Vascular Cells Proteome Associated with Bradykinin and Leptin Inflammation and Oxidative Stress Signals


 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Primary Cell Extraction
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Extraction and Tryptic Digestion of Proteins
2.5. Liquid Chromatography-Mass Spectroscopy/Mass Spectroscopy Analysis
2.6. Statistical Analysis and LC-MS/MS Data Analysis
2.7. Systems Biology Assessment
2.8. RNA extraction and Real-Time PCR
2.9. Statistical Analysis
3. Results
3.1. RASMC Proteomic Analysis
3.2. Effect of BK Stimulation on the Proteome Profile in RASMC
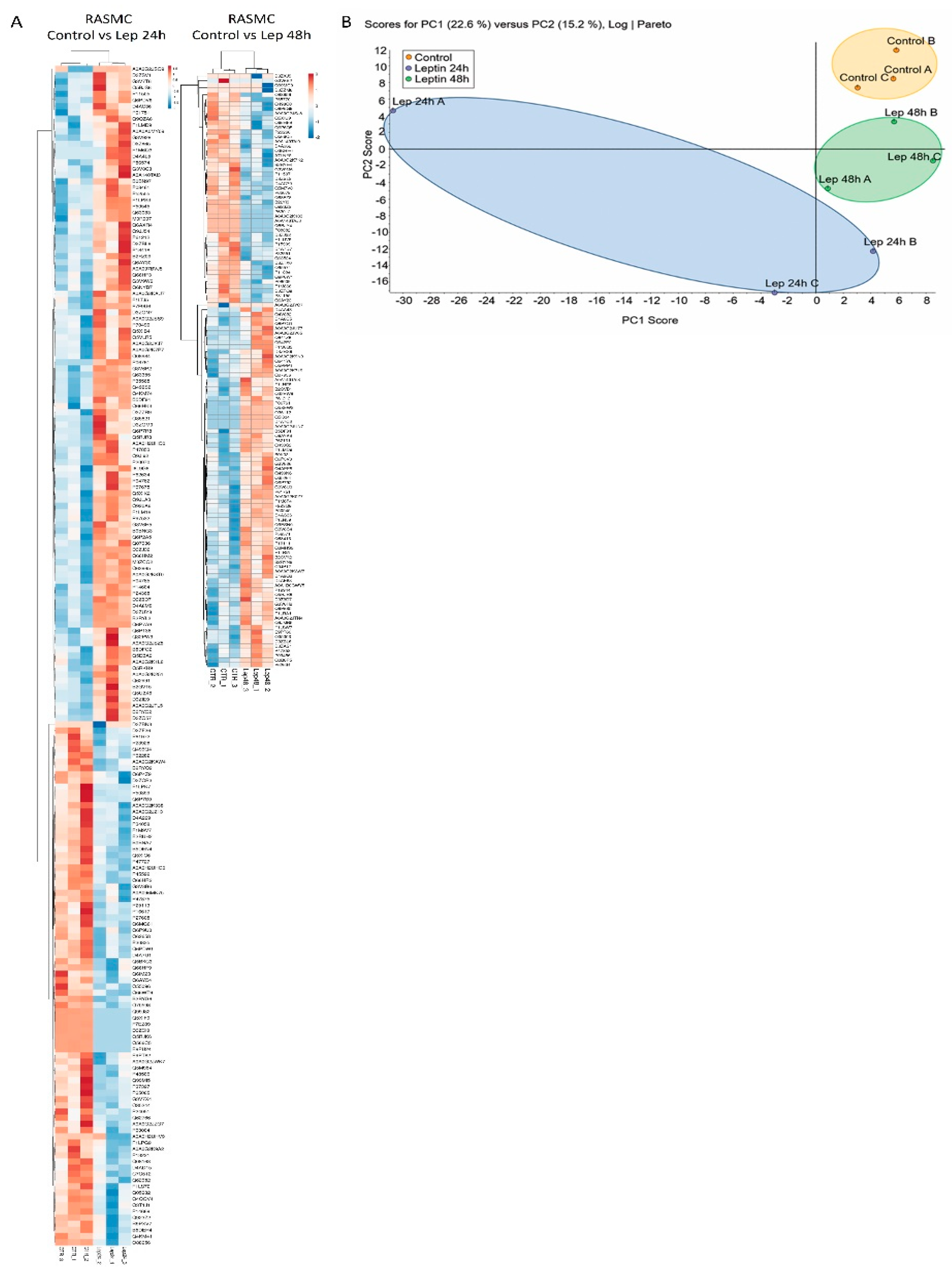
3.3. Principal Component Analysis (PCA)
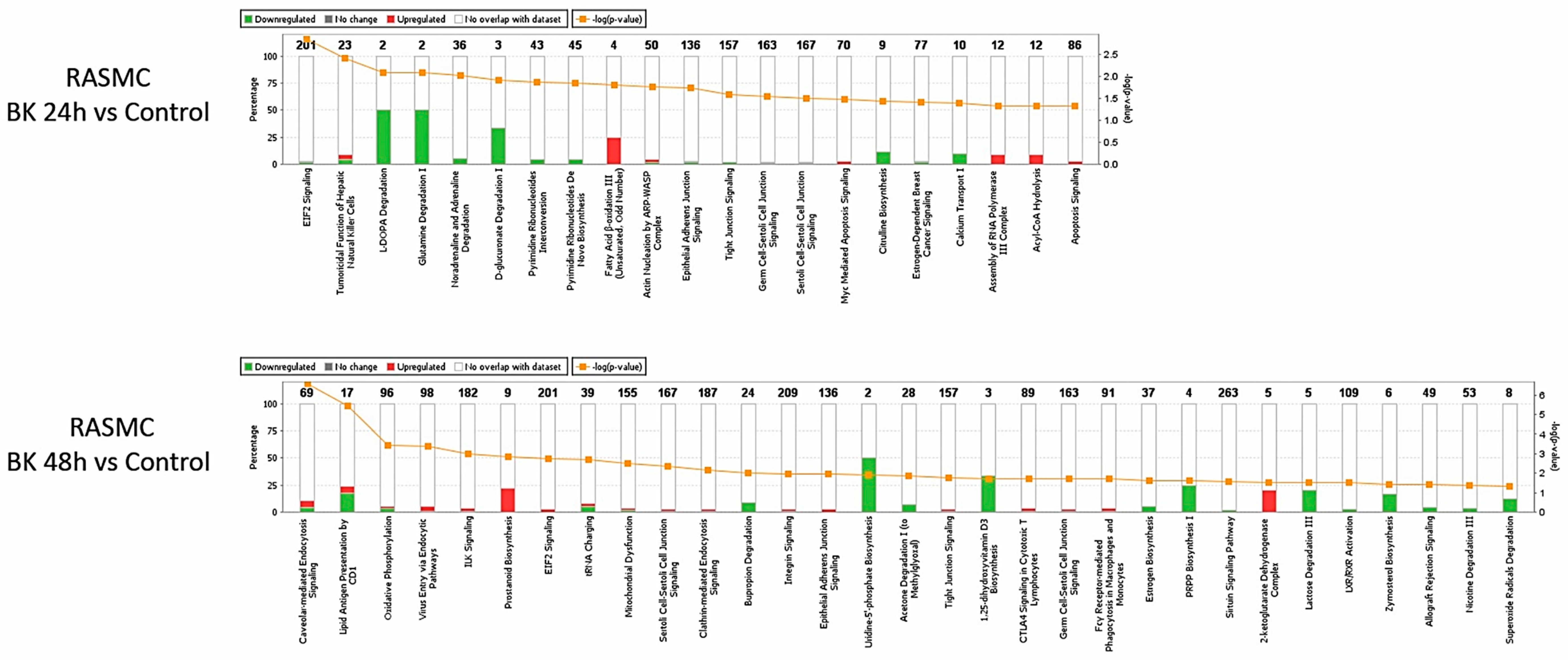
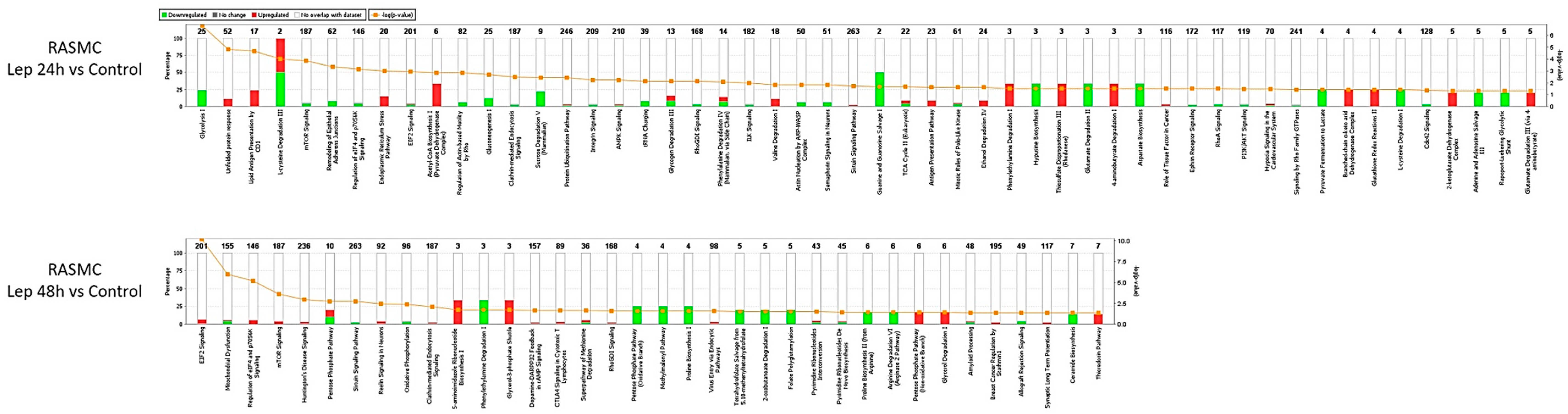
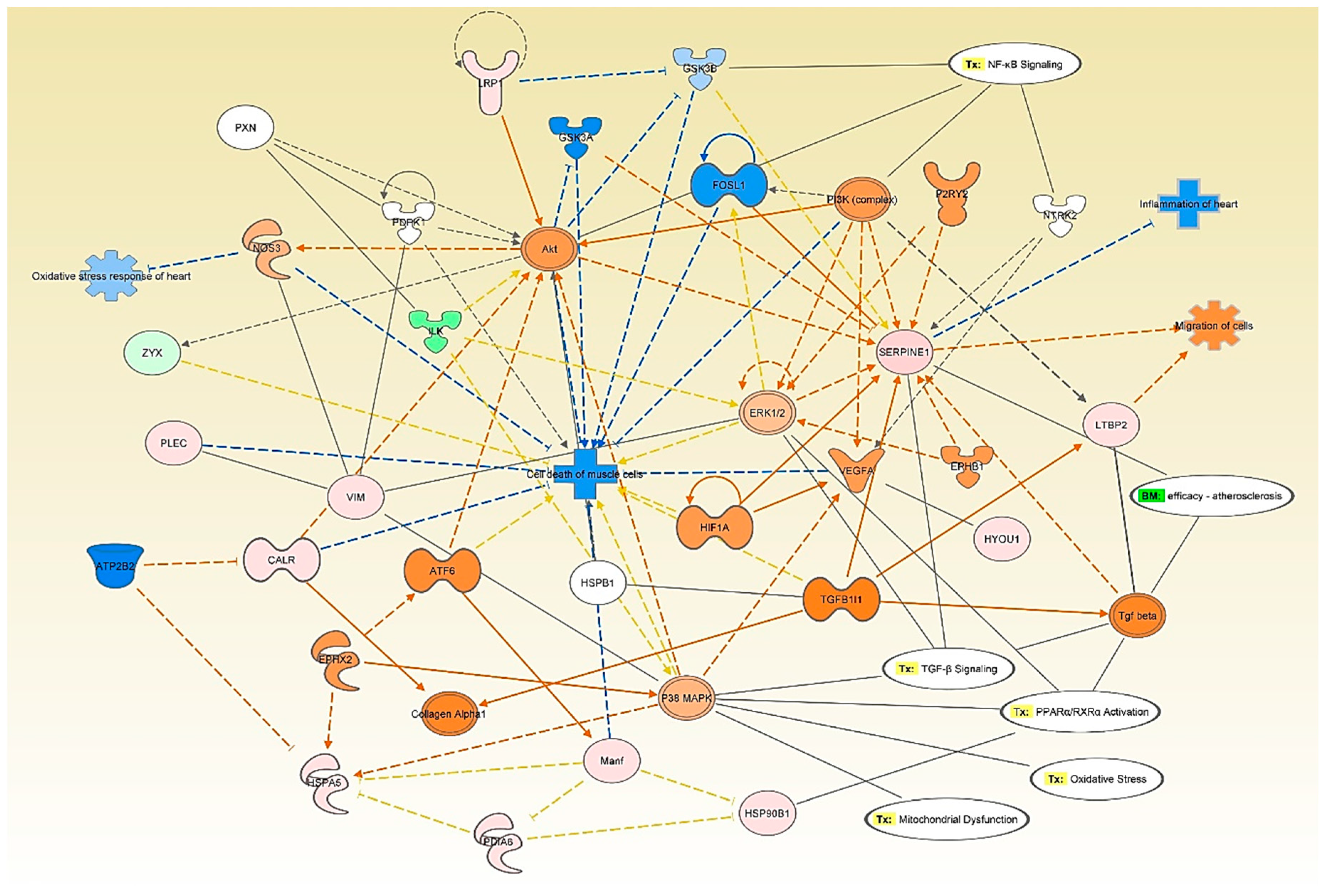
3.4. Network and Pathway Analysis of Proteomic Profiles
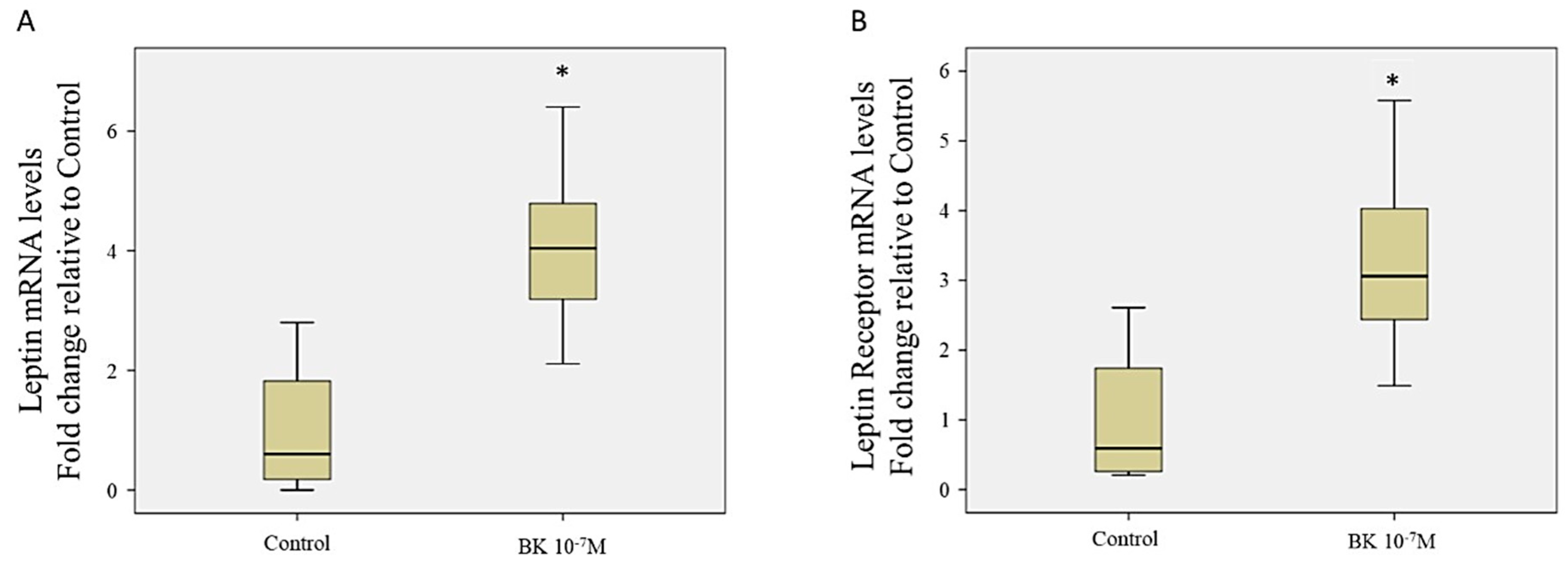
3.5. Effect of Leptin Stimulation on the Proteome Profile in RASMC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Coussens, L. Session 2: Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear Factor κB (NF-κB)–Mediated Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [Green Version]
- Fiordelisi, A.; Eiaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxidative Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Infusino, F.; Maestrini, V.; Mancone, M.; Fedele, F. Myocardial Ischemia and Diabetes Mellitus: Role of Oxidative Stress in the Connection between Cardiac Metabolism and Coronary Blood Flow. J. Diabetes Res. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Pucci, M.; Infusino, F.; Birtolo, L.I.; Mariani, M.V.; LaValle, C.; Maestrini, V.; Mancone, M.; Fedele, F. Ischemic Heart Disease and Heart Failure: Role of Coronary Ion Channels. Int. J. Mol. Sci. 2020, 21, 3167. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Executive summary: Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 2013, 127, 143–152. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Tavori, H.; Su, Y.R.; Yancey, P.G.; Giunzioni, I.; Wilhelm, A.J.; Blakemore, J.L.; Zabalawi, M.; Linton, M.F.; Sorci-Thomas, M.G.; Fazio, S. Macrophage apoAI protects against dyslipidemia-induced dermatitis and atherosclerosis without affecting HDL. J. Lipid Res. 2015, 56, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.W.; Lim, L.; Ju, S.; Choi, D.-H.; Song, H. Effects of matrix metalloproteinase 13 on vascular smooth muscle cells migration via Akt–ERK dependent pathway. Tissue Cell 2014, 47, 115–121. [Google Scholar] [CrossRef]
- Webb, R.C. Smooth muscle contraction and relaxation. Adv. Physiol. Educ. 2003, 27, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Salabei, J.K.; Hill, B.G. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free. Radic. Biol. Med. 2013, 65, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Jaffa, A.A.; Durazo-Arvizu, R.; Zheng, D.; Lackland, D.T.; Srikanth, S.; Garvey, W.T.; Schmaier, A.H. Plasma prekallikrein: A risk marker for hypertension and nephropathy in type 1 diabetes. Diabetes 2003, 52, 1215–1221. [Google Scholar] [CrossRef] [Green Version]
- Al Hariri, M.; Elmedawar, M.; Zhu, R.; Jaffa, M.A.; Zhao, J.; Mirzaei, P.; Ahmed, A.; Kobeissy, F.; Ziyadeh, F.N.; Mechref, Y.; et al. Proteome profiling in the aorta and kidney of type 1 diabetic rats. PLoS ONE 2017, 12, e0187752. [Google Scholar] [CrossRef] [Green Version]
- Frigolet, M.E.; Thomas, G.; Beard, K.; Lu, H.; Liu, L.; Fantus, I.G. The bradykinin-cGMP-PKG pathway augments insulin sensitivity via upregulation of MAPK phosphatase-5 and inhibition of JNK. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E321–E334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofman, Z.; De Maat, S.; Hack, C.E.; Maas, C. Bradykinin: Inflammatory Product of the Coagulation System. Clin. Rev. Allergy Immunol. 2016, 51, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Wang, B.; Keum, J.-S.; Jaffa, A.A. Mechanisms through which bradykinin promotes glomerular injury in diabetes. Am. J. Physiol. Renal. Physiol. 2005, 288, F483–F492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzuoli, E.; Meini, S.; Cucchi, P.; Catalani, C.; Cialdai, C.; Maggi, C.A.; Giachetti, A.; Ziche, M.; Donnini, S. Antagonism of Bradykinin B2 Receptor Prevents Inflammatory Responses in Human Endothelial Cells by Quenching the NF-kB Pathway Activation. PLoS ONE 2014, 9, e84358. [Google Scholar] [CrossRef] [PubMed]
- Velarde, V.; De La Cerda, P.M.; Duarte, C.; Arancibia, F.; Abbott, E.; González, A.; Moreno, F.; Jaffa, A.A. Role of reactive oxygen species in bradykinin-induced proliferation of vascular smooth muscle cells. Biol. Res. 2004, 37, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Greene, E.L.; Velarde, V.; Jaffa, A.A. Role of reactive oxygen species in bradykinin-induced mitogen-activated protein kinase and c-fos induction in vascular cells. Hypertension 2000, 35, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Jaffa, M.A.; Kobeissy, F.; Al Hariri, M.; Chalhoub, H.; Eid, A.; Ziyadeh, F.N.; Jaffa, A.A. Global Renal Gene Expression Profiling Analysis in B2-Kinin Receptor Null Mice: Impact of Diabetes. PLoS ONE 2012, 7, e44714. [Google Scholar] [CrossRef]
- Ferroni, P.; Basili, S.; Falco, A.; Davì, G. Inflammation, insulin resistance, and obesity. Curr. Atheroscler. Rep. 2004, 6, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Mantzoros, C. The Role of Leptin in Human Obesity and Disease: A Review of Current Evidence. Ann. Intern. Med. 1999, 130, 671–680. [Google Scholar] [CrossRef]
- Matarese, G.; Moschos, S.; Mantzoros, C.S. Leptin in Immunology. J. Immunol. 2005, 174, 3137–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.W.; Peskind, E.; Raskind, M.; Boyko, E.J.; Porte, D. Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nat. Med. 1996, 2, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Li, M.; Duan, C.; Rui, L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. 2005, 2, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majack, R.A.; Clowes, A.W. Inhibition of vascular smooth muscle cell migration by heparin-like glycosaminoglycans. J. Cell. Physiol. 1984, 118, 253–256. [Google Scholar] [CrossRef]
- Wilson, P.C.; Lee, M.-H.; Appleton, K.M.; El-Shewy, H.M.; Morinelli, T.A.; Peterson, Y.K.; Luttrell, L.M.; Jaffa, A.A. The Arrestin-selective Angiotensin AT1 Receptor Agonist [Sar1,Ile4,Ile8]-AngII Negatively Regulates Bradykinin B2 Receptor Signaling via AT1-B2 Receptor Heterodimers*. J. Biol. Chem. 2013, 288, 18872–18884. [Google Scholar] [CrossRef] [Green Version]
- Soudani, N.; Ghantous, C.M.; Farhat, Z.; Shebaby, W.N.; Zibara, K.; Zeidan, A. Calcineurin/NFAT Activation-Dependence of Leptin Synthesis and Vascular Growth in Response to Mechanical Stretch. Front. Physiol. 2016, 7, 433. [Google Scholar] [CrossRef] [Green Version]
- Dagher, O.K.; Jaffa, M.A.; Habib, A.; Ziyadeh, F.N.; Jaffa, A.A. Heteromerization fingerprints between bradykinin B2 and thromboxane TP receptors in native cells. PLoS ONE 2019, 14, e0216908. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Mayr, M.; Gomes, A.V.; Delles, C.; Arrell, D.K.; Murphy, A.M.; Lange, R.A.; Costello, C.E.; Jin, Y.-F.; Laskowitz, D.T.; et al. Transformative Impact of Proteomics on Cardiovascular Health and Disease: A Scientific Statement From the American Heart Association. Circulation 2015, 132, 852–872. [Google Scholar] [CrossRef] [Green Version]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Douillet, C.D.; Velarde, V.; Christopher, J.T.; Mayfield, R.K.; Trojanowska, M.E.; Jaffa, A.A. Mechanisms by which bradykinin promotes fibrosis in vascular smooth muscle cells: Role of TGF-beta and MAPK. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 2829–2837. [Google Scholar] [CrossRef] [Green Version]
- Nevelsteen, I.; Bergh, A.V.D.; Van Der Mieren, G.; Vanderper, A.; Mubagwa, K.; Bult, H.; Herijgers, P. NO-Dependent Endothelial Dysfunction in Type II Diabetes Is Aggravated by Dyslipidemia and Hypertension, but Can Be Restored by Angiotensin-Converting Enzyme Inhibition and Weight Loss. J. Vasc. Res. 2013, 50, 486–497. [Google Scholar] [CrossRef]
- Ramírez-Sánchez, I.; Maya-Ramos, L.; Ceballos-Reyes, G.; Villarreal, F. (-)-Epicatechin induces calcium and translocation independent eNOS activation in arterial endothelial cells. Am. J. Physiol. Cell Physiol. 2011, 300, C880–C887. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-M.; Kreipe, B.A.; Trzeciakowski, J.; Dangott, L.; Trache, A. Extracellular matrix effect on RhoA signaling modulation in vascular smooth muscle cells. Exp. Cell Res. 2010, 316, 2833–2848. [Google Scholar] [CrossRef]
- Maya-Monteiro, C.M.; Bozza, P.T. Leptin and mTOR: Partners in metabolism and inflammation. Cell Cycle 2008, 7, 1713–1717. [Google Scholar] [CrossRef] [Green Version]
- Fazolini, N.P.B.; Cruz, A.L.S.; Werneck, M.B.F.; Viola, J.P.B.; Maya-Monteiro, C.M.; Bozza, P.T. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 2015, 14, 2667–2676. [Google Scholar] [CrossRef]
- Sun, Z.; Li, Z.; Meininger, G.A. Mechanotransduction through fibronectin-integrin focal adhesion in microvascular smooth muscle cells: Is calcium essential? Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1965–H1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciver, S.K.; Hussey, P.J. The ADF/cofilin family: Actin-remodeling proteins. Genome Biol. 2002, 3, 3007. [Google Scholar] [CrossRef] [PubMed]
- Galkin, V.E.; Orlova, A.; Kudryashov, D.S.; Solodukhin, A.; Reisler, E.; Schröder, G.F.; Egelman, E.H. Remodeling of actin filaments by ADF/cofilin proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 20568–20572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free. Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- King-Briggs, K.E.; Shanahan, C.M. TGF-beta superfamily members do not promote smooth muscle-specific alternative splicing, a late marker of vascular smooth muscle cell differentiation. Differentiation 2000, 66, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Jaffa, M.A.; Luttrell, D.; Schmaier, A.H.; Klein, R.L.; Lopes-Virella, M.; Luttrell, L.M.; Jaffa, A.A.; The DCCT/EDIC Research Group. Plasma Prekallikrein Is Associated With Carotid Intima-Media Thickness in Type 1 Diabetes. Diabetes 2016, 65, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Medina, I.; Cougoule, C.; Drechsler, M.; Bermudez, B.; Koenen, R.R.; Sluimer, J.; Wolfs, I.; Döring, Y.; Herias, V.; Gijbels, M.; et al. Hck/Fgr Kinase Deficiency Reduces Plaque Growth and Stability by Blunting Monocyte Recruitment and Intraplaque Motility. Circulation 2015, 132, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Page, T.H.; Smolinska, M.; Gillespie, J.; Urbaniak, A.M.; Foxwell, B.M.J. Tyrosine kinases and inflammatory signalling. Curr. Mol. Med. 2009, 9, 69–85. [Google Scholar] [CrossRef]
- Han, M.H.; Lee, W.S.; Nagappan, A.; Hong, S.H.; Jung, J.H.; Park, C.; Kim, H.J.; Kim, G.-Y.; Kim, G.; Jung, J.-M.; et al. Flavonoids Isolated from Flowers of Lonicera japonica Thunb. Inhibit Inflammatory Responses in BV2 Microglial Cells by Suppressing TNF-alpha and IL-beta Through PI3K/Akt/NF-kb Signaling Pathways. Phytother. Res. 2016, 30, 1824–1832. [Google Scholar] [CrossRef]
- Srivastava, S.; Sharma, K.; Kumar, N.; Roy, P. Bradykinin regulates osteoblast differentiation by Akt/ERK/NFkappaB signaling axis. J. Cell. Physiol. 2014, 229, 2088–2105. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hariri, M.A.; Jaffa, M.A.; Saoud, R.; Zhao, J.; Zhu, R.; Jaffa, A.A.; El-Achkar, G.A.; Moussa, M.; Kobeissy, F.; Hassan, A.; et al. Vascular Cells Proteome Associated with Bradykinin and Leptin Inflammation and Oxidative Stress Signals. Antioxidants 2020, 9, 1251. https://doi.org/10.3390/antiox9121251
Hariri MA, Jaffa MA, Saoud R, Zhao J, Zhu R, Jaffa AA, El-Achkar GA, Moussa M, Kobeissy F, Hassan A, et al. Vascular Cells Proteome Associated with Bradykinin and Leptin Inflammation and Oxidative Stress Signals. Antioxidants. 2020; 9(12):1251. https://doi.org/10.3390/antiox9121251
Chicago/Turabian StyleHariri, Moustafa Al, Miran A. Jaffa, Richard Saoud, Jingfu Zhao, Rui Zhu, Aneese A. Jaffa, Ghewa A. El-Achkar, Mayssam Moussa, Firas Kobeissy, Anwarul Hassan, and et al. 2020. "Vascular Cells Proteome Associated with Bradykinin and Leptin Inflammation and Oxidative Stress Signals" Antioxidants 9, no. 12: 1251. https://doi.org/10.3390/antiox9121251