1. Introduction
Ischemia/reperfusion (I/R) installs a cascade of pathological events that culminate in several clinical conditions, including ischemic stroke and degenerative brain disease [
1]. The I/R provokes a deleterious mechanism that involves excess production of reactive oxygen species (ROS), resulting in the oxidative stress in cells [
2]. With high metabolic demand and peroxidizable lipid contents, neurons are potentially vulnerable to I/R-induced oxidative injury, which is accompanied by apoptosis, necrosis, and DNA damage. In reperfused cells, mitochondria play a critical role in initiating apoptosis through contributing excess ROS generation. During I/R, there is an involvement of glutamate excitotoxicity, to which the expression of extrasynaptic glutamate receptors such as GluN2B has been crucially implicated. This I/R-induced excitotoxicity initiates a cascade mechanism that results in the cellular oxidative stress and loss of mitochondrial membrane potential (ΔΨ
m). Targeting excitotoxicity-mediated oxidative stress might, therefore, be a potential therapeutic strategy in neurodegenerative diseases.
A neuronal model of hypoxia/reoxygenation (H/R) provides a useful tool for the study of ROS-mediated mechanisms of cellular dysfunction in I/R injury in the brain [
1]. In this study, a cellular model of H/R-induced oxidative injury that can simulate the pathophysiological events of cerebral I/R was, therefore, employed to investigate the neuroprotective effect of GAE in primary hippocampal neurons. In several of our previous studies, we reported a very potential neurotrophic activity of GAE that spans every stage of neuronal developments [
3], including early neuronal differentiation, axonal sprouting, dendritic arborization, axonal maturation and synaptic modulation [
4,
5,
6]. In addition, GAE has shown various other pharmacological activities, such as immunomodulation [
7] and antioxidation [
8]. Having all these pharmacological effects, we hypothesized that GAE could protect against H/R-induced oxidative injury in primary hippocampal neurons.
2. Materials and Methods
2.1. Sample Collection and Extract Preparation
The mature thalli of
G. amansii were collected along the coast of the southern part of the Korean peninsula and processed as described in our previous study [
4]. An ethanolic extract of
G. amansii (GAE) was then prepared following the protocol as previously described [
4]. The extract was reconstituted in dimethyl sulfoxide (DMSO) to make an aliquot of 8 mg/mL.
2.2. Primary Neuronal Culture and GAE Treatment
All the reagents used for cell cultures were purchased from Invitrogen (Carlsbad, CA, USA) unless otherwise stated. The animal experiment was approved by the Institutional Animal Care and Use Committee of the Dongguk University College of Medicine (approval certificate number IACUC-2016-001). Time-pregnant rats (Sprague-Dawley) were ordered on the 13th day of pregnancy and housed in controlled temperature with a light/dark cycle of 12/12 h and with access to food and water
ad libitum. On the 19th day of pregnancy, the pregnant rat was euthanized with isofluorane, and the fetuses were collected. The dissociated cultures of primary hippocampal neurons from the fetal brain were prepared as previously described [
9]. The dissected hippocampi were collected in Hank’s balanced salt solution (HBSS), and the tissues were dissociated by digestion with 0.25% trypsin in HBSS for 12 min at 37 °C and trituration with fire-polished graded Pasteur pipettes. The dissociated cells were seeded at a density of 3.0 × 10
4 cells/cm
2 onto poly-
dl-lysine-coated (PDL, Sigma-Aldrich, St. Louis, MO, USA) 12-mm glass coverslips in 24-well culture plates for morphological and viability analysis, or 3.0 × 10
6 cells/cm
2 and 1.2 × 10
5 cells/cm
2 onto PDL-coated six-well culture plates for agarose gel electrophoresis and Western blot, respectively. Cultures were maintained in a defined serum-free neurobasal media supplemented with B27 and incubated at 37 °C under 5% CO
2 and 95% air. The culture medium was preincubated with GAE or vehicle (DMSO, final concentration <0.5%).
2.3. Hypoxia/Reoxygenation (H/R) Injury
Neurons were exposed to H/R following the protocol as described previously [
10], with a slight modification. Briefly, at the indicated time, cultured neurons were transferred to a hypoxic chamber (Modular Incubator Chamber MIC-101; Billups-Rothenberg Inc., Del Mar, CA, USA) containing 94% N
2, 5% CO
2, and 1% O
2 and incubated for 4 h at 37 °C. The culture plates were then returned to normoxic conditions (95% air and 5% CO
2 at 37 °C) and incubated for the indicated time.
2.4. Assessment of Neuronal Viability and Cytotoxicity
Trypan blue exclusion assay. Neuronal viability was determined by trypan blue exclusion assay in cultures maintained in normoxic and hypoxic conditions. The cultures were stained with 0.4% trypan blue for 15 min at room temperature and then washed with Dulbecco’s phosphate-buffered saline (D-PBS). The neurons were then quantified under a microscope (Leica Microsystems AG, Wetzlar, Germany). Dead neurons are compromised to membrane permeability, and thus, uptake dye and appeared dark-blue in phase-contrast images. Live neurons have intact membrane integrity, and thus, exclude dye. The viability was expressed as the percentage of trypan blue-impermeable cells (live neurons) and results were normalized versus trypan blue-stained non-H/R exposed controls. In each experiment, cells on three coverslips, each with 500 cells, were counted randomly.
Measurement of lactate dehydrogenase (LDH) release. The cellular injury was evaluated by measuring the LDH released in the culture media through the damaged cell membrane using the CytoTox96 nonradioactive assay (Promega, Madison, WI, USA) and quantitated by measuring the wavelength at 490 nm using a microplate reader (Molecular Devices, San Jose, CA, USA). LDH activity is the percentage of the ratio of experimental LDH release with maximum LDH release. Data are normalized to the amount of LDH released from vehicle-treated cells (100%).
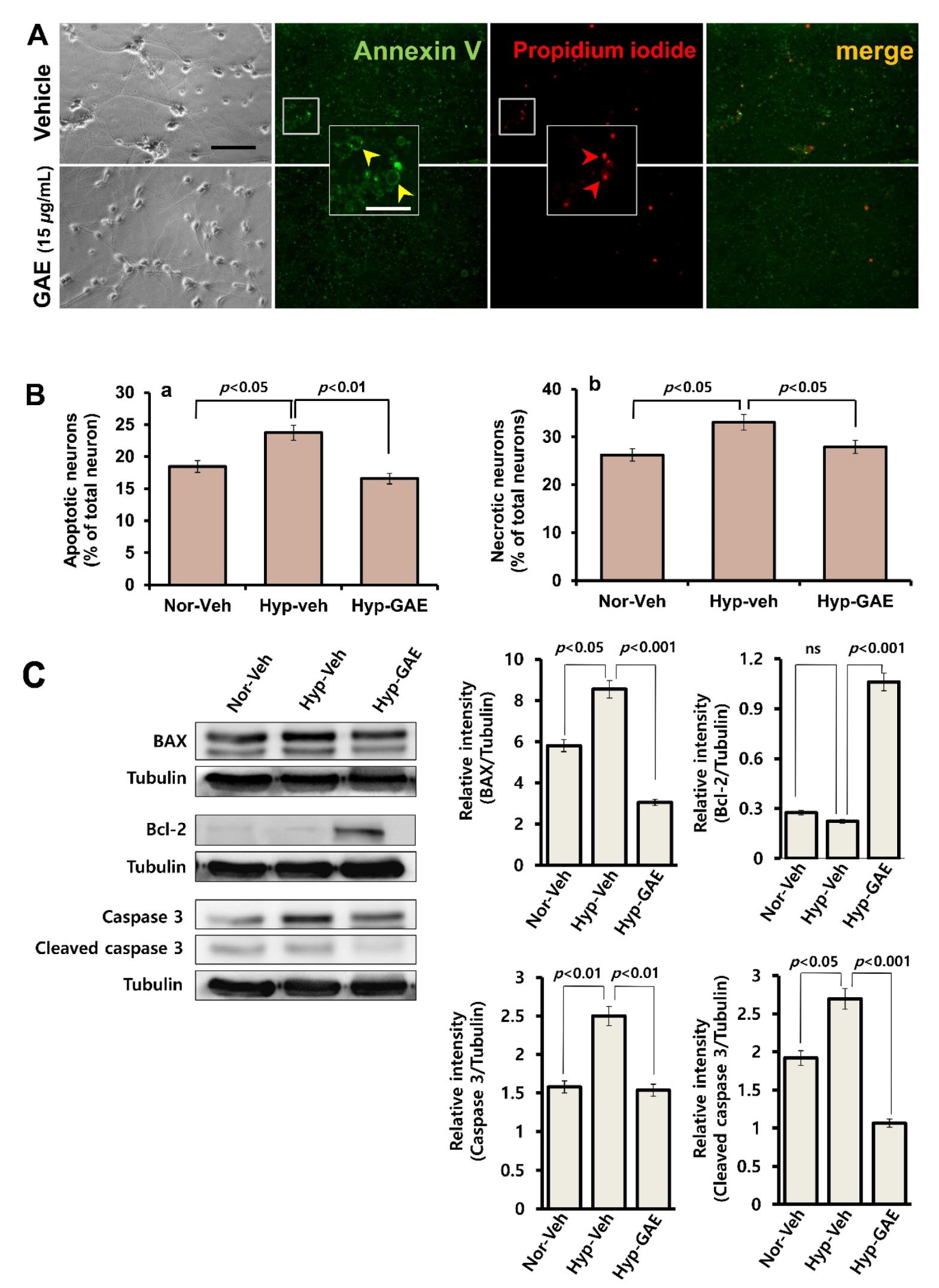
2.5. Measurement of Apoptotic Cell Death
Neurons that underwent apoptotic and necrotic death were determined by Annexin V binding and propidium iodide (PI) uptake, respectively. Annexin V shows a high affinity for phosphatidylserine, which translocates from the internal to the external surface of the plasma membrane as a characteristic feature of apoptosis. Necrotic cells take up PI due to increased permeability of the damaged cell membrane for this molecule. Neuronal cultures maintained in normoxic and hypoxic conditions were washed with binding buffer and incubated for 15 min in the dark with Annexin V and PI. Apoptotic and necrotic cells were quantitated under a fluorescence microscope and expressed as percentages of total neurons in culture.
2.6. Analysis of DNA Fragmentation by Agarose Gel Electrophoresis
Neuronal cells (3 × 106) were rinsed twice with DPBS and lysed in 700 μL of DNA extraction solution (20 mM Tris-HCl, pH 7.4, 0.1M NaCl, 5 mM EDTA, and 0.5% sodium dodecyl sulfate). The lysates were incubated with DNAse free RNAse A (100 μg/mL) and proteinase K (200 μg/mL) at 37 °C in a shaking incubator overnight. After incubation, cell lysates were mixed well with 700 μL of phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v), and then centrifuged at 13,000 rpm for 10 min at 4 °C. DNA that remained in the aqueous phase was extracted twice more with phenol/chloroform/isoamyl alcohol and then with chloroform. DNA was then precipitated overnight at −80 °C with two volumes of absolute ethanol in the presence of 1/10th volume of 3M sodium acetate. After centrifugation, the DNA pellets were washed with 70% ethanol and air-dried. The DNA was dissolved in TE buffer (10 mM Tris-HCl and 1 mM EDTA). DNA was electrophoresed on 1.5% agarose gel containing 1 μg/mL ethidium bromide, and DNA fragments were visualized by exposing the gel to UV light.
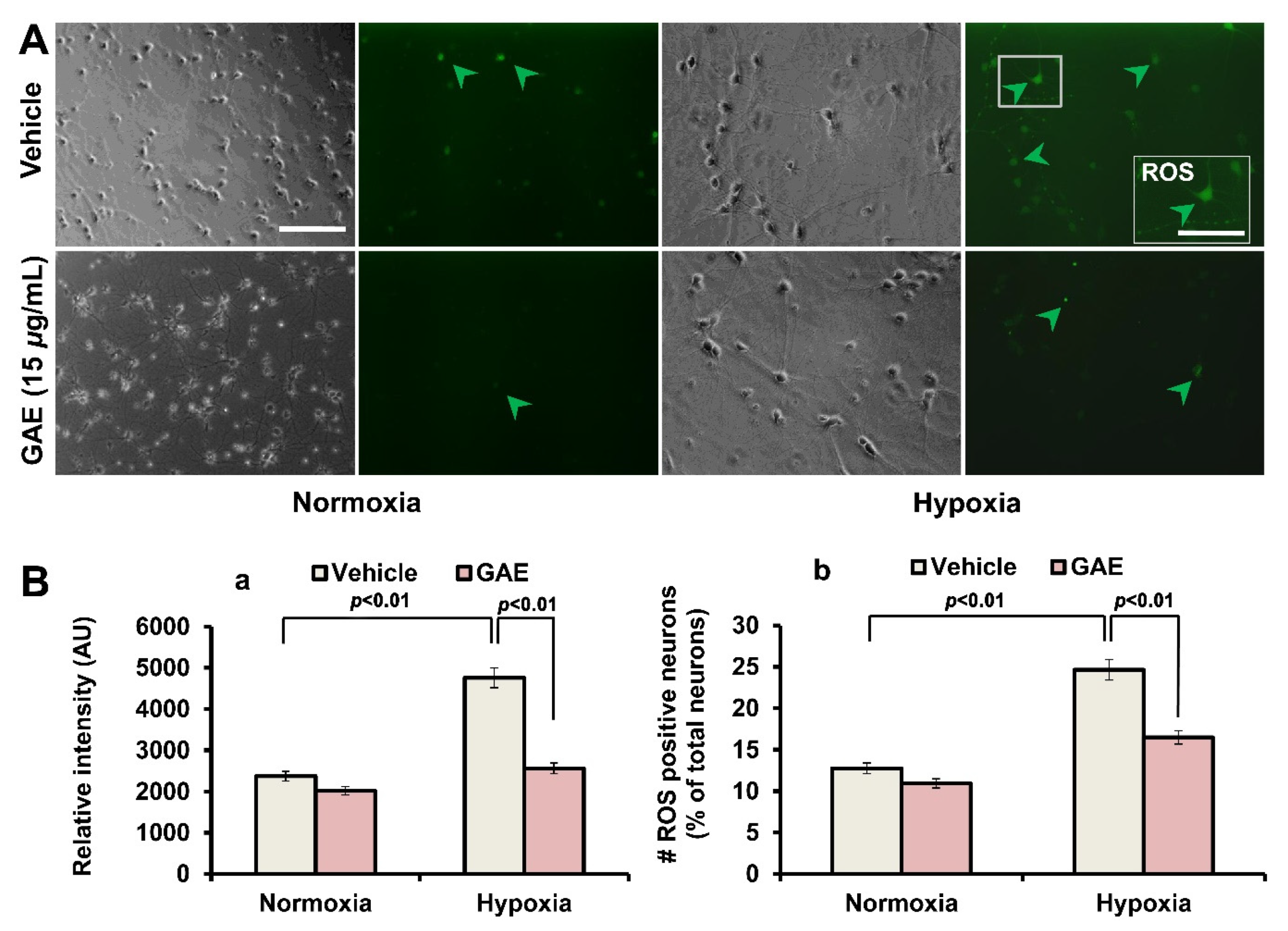
2.7. Measurement of Reactive Oxygen Species (ROS) Generation
Cellular ROS production was confirmed by DCFH-DA staining. DCFH-DA freely crosses cell membranes and is hydrolyzed by cellular esterases to 2’,7’-dichlorodihydrofluorescein (DCFH2). DCFH2 is a non-fluorescent molecule; however, it is oxidized to the fluorescent 2’,7’-dichlorofluorescein (DCF) in the presence of peroxides. To measure ROS production under normoxic and hypoxic conditions, neurons grown in the presence or absence of GAE were rinsed with fresh media and incubated with the fluorescent probe 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA, 10 nM; Molecular Probes Inc., Eugene, OR, USA). Cells were then incubated in a CO2 incubator for 15 min and observed under a fluorescence microscope after rinsing with culture media. The relative fluorescent intensity was measured. The number of ROS positive neurons was also quantitated.
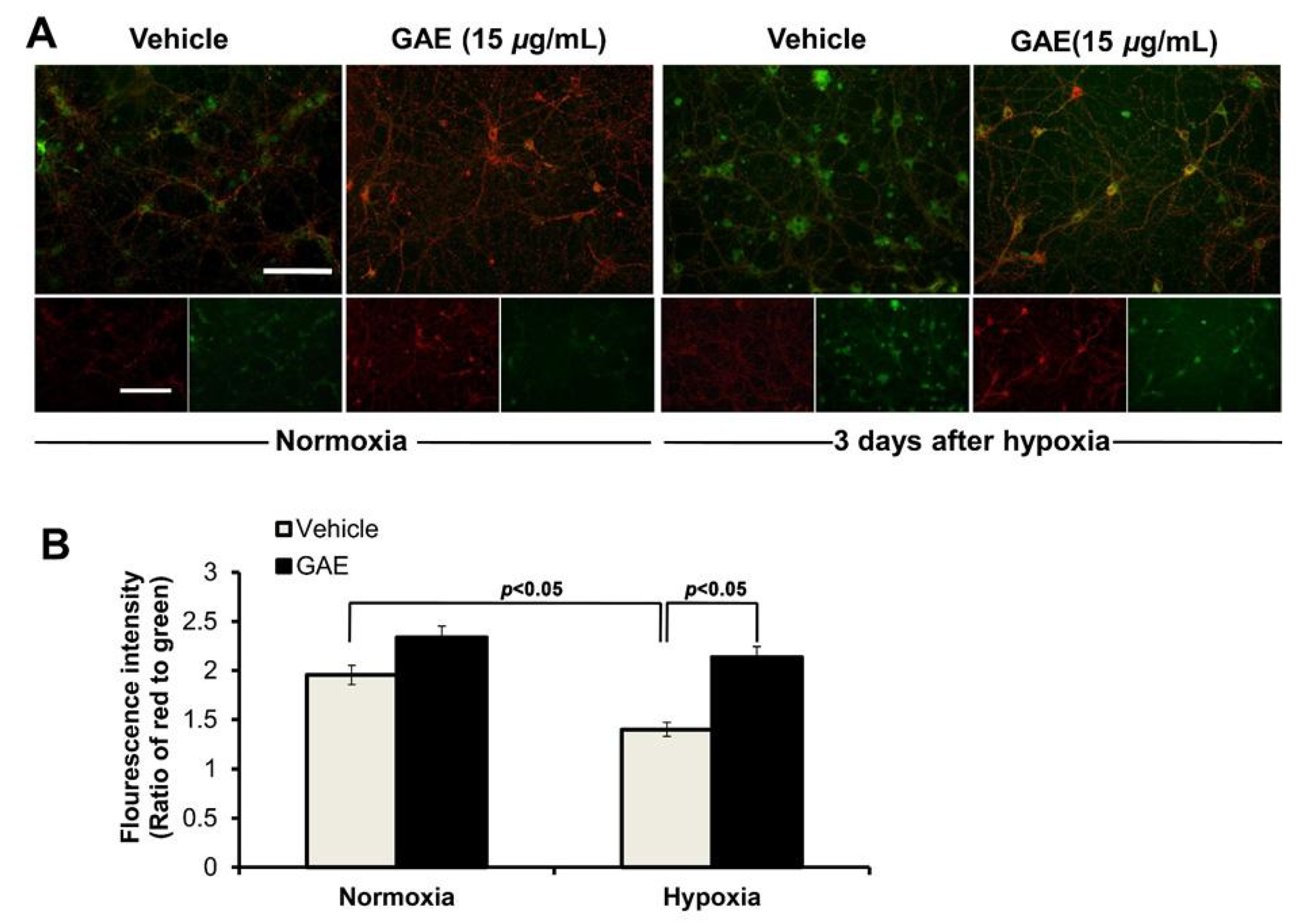
2.8. Determination of Mitochondrial Membrane Potential (ΔΨm)
ΔΨ
m was measured using 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl benzimidazolyl carbocyanine iodide (JC-1), as previously described [
10]. To determine the changes in the ΔΨ
m under normoxic and hypoxic conditions, neurons cultured in the presence or absence of GAE were rinsed with fresh media and incubated with the JC-1 (1μg/mL; Molecular Probes Inc., Eugene, OR, USA) in a CO
2 incubator for 20 min and observed under a fluorescence microscope. ΔΨ
m was determined as the proportion of red to green fluorescent intensity.
2.9. Western Blot
Hippocampal cells (1.2 × 10
5 cells/cm
2, DIV13) were harvested after H/R treatment and lysed in ice-cold RIPA buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% (
v/
v) NP-40, 0.5% (
w/
v) sodium deoxycholate, 1% (
w/
v) sodium dodecyl sulfate, and protease inhibitor cocktail (Thermo Scientific, Rockford, IL, USA)]. Protein concentrations were measured using the Bradford method [
11]. Equal amounts of protein were separated by 6%, or 12% SDS-PAGE and transferred to PVDF membranes [
12], which were incubated with primary antibodies: GluN2A and GluN2B (1:1000; rabbit polyclonal), phospho-H2AX antibody (1:500; mouse monoclonal; Millipore, Billerica, MA, USA), BAX (1:1000; rabbit polyclonal; Santa Cruz Biotechnology, Dallas, TX, USA) and Bcl-2 (1:1000; mouse polyclonal; Santa Cruz Biotechnology, Dallas, TX, USA), Caspase 3 (1:2500; rabbit polyclonal, Cell signaling, Danvers, MA, USA), actin and tubulin (JLA20 and 12G10, respectively; 1:1500, mouse monoclonal, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA). After rinsing with TTBS (0.05% Tween-20 in TBS), membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:1000; anti-mouse or -rabbit IgG; Amersham Biosciences, Buckinghamshire, UK). Signals were detected using an ECL detection kit (Invitrogen, Waltham, MA, USA).
2.10. Image Acquisition and Analysis
A Leica Research Microscope DM IRE2 equipped with I3 S, N2.1 S, and Y5 filter systems (Leica Microsystems AG, Wetzlar, Germany) was used for phase-contrast and epifluorescence microscopy. Images (1388 × 1039 pixels) were acquired using a high-resolution CoolSNAP
TM CCD camera (Photometrics, Inc., Tucson, AZ, USA) under the control of a computer running Leica FW4000 software (Leica Microsystems AG, Wetzlar, Germany). Digital images were processed using Adobe Illustrator CC 2015 (Adobe Systems, Inc., San Jose, CA, USA). The quantifications of cells or puncta were performed using ImageJ (version 1.49, National Institute of Health, Bethesda, MA, USA) software with the cell counter plugin (National Institute of Health, Bethesda, MA, USA). Gel imaging was processed using the AlphaImager
TM HP system (
www.alphainnotech.com).
2.11. Statistical Analysis
All data are expressed as the mean ± SEM with at least three independent experiments. Statistical comparisons were made by Student’s t-test and one-way analysis of variance (ANOVA) with post hoc Duncan multiple comparisons (SPSS software, version 16.0, IBM, NY, USA). Predetermined p-values ≤ 0.05 were considered statistically significant.
4. Discussion
Oxidative stress-induced neuronal damage following ischemia/reperfusion (I/R) in the brain has been crucially implicated in the pathophysiology of many neurological disorders [
14,
15]. The neurotrophic support compromised in the aging brain also affects the survival of brain neurons [
16]. In light of these phenomena, pharmacological agents that could help overcome these pathological consequences might hold therapeutic promise against the associated brain disorders. In this context, we asked whether an ethanolic extract of
G. amansii (GAE) that has already proved its neurotrophic potentials in several of our previous investigations [
4,
5,
6] could protect against oxidative injury induced by hypoxia/reoxygenation (H/R, an in vitro replica of I/R) in hippocampal neurons. Remarkably, in the present study, GAE also exhibited its potential as a promising neuroprotective substance through defending against H/R-induced oxidative damage.
The massive stroke, referred to as an ischemic stroke, following myocardial infarction or coronary occlusion, very often results in the instant demise of the patients. However, if the patients survive, the damage caused by the reperfusion is much higher than that by ischemia itself [
17]. The sudden oxygen supply following reperfusion leads to excessive generation of ROS that results in cellular oxidative stress, which follows a cascade of pathological events, such as mitochondrial dysfunction, DNA disintegration, apoptosis and necrosis, and ultimately death of neurons [
18,
19]. In this study, GAE significantly suppressed ROS accumulation in cultured neurons as opposed to the amount of ROS in untreated neurons and successfully attenuated these pathological consequences. Moreover, GAE attenuated H/R-mediated increase of BAX and caspase 3 expressions while promoting Bcl-2 expression, indicating that GAE helps maintain a balance between proapoptotic and antiapoptotic proteins. A previous report demonstrating that
G. amansii extract suppresses ROS production, and protects against oxidative stress by activating ROS-scavenging enzymes in 3T3-L1 cells [
20], also suggests that antioxidant property might contribute, at least in part, to GAE-mediated neuroprotection in our study.
Mitochondria is the powerhouse of cells that fuel every cellular process, including synaptic transmission. However, being a vulnerable target of free radical-induced damage, this essential organelle is also intimately associated with cellular death [
21]. Moreover, mitochondrial dysfunction has been crucially implicated in the pathogenesis of several neurodegenerative diseases [
22]. In the current study, mitochondrial membrane potential, ΔΨ
m, a key indicator of mitochondrial function, was compromised in cultured neurons following H/R. In contrast, neurons treated with GAE successfully attenuated ΔΨ
m dissipation. No previous report supporting the protective action of GAE against ΔΨ
m loss is available; however, an ethanolic extract of
Gracilariopsis chorda, also an edible red alga, has been shown to preserve ΔΨ
m [
10], contributing a similar neuroprotective mechanism.
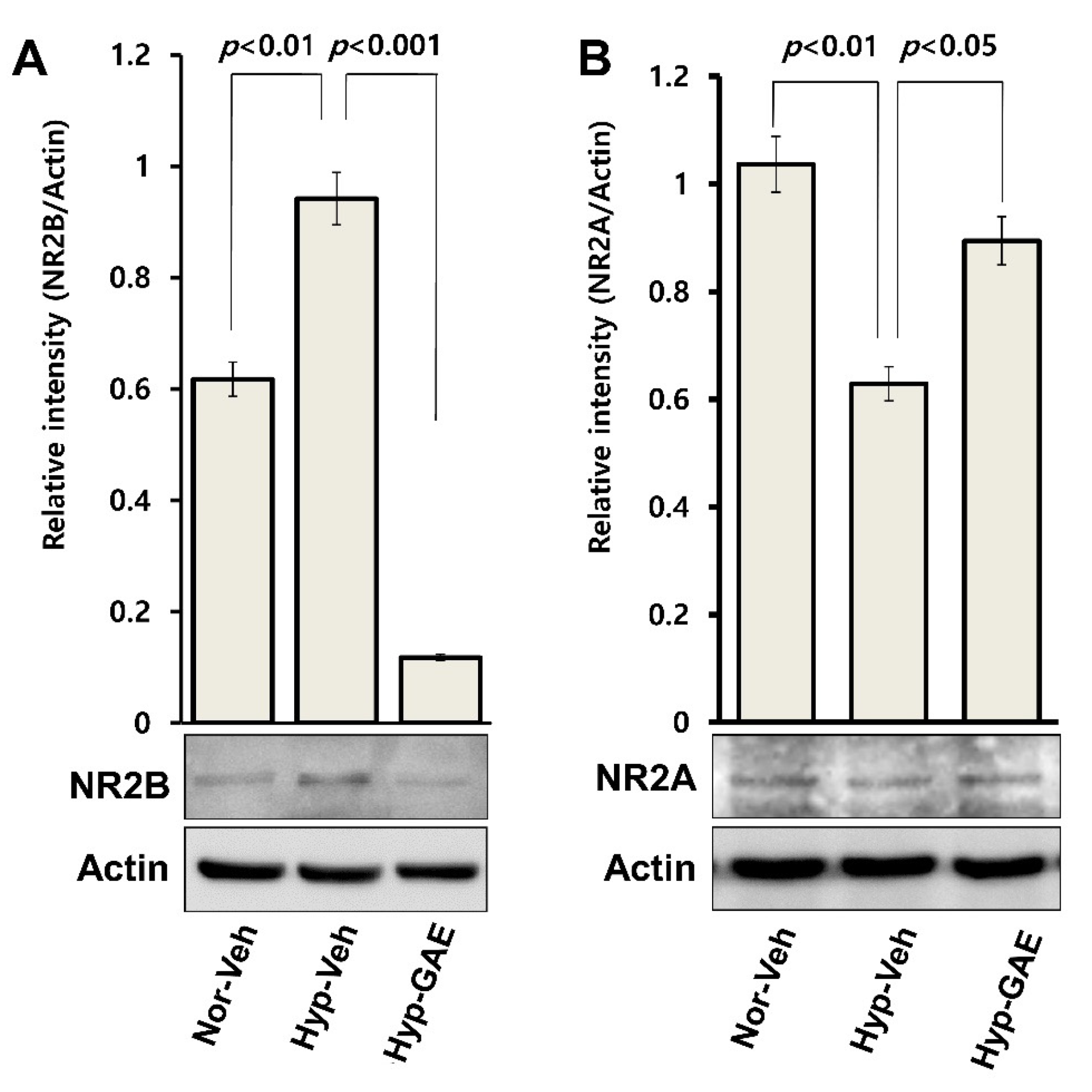
Primarily expressed in the extrasynaptic domain of postsynaptic neurons, GluN2B takes part in neuronal activity during physiological brain function as well as I/R-induced excitotoxicity [
23,
24]. During I/R, there is an accumulation of excitatory neurotransmitter glutamate in the synaptic cleft due to the ionic imbalance (particularly, Ca
2+ dyshomeostasis) [
25] and failure of excess glutamate clearance by reuptake transporters [
26]. As a consequence, there is an over-activation of GluN2B that leads to Ca
2+ overload inside the postsynaptic neurons, which induces downstream pro-death signaling cascades, such as ROS generation [
27] and mitochondrial damage [
25] resulting in neuronal apoptosis. In the present study, GAE significantly suppressed the expression of GluN2B compared to both treatment-naïve and normoxic cultures, indicating that GAE-mediated neuroprotection might be due, at least in part, to attenuation of GluN2B-mediated excitotoxicity following H/R.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






