Redox Potential of Antioxidants in Cancer Progression and Prevention



Abstract
1. Introduction
2. Endogenous Antioxidants







3. Exogenous Antioxidants

4. Redox Potential of Antioxidants
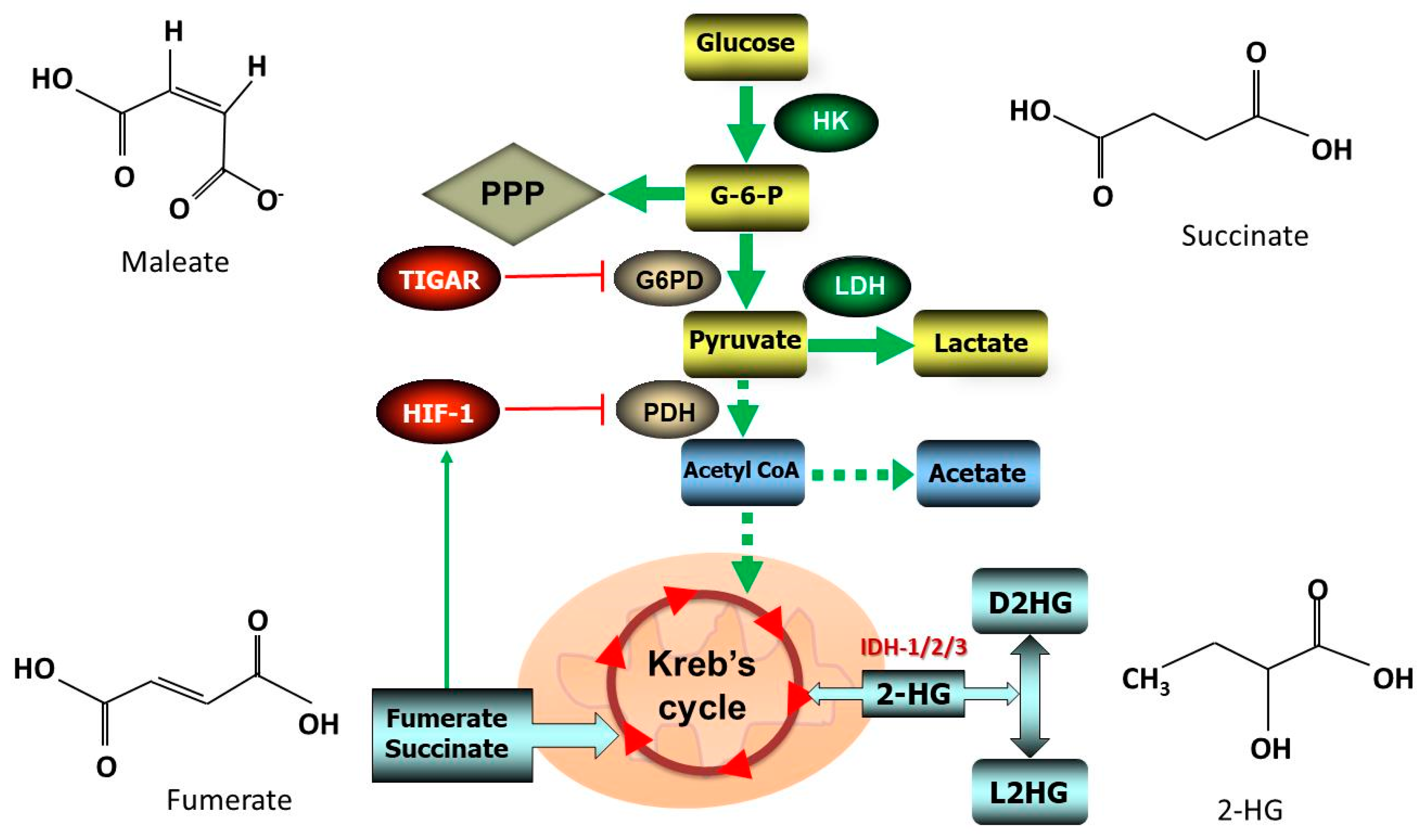
5. Metabolic Effects of Antioxidants
6. Antioxidants in Chemotherapy
7. Role of Photodynamic Therapy
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The Role of Oxidized Low-Density Lipoproteins in Atherosclerosis: The Myths and the Facts. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tinkel, J.; Hassanain, H.; Khouri, S.J. Cardiovascular Antioxidant Therapy. Cardiol. Rev. 2012, 20, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cook, N.R.; Albert, C.M.; Van Denburgh, M.; Manson, J.E. Effects of vitamins C and E and β-carotene on the risk of type 2 diabetes in women at high risk of cardiovascular disease: A randomized controlled trial. Am. J. Clin. Nutr. 2009, 90, 429–437. [Google Scholar] [CrossRef]
- Kunwar, A.; Priyadarsini, K.I. Free radicals, oxidative stress and importance of antioxidants in human health. J. Med. Allied Sci. 2011, 1, 53–60. [Google Scholar]
- Singh, K.; Bhori, M.; Kasu, Y.A.; Bhat, G.; Marar, T. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity—Exploring the armoury of obscurity. Saudi Pharm. J. 2018, 26, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-Associated Oxidative Stress: Impact on Chemotherapeutic Effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Oxidative Decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef]
- Sarangarajan, R.; Meera, S.; Rukkumani, R.; Sankar, P.; Anuradha, G. Antioxidants: Friend or foe? Asian Pac. J. Trop. Med. 2017, 10, 1111–1116. [Google Scholar] [CrossRef]
- Bergendi, L.; Beneš, L.; Ďuračková, Z.; Ferenčik, M. Chemistry, physiology and pathology of free radicals. Life Sci. 1999, 65, 1865–1874. [Google Scholar] [CrossRef]
- George, S.; Hamblin, M.R.; Abrahamse, H. Differentiation of Mesenchymal Stem Cells to Neuroglia: In the Context of Cell Signalling. Stem Cell Rev. Rep. 2019, 15, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hashimoto, J.; Suzuki, T.; Satoh, A. The effects of exercise load during development on oxidative stress levels and antioxidant potential in adulthood. Free. Radic. Res. 2017, 51, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free Radicals, Antioxidants in Disease and Health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Ighodaro, O.; Akinloye, O. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef]
- Radi, R.; Turrens, J.F.; Chang, L.Y.; Bush, K.M.; Crapo, J.D.; Freeman, B.A. Detection of catalase in rat heart mitochondria. J. Biol. Chem. 1991, 266, 22028–220234. [Google Scholar]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxidative Med. Cell. Longev. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free. Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBio. Med. 2019, 39, 239–254. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial Thiols in Antioxidant Protection and Redox Signaling: Distinct Roles for Glutathionylation and Other Thiol Modifications. Antiox. Redox. Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Molavian, H.; Goldman, A.; Phipps, C.J.; Kohandel, M.; Wouters, B.G.; Sengupta, S.; Sivaloganathan, S. Drug-induced reactive oxygen species (ROS) rely on cell membrane properties to exert anticancer effects. Sci. Rep. 2016, 6, 27439. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antiox. Redox. Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M.K. Heme Oxygenase-1: Redox Regulation of a Stress Protein in Lung and Cell Culture Models. Antiox. Redox. Signal. 2005, 7, 80–91. [Google Scholar] [CrossRef]
- Kim, J.-S.; Roberts, J.M.; Bingman, W.E.; Shao, L.; Wang, J.; Ittmann, M.M.; Weigel, N. The Prostate Cancer TMPRSS2:ERG Fusion Synergizes With the Vitamin D Receptor (VDR) to Induce CYP24A1 Expression-Limiting VDR Signaling. Endocrinology 2014, 155, 3262–3273. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide Radical and Superoxide Dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Tainer, J.A.; Getzoff, E.D.; Malencik, D.A.; Anderson, S.R.; Bomben, V.C.; Meyers, K.R.; Karplus, P.A.; Beckman, J.S. Structural Characterization of Zinc-deficient Human Superoxide Dismutase and Implications for ALS. J. Mol. Biol. 2007, 373, 877–890. [Google Scholar] [CrossRef]
- Dayal, S.; Baumbach, G.L.; Arning, E.; Bottiglieri, T.; Faraci, F.M.; Lentz, S.R. Deficiency of superoxide dismutase promotes cerebral vascular hypertrophy and vascular dysfunction in hyperhomocysteinemia. PLoS ONE 2017, 12, e0175732. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Emadi, E.M.A.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer. Res. 2003, 23, 2425–2433. [Google Scholar]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Young, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Fiedor, J.; Burda, K. Potential Role of Carotenoids as Antioxidants in Human Health and Disease. Nutrients 2014, 6, 466–488. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 1–22. [Google Scholar] [CrossRef]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free. Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free. Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Yu, G.-M.; Kubota, H.; Okita, M.; Maeda, T. The anti-inflammatory and antioxidant effects of melatonin on LPS-stimulated bovine mammary epithelial cells. PLoS ONE 2017, 12, e0178525. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Yamada, K.; Yamashita, N.; Tada-Oikawa, S.; Kawanishi, S. N-acetylcysteine, a cancer chemopreventive agent, causes oxidative damage to cellular and isolated DNA. Carcinogenesis 1999, 20, 1485–1490. [Google Scholar] [CrossRef]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef]
- Sagun, K.C.; Càrcamo, J.M.; Golde, D.W. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Gluti) and confers mitochondrial protection against oxidative injury. FASEB J. 2005, 19, 1657–1667. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef]
- Wilson, J.X. Regulation of Vitamin C Transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1–10. [Google Scholar] [CrossRef]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.-L.; Athineos, D.; et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell. Stem Cell. 2013, 12, 761–773. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Han, D. Redox regulation of NF-kappaB: From basic to clinical research. Antiox. Redox. Signal 2009, 11, 2055–2056. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2010, 21, 103–115. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-Induced Up-Regulation of MnSOD and GPx but not Catalase Increases Oxidative Stress and Apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef]
- Nam, S.Y.; Sabapathy, K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene 2011, 30, 4476–4486. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nat. Cell Biol. 1997, 389, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Hu, M.; Liu, L.; Yao, W. Activation of p53 by costunolide blocks glutaminolysis and inhibits proliferation in human colorectal cancer cells. Gene 2018, 678, 261–269. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Cebula, M.; Schmidt, E.E.; Arnér, E.S.J. TrxR1 as a Potent Regulator of the Nrf2-Keap1 Response System. Antiox. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.; Thimmulappa, R.; Singh, A.; Blake, D.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free. Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antiox. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Dev. Ther. 2018, 12, 3181–3197. [Google Scholar] [CrossRef]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 2007, 362, 816–821. [Google Scholar] [CrossRef]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, G.S.; Biswal, S. Loss of Keap1 function in prostate cancer cells causes chemo- and radio-resistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336. [Google Scholar] [CrossRef]
- Chu, X.Y.; Li, Z.J.; Zheng, Z.W.; Tao, Y.L.; Zou, F.X.; Yang, X.F. KEAP1/NRF2 signaling pathway mutations in cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4458–4466. [Google Scholar]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 Mutations and Nrf2 Pathway Activation in Epithelial Ovarian Cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic Alteration of Keap1 Confers Constitutive Nrf2 Activation and Resistance to Chemotherapy in Gallbladder Cancer. Gastroenterology 2008, 135, 1358–1368.e4. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.-I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural Basis for Defects of Keap1 Activity Provoked by Its Point Mutations in Lung Cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1–NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, X.; Zheng, C. Loss-of-Function Mutations in KEAP1 Drive Lung Cancer Progression via KEAP1/NRF2 Pathway Activation. Available online: https://assets.researchsquare.com/files/rs-8944/v2/manuscript.pdf (accessed on 1 October 2020).
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 Mutation Confers Malignant Potential and Resistance to Chemoradiation Therapy in Advanced Esophageal Squamous Cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2009, 220, 446–451. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-Associated Protein Kinase C Mutations Reveal Kinase’s Role as Tumor Suppressor. Cell 2015, 160, 489–502. [Google Scholar] [CrossRef]
- Leslie, N.R. The Redox Regulation of PI 3-Kinase–Dependent Signaling. Antioxidants Redox Signal. 2006, 8, 1765–1774. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of epithelial–mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 4, e875. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Goh, J.; Enns, L.C.; Fatemie, S.; Hopkins, H.; Morton, J.; Pettan-Brewer, C.; Ladiges, W.C. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 2011, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nat. Cell Biol. 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; DeMatteo, R.G.; Venneti, S.; Finley, L.W.S.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef]
- Corrado, M.; Scorrano, L.; Campello, S. Changing perspective on oncometabolites: From metabolic signature of cancer to tumorigenic and immunosuppressive agents. Oncotarget 2016, 7, 46692–46706. [Google Scholar] [CrossRef]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Frezza, C.; Pollard, P.J.; Gottlieb, E. Inborn and acquired metabolic defects in cancer. J. Mol. Med. 2011, 89, 213–220. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.Y.; Lu, G.; Ramkissoon, S.H.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nat. Cell Biol. 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; DeBerardinis, R.J.; Chandel, N.S. The Proto-oncometabolite Fumarate Binds Glutathione to Amplify ROS-Dependent Signaling. Mol. Cell 2013, 51, 273. [Google Scholar] [CrossRef]
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.G.; Edrada-Ebel, R.; et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 2015, 6, 6001. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—A review. Int. J. Biochem. Cell Biol. 2014, 53, 514–519. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.-W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine Catabolism Regulates Mitochondrial Redox Control during Hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef]
- Nguyen, T.-L.; Durán, R.V. Glutamine metabolism in cancer therapy. Cancer Drug Resist. 2018, 1, 126–138. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Lamson, D.W.; Brignall, M.S. Antioxidants in cancer therapy; their actions and interactions with oncologic therapies. Altern. Med. Rev. J. Clin. Ther. 1999, 4, 304–329. [Google Scholar]
- Hennekens, C.H.; Buring, J.E.; Manson, J.E.; Stampfer, M.; Rosner, B.; Cook, N.R.; Belanger, C.; LaMotte, F.; Gaziano, J.M.; Ridker, P.M.; et al. Lack of Effect of Long-Term Supplementation with Beta Carotene on the Incidence of Malignant Neoplasms and Cardiovascular Disease. N. Engl. J. Med. 1996, 334, 1145–1149. [Google Scholar] [CrossRef]
- Pantuck, A.J.; Leppert, J.T.; Zomorodian, N.; Aronson, W.; Hong, J.; Barnard, R.J.; Seeram, N.; Liker, H.; Wang, H.; Elashoff, R.; et al. Phase II Study of Pomegranate Juice for Men with Rising Prostate-Specific Antigen following Surgery or Radiation for Prostate Cancer. Clin. Cancer Res. 2006, 12, 4018–4026. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.O.; Shamma, R.N.; Elbatanony, R.S.E.; Khater, B. Antioxidants in Cancer Therapy: Recent Trends in Application of Nanotechnology for Enhanced Delivery. Sci. Pharm. 2020, 88, 5. [Google Scholar] [CrossRef]
- Cooke, M.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Pieniazek, A.; Czepas, J.; Piasecka-Zelga, J.; Gwozdzinski, K.; Koceva-Chyła, A. Oxidative stress induced in rat liver by anticancer drugs doxorubicin, paclitaxel and docetaxel. Adv. Med. Sci. 2013, 58, 104–111. [Google Scholar] [CrossRef]
- Khan, H.Y.; Zubair, H.; Ullah, M.F.; Ahmad, A.; Hadi, S.M. A prooxidant mechanism for the anticancer and chemopreventive properties of plant polyphenols. Curr. Drug Targ. 2012, 13, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Hadi, N.; Khan, N.; Hadi, S. Prooxidant property of green tea polyphenols epicatechin and epigallocatechin-3-gallate: Implications for anticancer properties. Toxicol. Vitr. 2004, 18, 555–561. [Google Scholar] [CrossRef]
- Zhong, J.; Rajaram, N.; Brizel, D.M.; Frees, A.E.; Ramanujam, N.; Batinic-Haberle, I.; Dewhirst, M.W. Radiation induces aerobic glycolysis through reactive oxygen species. Radiother. Oncol. 2013, 106, 390–396. [Google Scholar] [CrossRef]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Greenlee, H.; Kwan, M.L.; Kushi, L.H.; Song, J.; Castillo, A.; Weltzien, E.; Quesenberry, C.P., Jr.; Caan, B.J. Antioxidant supplement use after breast cancer diagnosis and mortality in the Life After Cancer Epidemiology (LACE) cohort. Cancer 2011, 118, 2048–2058. [Google Scholar] [CrossRef]
- Yang, C.S.; Luo, P.; Zeng, Z.; Wang, H.; Malafa, M.; Suh, N. Vitamin E and cancer prevention: Studies with different forms of tocopherols and tocotrienols. Mol. Carcinog. 2020, 59, 365–389. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Vile, G.F.; Tyrrell, R.M. Uva radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free. Radic. Biol. Med. 1995, 18, 721–730. [Google Scholar] [CrossRef]
- Moss, R.W. Do Antioxidants Interfere with Radiation Therapy for Cancer? Integr. Cancer Ther. 2007, 6, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Weijer, R.; Van Gulik, T.M.; Hamblin, M.R.; Heger, M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metast. Rev. 2015, 34, 643–690. [Google Scholar] [CrossRef]
- Jakus, J.; Farkas, O. Photosensitizers and antioxidants: A way to new drugs? Photochem. Photobiol. Sci. 2005, 4, 694. [Google Scholar] [CrossRef]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Simultaneous Photodiagnosis and Photodynamic Treatment of Metastatic Melanoma. Molecules 2019, 24, 3153. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kajimoto, Y.; Sun, W.; Nakagawa, H.; Inoue, Y.; Ikegami, Y.; Miyatake, S.-I.; Kuroiwa, T. Role of Nrf2 in Cancer Photodynamic Therapy: Regulation of Human ABC Transporter ABCG2. J. Pharm. Sci. 2013, 102, 3058–3069. [Google Scholar] [CrossRef]
- Piette, J. Signalling pathway activation by photodynamic therapy: NF-κB at the crossroad between oncology and immunology. Photochem. Photobiol. Sci. 2015, 14, 1510–1517. [Google Scholar] [CrossRef]
- Tynga, I.M.; Abrahamse, H. Nano-Mediated Photodynamic Therapy for Cancer: Enhancement of Cancer Specificity and Therapeutic Effects. Nanomaterials 2018, 8, 923. [Google Scholar] [CrossRef]
- Chung, M.K.; Kim, D.H.; Ahn, Y.C.; Choi, J.Y.; Kim, E.H.; Son, Y.-I. Randomized Trial of Vitamin C/E Complex for Prevention of Radiation-Induced Xerostomia in Patients with Head and Neck Cancer. Otolaryngol. Neck Surg. 2016, 155, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Dey, N.; Ghosh, A.; Das, S.; Chattopadhyay, D.J.; Chatterjee, I.B. Molecular and Cellular Mechanisms of Cigarette Smoke-Induced Myocardial Injury: Prevention by Vitamin C. PLoS ONE 2012, 7, e44151. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Somatic Mutations | Cancer Types | References |
|---|---|---|---|
| KeapI | C23Y, D256G | Breast cancer | [70,71] |
| R234W, T330I, V660M, | Colorectal adenocarcinoma | [70,72] | |
| R470C | Cervical squamous carcinoma | [73] | |
| F107L, A159T, A188V, P412S, E611K | Epithelial ovarian cancer | [74] | |
| Q82H, S233N, F280L | Gastric adenocarcinoma | [70] | |
| S338L, G379D, | Gall bladder adenocarcinoma | [75] | |
| N183S, D236Y, L342M | Hepatocellular carcinoma | [70] | |
| C249Y, W554X | Liver cholangiocarcinoma | [75] | |
| E244K, G364C, R415G, G430C, W591L | Lung adenocarcinoma | [70,76] | |
| G333C | Lung non-small-cell carcinoma | [77] | |
| V42A, S45F, R320Q | Lung squamous cell carcinoma | [70,78] | |
| Y255H, T314M, D357N, | Prostrate adenocarcinoma | [70,75] | |
| Nrf2 | W24C, D29Y, R34Q, D77A, G81D | Cervical squamous carcinoma | [73,79] |
| D27Y, E78K, E79K, T80K, G81V, E82L | Esophagus squamous carcinoma | [79,80] | |
| D29G, Q75H, T80I | Head and neck cancer | [75] | |
| D29H, D77N, E79G | Larynx carcinoma | [80] | |
| E79Q, | Lung adenocarcinoma | [75] | |
| D77V, E79K, R34L, R34Q, R34Q | Lung non-small-cell carcinoma | [75,81] | |
| D77A, E79K, T80R, G81D, E82G | Lung squamous cell carcinoma | [75,76,80] | |
| L30F | Malignant melanoma | [79] | |
| G31A | Skin squamous cell carcinoma | [80] |
| Advantages | Disadvantages | References | |
|---|---|---|---|
| Antioxidants | Antagonize excess free radicals in healthy cells | Cancers may also benefit from the use of antioxidants | [13,45,58] |
| Enhancement of host antitumor immunity | Antioxidants might prevent cancers than curing them | [47,108] | |
| Available in a wide variety of fruits and vegetables | Difficulty in delivery to tumor mass or internal organs | [109,110] | |
| Short-lived and ultra-fast acting oxidizing biomolecules | Acts on DNA causing mutations and increasing cancer | [111,112,113] | |
| Prooxidants | Increase oxidative stress hindering cancer growth | Damage healthy tissues, especially the internal organs | [23,114] |
| Synergistic action with many anti-cancer drugs | Increase cytotoxicity during radiotherapy | [115,116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. https://doi.org/10.3390/antiox9111156
George S, Abrahamse H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants. 2020; 9(11):1156. https://doi.org/10.3390/antiox9111156
Chicago/Turabian StyleGeorge, Sajan, and Heidi Abrahamse. 2020. "Redox Potential of Antioxidants in Cancer Progression and Prevention" Antioxidants 9, no. 11: 1156. https://doi.org/10.3390/antiox9111156
APA StyleGeorge, S., & Abrahamse, H. (2020). Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants, 9(11), 1156. https://doi.org/10.3390/antiox9111156