Global Evolutionary Analysis of 11 Gene Families Part of Reactive Oxygen Species (ROS) Gene Network in Four Eucalyptus Species



Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of Genomic and Protein Sequences
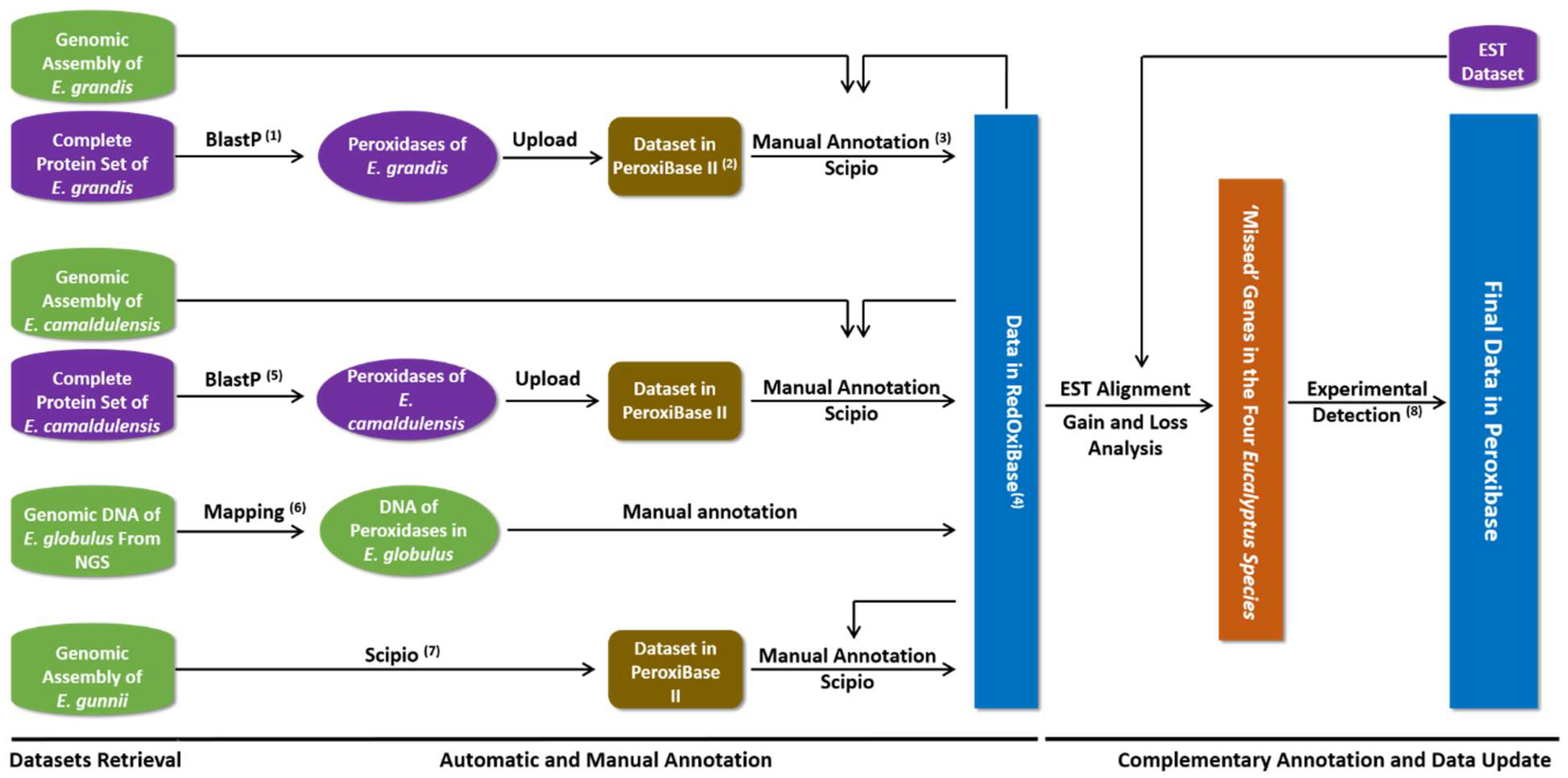
2.2. Data Mining and Expert Annotation
2.3. Pairwise Comparison and Search for Missed Peroxidase Sequences
2.4. Analysis of Phylogeny, Chromosomal Localization and Duplication Events
2.5. Expression Analysis Based on ESTs and RNA-Seq Data
2.6. Analysis of Evolutionary Rate and Divergence Time
3. Results and Discussion
3.1. Data Retrieval, Semi-Automatic Annotation and Statistics
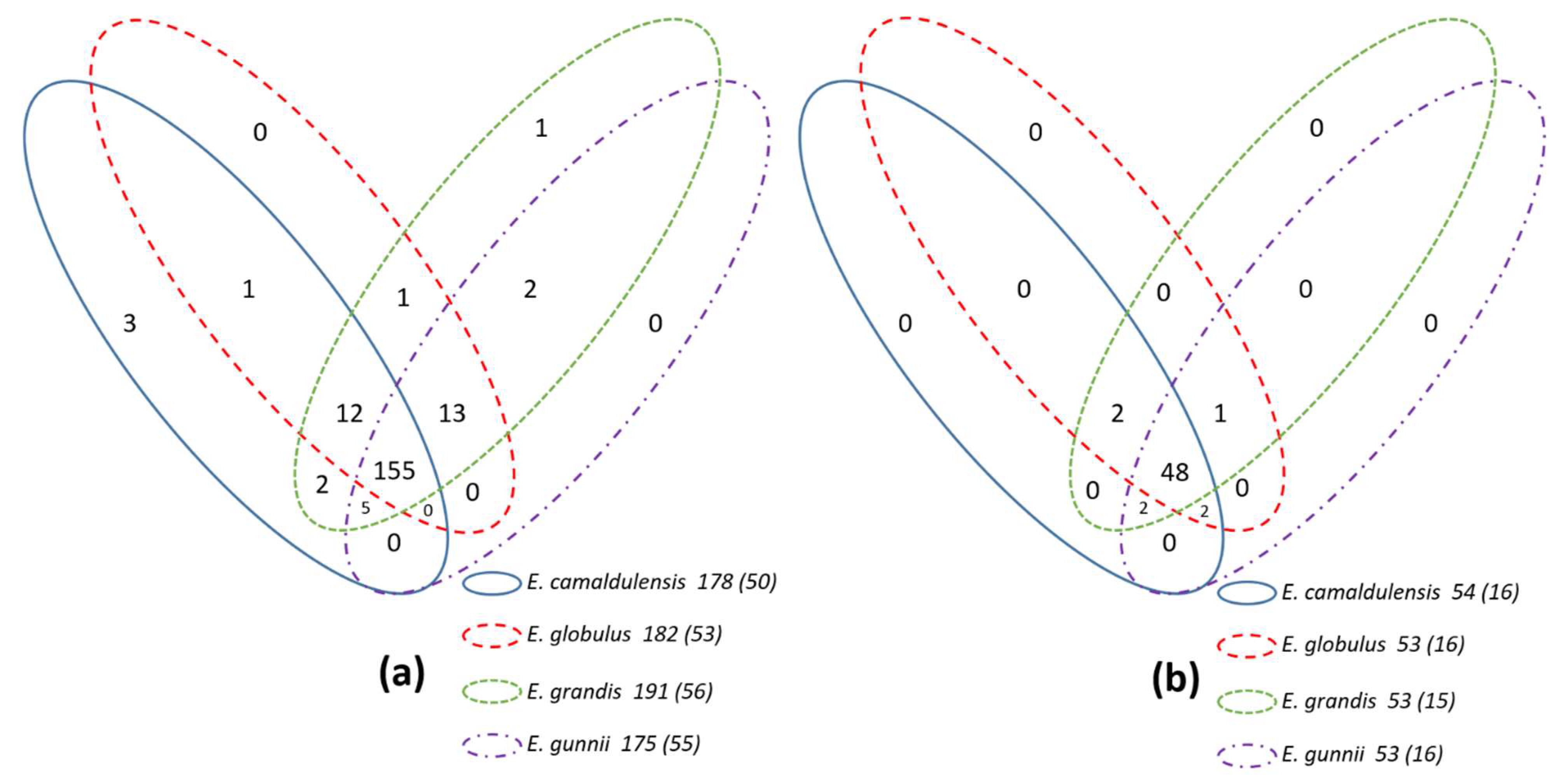
3.2. Necessary and Effective Detection of Missed Genes
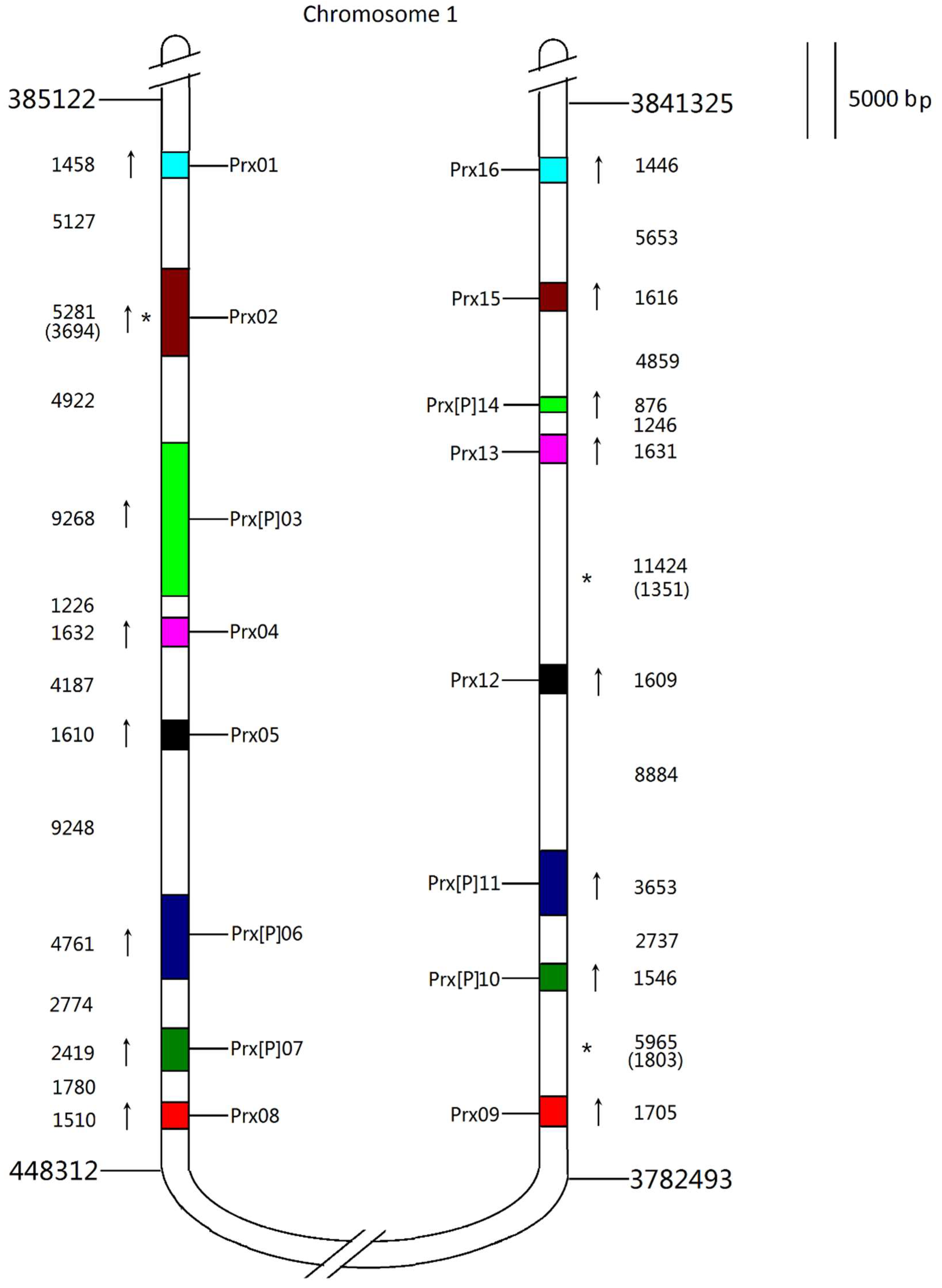
3.3. Phylogeny and Chromosomal Localisation of ROS Genes Network
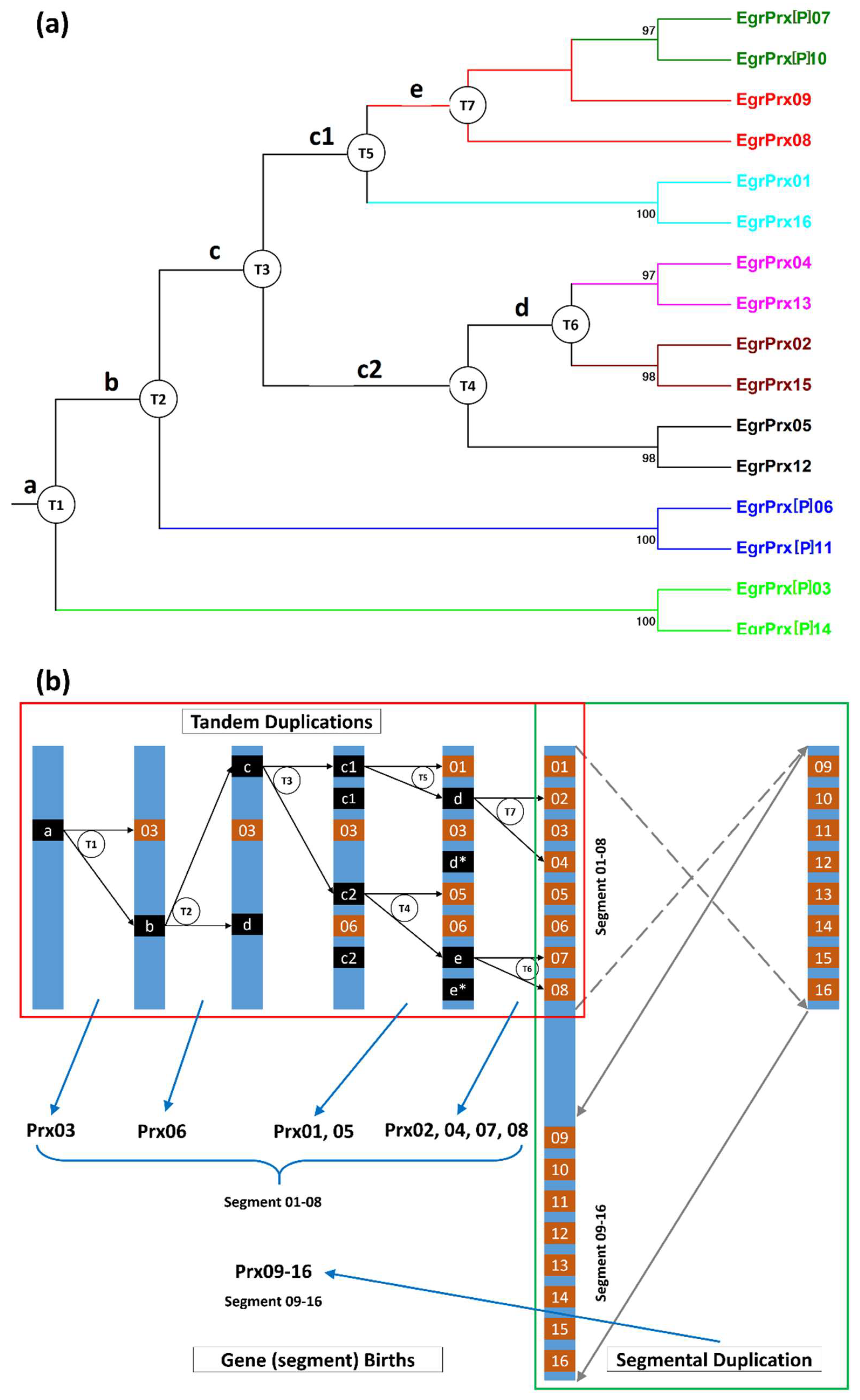
3.4. Gene Gain and Loss Events during the Evolutionary Process
3.5. ROS Gene Families Possess Different Features of Conservation
3.6. Families with Size Variation Contain a Lot of Gene Duplication Events
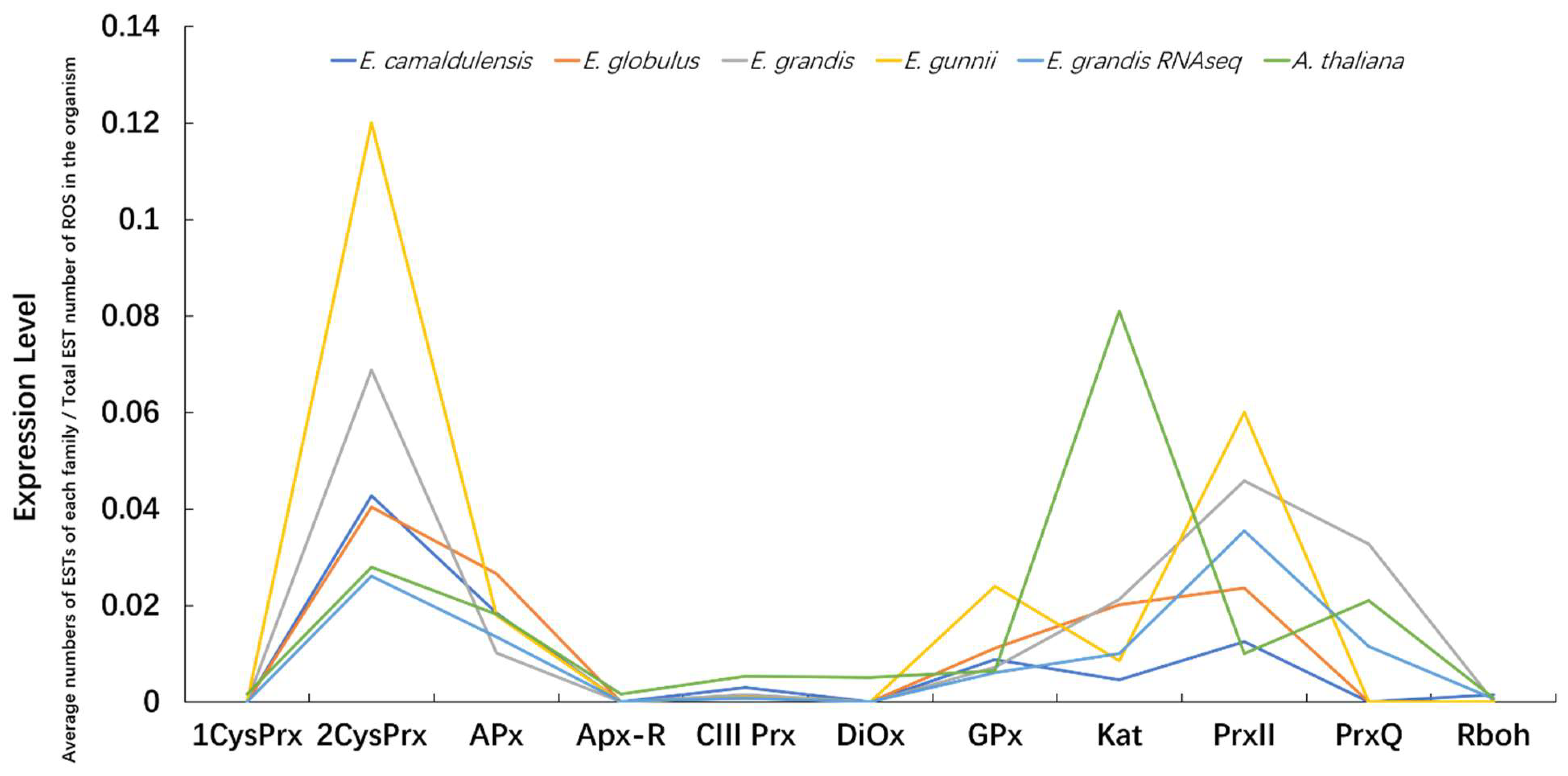
3.7. Expression Profiles of ROS Gene Families within and among Species
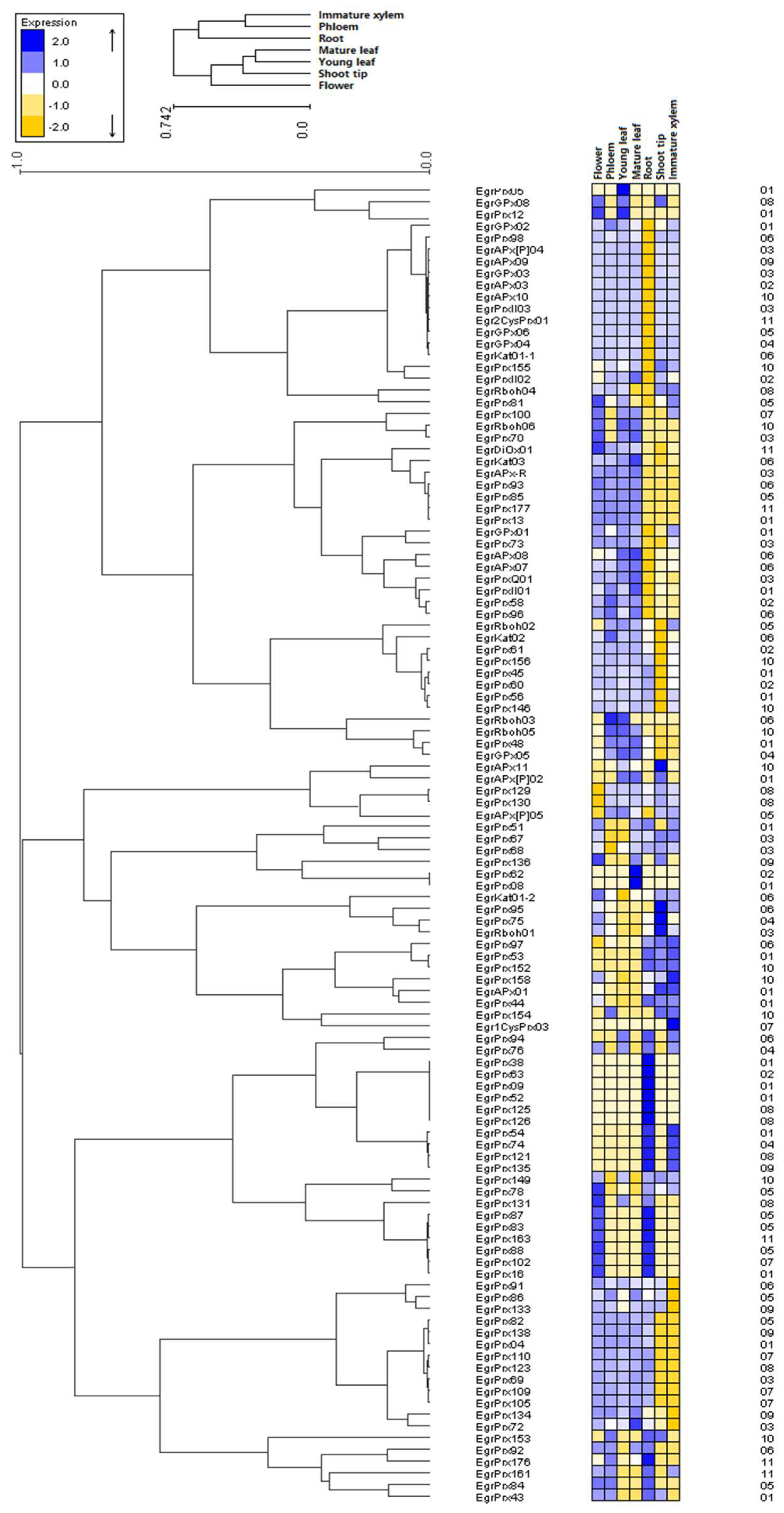
3.8. Duplicated Genes Possess Different Expression Profiles
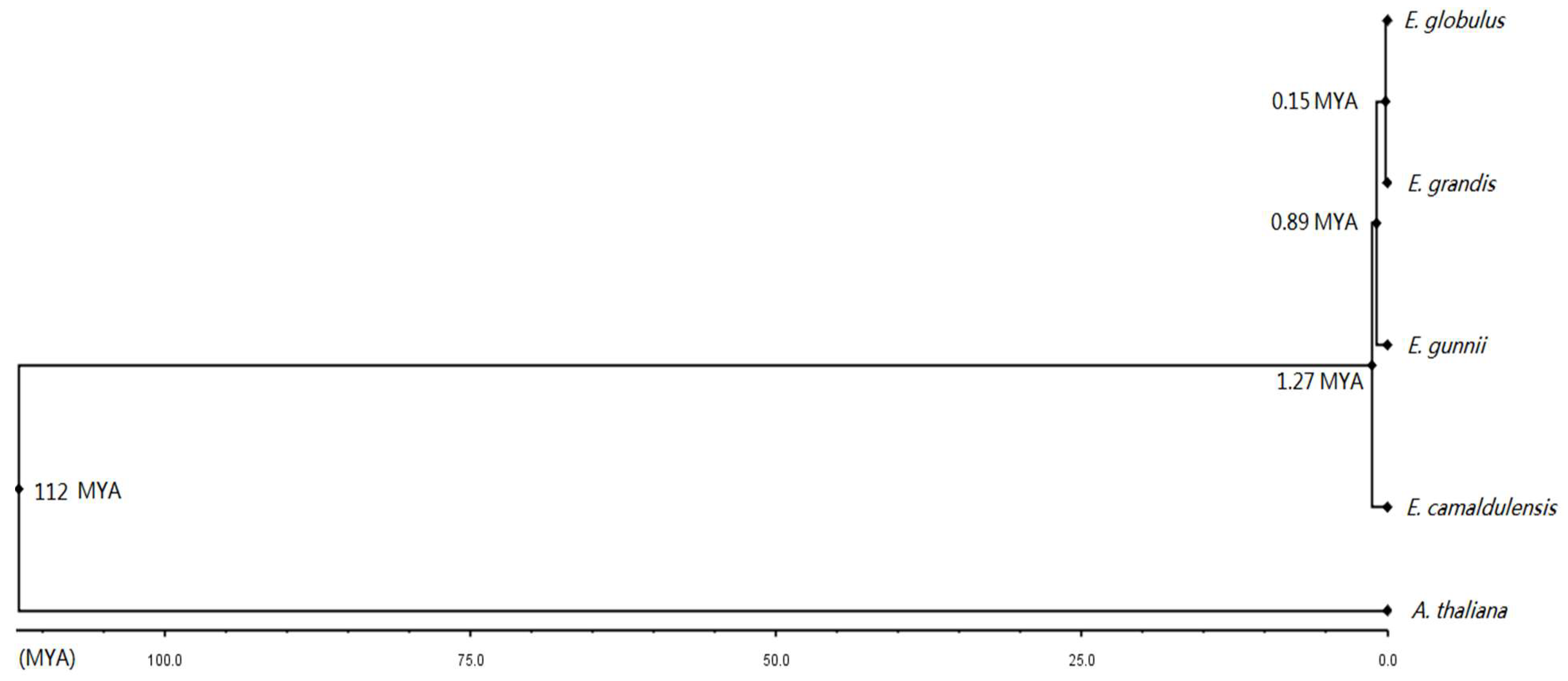
3.9. Divergence Dates of the Four Eucalyptus Species
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tournier, V.; Grat, S.; Marque, C.; El Kayal, W.; Penchel, R.; de Andrade, G.; Boudet, A.M.; Teulières, C. An efficient procedure to stably introduce genes into an economically important pulp tree (Eucalyptus grandis × Eucalyptus urophylla). Transgenic Res. 2003, 12, 403–411. [Google Scholar] [CrossRef]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 509, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Foreman, J.; Demidchik, V.; Bothwell, J.H.; Mylona, P.; Miedema, H.; Torres, M.A.; Linstead, P.; Costa, S.; Brownlee, C.; Jones, J.D.; et al. Reactive oxygen species produced by NADPH oxidase regulate plant cell growth. Nature 2003, 422, 442–446. [Google Scholar] [CrossRef]
- Overmyer, K.; Brosché, M.; Kangasjärvi, J. Reactive oxygen species and hormonal control of cell death. Trends Plant Sci. 2003, 8, 335–342. [Google Scholar] [CrossRef]
- Neill, S.; Desikan, R.; Hancock, J. Hydrogen peroxide signalling. Curr. Opin. Plant Biol. 2002, 5, 388–395. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, F.; Teixeira, F.K.; Rosa, S.B.; Dunand, C.; Fernandes, C.L.; Fontenele, A.V.; Silveira, J.A.; Verli, H.; Margis, R.; Margis-Pinheiro, M. Ascorbate peroxidase-related (APx-R) is a new heme-containing protein functionally associated with ascorbate peroxidase but evolutionarily divergent. New Phytol. 2011, 191, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Passardi, F.; Longet, D.; Penel, C.; Dunand, C. The class III peroxidase multigenic family in rice and its evolution in land plants. Phytochemistry 2004, 65, 1879–1893. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, H.; Cao, P.B.; Fawal, N.; Mathé, C.; Azar, S.; Cassan-Wang, H.; Myburg, A.A.; Grima-Pettenati, J.; Marque, C.; et al. Explosive Tandem and Segmental Duplications of Multigenic Families in Eucalyptus grandis. Genome Biol. Evol. 2015, 7, 1068–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Dou, W.; Qi, J.; Qin, X.; Chen, S.; He, Y. Genomewide analysis of the CIII peroxidase family in sweet orange (Citrus sinensis ) and expression profiles induced by Xanthomonas citri subsp. citri and hormones. J. Genet. 2020, 99, 10. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Cerutti, L.; Falquet, L.; Sigrist, C.J.; Theiler, G.; Hulo, N.; Dunand, C. PeroxiBase: A database with new tools for peroxidase family classification. Nucleic Acids Res. 2009, 37, D261–D266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Miller, G.; Morales, J.; Shulaev, V.; Torres, M.A.; Mittler, R. Respiratory burst oxidases: The engines of ROS signaling. Curr. Opin. Plant Biol. 2011, 14, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Kettler, G.C.; Martiny, A.C.; Huang, K.; Zucker, J.; Coleman, M.L.; Rodrigue, S.; Chen, F.; Lapidus, A.; Ferriera, S.; Johnson, J.; et al. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 2007, 3, e231. [Google Scholar] [CrossRef] [PubMed]
- Filipski, E.; Innominato, P.F.; Wu, M.; Li, X.M.; Iacobelli, S.; Xian, L.J.; Lévi, F. Effects of light and food schedules on liver and tumor molecular clocks in mice. J. Natl. Cancer Inst. 2005, 97, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawal, N.; Li, Q.; Mathé, C.; Dunand, C. Automatic multigenic family annotation: Risks and solutions. Trends Genet. 2014, 30, 325. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Swanson, W.; Vacquier, V. Maximum-likelihood analysis of molecular adaptation in abalone sperm lysin reveals variable selective pressures among lineages and sites. Mol. Biol. Evol. 2000, 17, 1446–1455. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Easteal, S.; Herbert, G. Molecular evidence from the nuclear genome for the time frame of human evolution. J. Mol. Evol. 1997, 44 (Suppl. 1), S121–S132. [Google Scholar] [CrossRef]
- Nei, M.; Xu, P.; Glazko, G. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc. Natl. Acad. Sci. USA 2001, 98, 2497–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doolittle, R.F.; Feng, D.F.; Tsang, S.; Cho, G.; Little, E. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science 1996, 271, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Hedges, S.B. A molecular timescale for vertebrate evolution. Nature 1998, 392, 917–920. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, M.; Yang, Z. Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol. 2011, 28, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Q.; Tank, D.C.; Sang, T. Phylogeny and divergence times in Pinaceae: Evidence from three genomes. Mol. Biol. Evol. 2000, 17, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Chen, X.; Hinds, D.A.; Pant, P.V.; Patil, N.; Cox, D.R. Genomic DNA insertions and deletions occur frequently between humans and nonhuman primates. Genome Res. 2003, 13, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Fawal, N.; Li, Q.; Savelli, B.; Brette, M.; Passaia, G.; Fabre, M.; Mathé, C.; Dunand, C. PeroxiBase: A database for large-scale evolutionary analysis of peroxidases. Nucleic Acids Res. 2013, 41, D441–D444. [Google Scholar] [CrossRef] [Green Version]
- Savelli, B.; Li, Q.; Webber, M.; Jemmat, A.M.; Robitaille, A.; Zamocky, M.; Mathé, C.; Dunand, C. RedoxiBase: A database for ROS homeostasis regulated proteins. Redox Biol. 2019, 26, 101247. [Google Scholar] [CrossRef]
- Keller, O.; Odronitz, F.; Stanke, M.; Kollmar, M.; Waack, S. Scipio: Using protein sequences to determine the precise exon/intron structures of genes and their orthologs in closely related species. BMC Bioinform. 2008, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Tsai, I.J.; Otto, T.D.; Berriman, M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010, 11, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet--next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Thilmony, R.; Gu, Y.Q. NetVenn: An integrated network analysis web platform for gene lists. Nucleic Acids Res. 2014, 42, W161–W166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tippmann, H.F. Analysis for free: Comparing programs for sequence analysis. Brief Bioinform. 2004, 5, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Maron-Katz, A.; Shavit, S.; Sagir, D.; Linhart, C.; Elkon, R.; Tanay, A.; Sharan, R.; Shiloh, Y.; Shamir, R. Expander: From expression microarrays to networks and functions. Nat. Protoc. 2010, 5, 303–322. [Google Scholar] [CrossRef]
- Rozas, J.; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 1995, 11, 621–625. [Google Scholar] [CrossRef]
- Hirakawa, H.; Nakamura, Y.; Kaneko, T.; Isobe, S.; Sakai, H.; Kato, T.; Hibino, T.; Sasamoto, S.; Watanabe, A.; Yamada, M.; et al. Survey of the genetic information carried in the genome of Eucalyptus camaldulensis. Plant Biotechnol. 2011, 28, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Nozawa, M. Roles of Mutation and Selection in Speciation: From Hugo de Vries to the Modern Genomic Era. Genome Biol. Evol. 2011, 3, 812–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.C.; Chen, C.J.; Li, W.H.; Chuang, T.J. Gene family size conservation is a good indicator of evolutionary rates. Mol. Biol. Evol. 2010, 27, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedges, S.B.; Dudley, J.; Kumar, S. TimeTree: A public knowledge-base of divergence times among organisms. Bioinformatics 2006, 22, 2971–2972. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organisms | E. camaldulensis | E. globulus | E. grandis | E. gunnii | ||||
|---|---|---|---|---|---|---|---|---|
| Data Sources | From Databases* | From PCR | From Databases | From PCR | From Databases | From PCR | From Databases | From PCR |
| 1CysPrx | 3 (2+0+1) | 1 (0+0+1) | 4 (0+2+2) | 0 | 3 (1+0+2) | 0 | 3 (1+0+2) | 1 (0+0+1) |
| 2CysPrx | 1 (0+1+0) | 0 | 1 (1+0+0) | 0 | 1 (1+0+0) | 0 | 1 (1+0+0) | 0 |
| APx | 10 (3+5+2) | 0 | 10 (7+0+3) | 1 (0+0+1) | 11 (7+0+4) | 0 | 10 (7+0+3) | 0 |
| Apx-R | 2 (1+0+1) | 0 | 1 (1+0+0) | 0 | 2 (1+0+1) | 0 | 2 (1+0+1) | 0 |
| CIII Prx | 163 (84+39+40) | 16 (0+6+10) | 180 (93+32+55) | 3 (0+2+1) | 179 (126+2+51) | 12 (2+5+5) | 159 (100+11+48) | 17 (1+8+8) |
| DiOx | 1 (1+0+0) | 0 | 1 (1+0+0) | 0 | 1 (1+0+0) | 0 | 1 (1+0+0) | 0 |
| GPx | 11 (3+5+3) | 0 | 10 (5+3+2) | 0 | 9 (9+0+0) | 1 (0+0+1) | 9 (7+1+1) | 1 (0+0+1) |
| Kat | 12 (1+4+7) | 2 (0+1+1) | 14 (3+3+8) | 0 | 12 (2+4+6) | 2 (0+1+1) | 13 (4+2+7) | 1 (0+1+0) |
| PrxII | 3 (3+0+0) | 0 | 3 (2+1+0) | 0 | 3 (3+0+0) | 0 | 3 (2+1+0) | 0 |
| PrxQ | 1 (1+0+0) | 0 | 1 (0+1+0) | 0 | 1 (1+0+0) | 0 | 1 (1+0+0) | 0 |
| Rboh | 7 (3+4+0) | 0 | 7 (5+2+0) | 0 | 7 (7+0+0) | 0 | 7 (6+1+0) | 0 |
| Total | 214 (102+58+54) | 19 (0+7+12) | 232 (118+44+70) | 4 (0+2+2) | 229 (159+6+64) | 15 (2+6+7) | 209 (131+16+62) | 19 (1+9+9) |
| Automatic correct prediction | 82 (35.19%) | na | 92 (37.70%) | na | ||||
| Coverage of Genomic Data | 91.8% | 98.3% | 93.9% | 91.7% | ||||
| Types of Missed Genes | E. camaldulensis | E. globulus | E. grandis | E. gunnii |
|---|---|---|---|---|
| Missed genes in clusters 1 | Prx19, Prx37, Prx64, Prx79, Prx83, Prx116, Prx127, Prx138, Prx139, Prx161, Prx164 | Apx-R[P], Prx19, Prx23, Prx37, Prx39, Prx50, Prx116, Prx129-2, GPx07 | 1CysPrx03-2, Prx129-2, Prx188 | Prx16, Prx18, Prx19, Prx20, Prx52, Prx53, Prx54, Prx64, Prx66-2, Prx106, Prx129-2, Prx130, Prx176, Prx188, GPx01 |
| Singletons 2 | APx04, Prx27, Prx28, Prx145, Prx155, Prx165, Prx168 | Prx89, Prx189, Prx194, Prx195, Prx197, Prx198 | Prx183, Prx189, Prx198, GPx11 | APx06, Prx81, Prx89, Prx108, Prx183, Prx189, Prx194 |
| Multigenic Families | A. thaliana | E. camaldulensis | E. globulus | E. grandis | E. gunnii | M. truncatula | P. trichocarpa | V. vinifera |
|---|---|---|---|---|---|---|---|---|
| 1CysPrx | 1 (0) | 4 (2) | 4 (2) | 3 (2) | 4 (3) | 1 (0) | 1 (0) | 2 (1) |
| 2CysPrx | 2 (0) | 1 (0) | 1 (0) | 1 (0) | 1 (0) | 2 (0) | 2 (0) | 1 (0) |
| APx | 8 (1) | 10 (2) | 11 (4) | 11 (4) | 10 (3) | 8 (1) | 10 (1) | 9 (2) |
| APx-R | 1 (0) | 2 (1) | 1 (0) | 2 (1) | 2 (1) | 1 (0) | 1 (0) | 1 (0) |
| CIII Prx | 75 (2) | 179 (50) | 183 (56) | 191 (56) | 176 (56) | 106 (8) | 101 (12) | 97 (10) |
| DiOx | 2 (0) | 1 (0) | 1 (0) | 1 (0) | 1 (0) | 2 (0) | 2 (0) | 3 (0) |
| GPx | 8 (0) | 11 (3) | 10 (2) | 10 (1) | 10 (2) | 7 (0) | 8 (2) | 5 (0) |
| Kat | 3 (0) | 14 (8) | 14 (8) | 14 (7) | 14 (7) | 1 (0) | 4 (1) | 2 (0) |
| PrxII | 6 (1) | 3 (0) | 3 (0) | 3 (0) | 3 (0) | 4 (0) | 5 (1) | 4 (0) |
| PrxQ | 1 (0) | 1 (0) | 1 (0) | 1 (0) | 1 (0) | 1 (0) | 2 (0) | 1 (0) |
| Rboh | 10 (0) | 7 (0) | 7 (0) | 7 (0) | 7 (0) | 10 (0) | 10 (0) | 9 (0) |
| Total | 117 (4) | 233 (66) | 236 (72) | 244 (71) | 228 (71) | 143 (9) | 146 (17) | 134 (13) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; San Clemente, H.; He, Y.; Fu, Y.; Dunand, C. Global Evolutionary Analysis of 11 Gene Families Part of Reactive Oxygen Species (ROS) Gene Network in Four Eucalyptus Species. Antioxidants 2020, 9, 257. https://doi.org/10.3390/antiox9030257
Li Q, San Clemente H, He Y, Fu Y, Dunand C. Global Evolutionary Analysis of 11 Gene Families Part of Reactive Oxygen Species (ROS) Gene Network in Four Eucalyptus Species. Antioxidants. 2020; 9(3):257. https://doi.org/10.3390/antiox9030257
Chicago/Turabian StyleLi, Qiang, Hélène San Clemente, Yongrui He, Yongyao Fu, and Christophe Dunand. 2020. "Global Evolutionary Analysis of 11 Gene Families Part of Reactive Oxygen Species (ROS) Gene Network in Four Eucalyptus Species" Antioxidants 9, no. 3: 257. https://doi.org/10.3390/antiox9030257
APA StyleLi, Q., San Clemente, H., He, Y., Fu, Y., & Dunand, C. (2020). Global Evolutionary Analysis of 11 Gene Families Part of Reactive Oxygen Species (ROS) Gene Network in Four Eucalyptus Species. Antioxidants, 9(3), 257. https://doi.org/10.3390/antiox9030257