is maintained in cancer cells by deubiquitinases. | [273] |
| Homocysteine induces NRF2, leading to increased GSH expression in liver cancer cells. | [282] | |
| Quinolone-indolone conjugate 2 decreases GSH. | [283] | |
| Nutrient deprivation promotes c-MYC expression, which upregulates the serine biosynthesis pathway, leading to increased GSH generation and supporting the survival and proliferation of tumor cells. | [284] | |
| GST | Overexpression of piR-31470 induces GSTP1 inactivation by the methylation of CpG island. | [277] |
| Long intergenic noncoding RNA 00844 recruits early B cell factor 1 to the GSTP1 promoter, inducing its expression and leading to the attenuated growth of prostate cancer. | [255] | |
| GSTM1 overexpression reduces ROS and elevates GSH and TP53. | [249] | |
| GSTZ1 downregulation reduces GSH, contributing to the promotion of oxidative stress and the constitutive activation of the KEAP1/NRF2 pathway, thus promoting cancer progression. | [250] | |
| GSTZ1-1 deficiency induces the accumulation of succinylacetone oncometabolite and alkylates KEAP1, leading to the activation of NRF2 signaling pathway and the transcription of insulin-like growth factor. This in turn promotes tumor growth. | [63] | |
| Long intergenic noncoding RNA 01419 overexpression promotes the methylation of GSTP1 in esophageal squamous cell carcinoma. | [285] | |
| MiR-133b targets 3’-UTR of GSTP1, downregulating its expression. | [276] | |
| GSTP1 overexpression upregulates p21 and p27, while it downregulates pAKT, inducing the G1/S cell cycle arrest of liver cancer cells. | [254] | |
| GSTA4 overexpression induces the AKT pathway, promoting the tumorigenesis of HCC. | [286] |
| Modulators | Examples |
|---|---|
| Vitamins | alpha-tocopherol |
| beta-carotene cancer | |
| Nrf2 activators | broccoli sprout extracts/sulforaphane |
| curcumin | |
| resveratrol | |
| bardoxolone-methyl (CDDO-Me) | |
| oltipraz | |
| RTA-408 (omaveloxolone) | |
| saxagliptin and sitagliptin | |
| Nrf2 inhibitors | all-trans retinoic acid (ATRA) |
| clobetasol propionate (CP) | |
| apigenin | |
| ARE expression modulator 1 (AEM1) | |
| ML385 | |
| 1-(2-cyclohexylethoxy)aniline (IM3829) | |
| malabaricone-A (MAL-A) | |
| TRX system inhibitor | auranofin |
| GSH system inhibitor | buthionine sulfoximine (BSO) |
| PI3K/AKT inhibitor | MK2206 (Merck) |
| Others | glutaminase inhibitors |
| immunotherapy for patients with mutations in NFE2L2/KEAP1 | |
| ARE-regulated lentiviral vector, expressing HSV-TK/GCV for suicide gene therapy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. https://doi.org/10.3390/antiox9111151
Jaganjac M, Milkovic L, Sunjic SB, Zarkovic N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants. 2020; 9(11):1151. https://doi.org/10.3390/antiox9111151
Chicago/Turabian StyleJaganjac, Morana, Lidija Milkovic, Suzana Borovic Sunjic, and Neven Zarkovic. 2020. "The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies" Antioxidants 9, no. 11: 1151. https://doi.org/10.3390/antiox9111151
APA StyleJaganjac, M., Milkovic, L., Sunjic, S. B., & Zarkovic, N. (2020). The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants, 9(11), 1151. https://doi.org/10.3390/antiox9111151