Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Cartilage Specimens
2.2. Destabilized Medial Meniscus (DMM)-Induced OA
2.3. Purification of Irisin Recombinant Protein
2.4. Intra-Articular Injection of Irisin
2.5. Gait Profile Analysis
2.6. Histomorphometry and Immunohistochemistry
2.7. Chondrocyte Cultures
2.8. Assessment of Chondrocytic Activity
2.9. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.10. Analysis of Autophagosome and Mitophagosome Formation
2.11. Mitochondrial Dynamics Assay
2.12. Mitochondrial Membrane Potential, ATP Production and Reactive Oxygen Radical (ROS) Level Assay
2.13. Immunoblotting
2.14. Statistical Analysis
3. Results
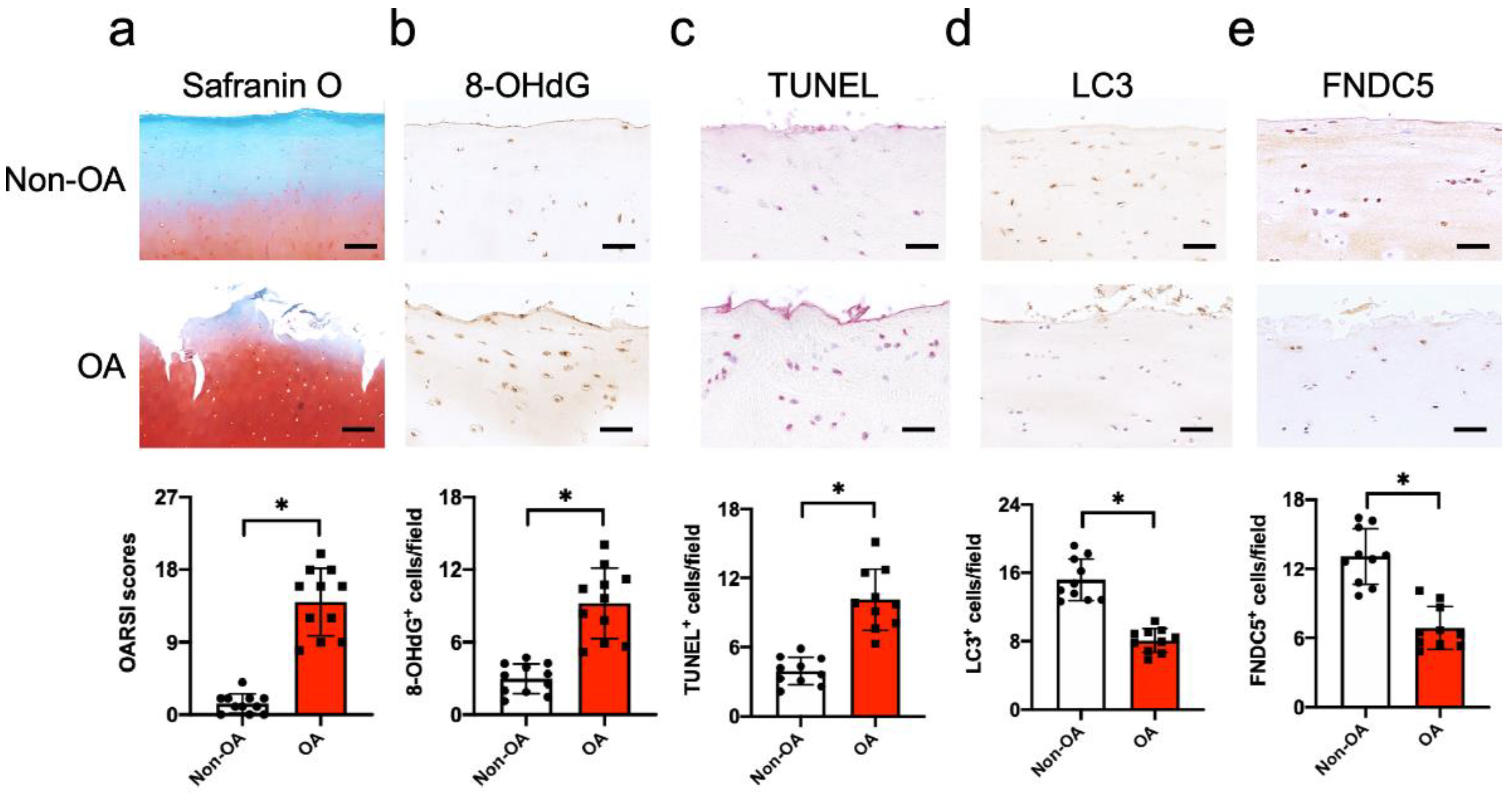
3.1. FNDC5 Loss and Oxidative DNA Damage in Chondrocytes in Human Knee OA
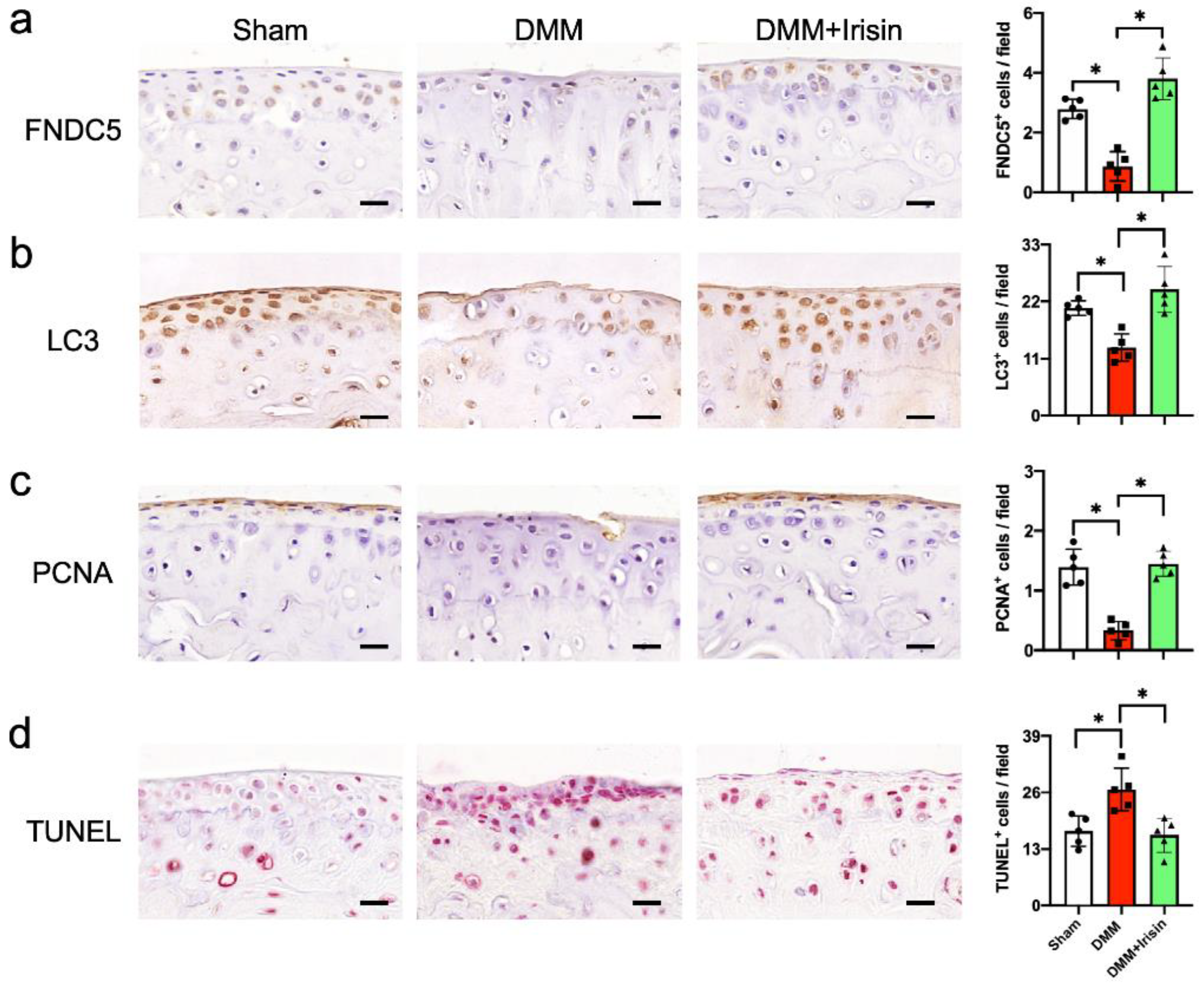
3.2. Irisin Attenuated Articular Cartilage Loss and Irregular Gait of DMM-Injured Knees
3.3. Irisin Attenuated DMM-Induced Defective Autophagy and Survival in Chondrocytes
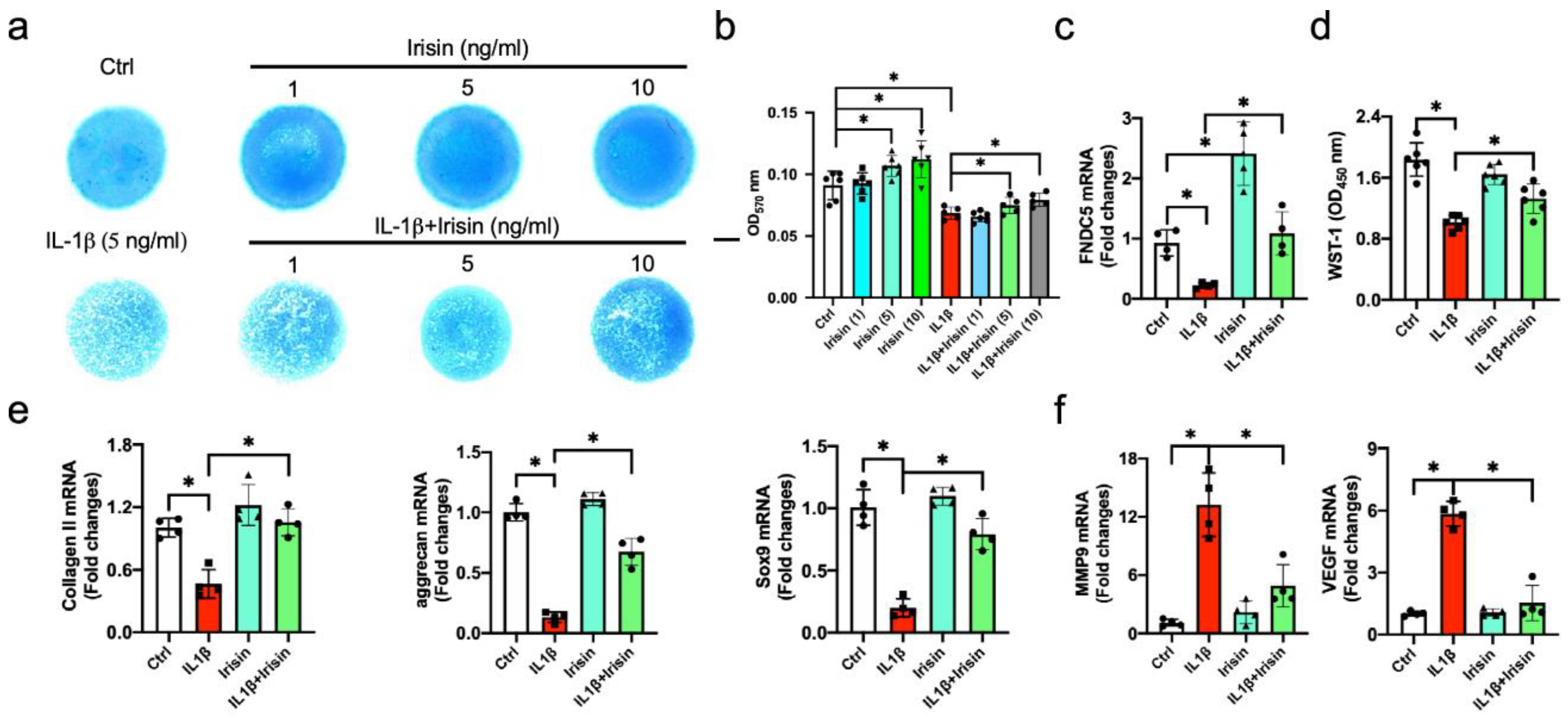
3.4. Irisin Alleviated ECM Production of Inflamed Chondrocytes
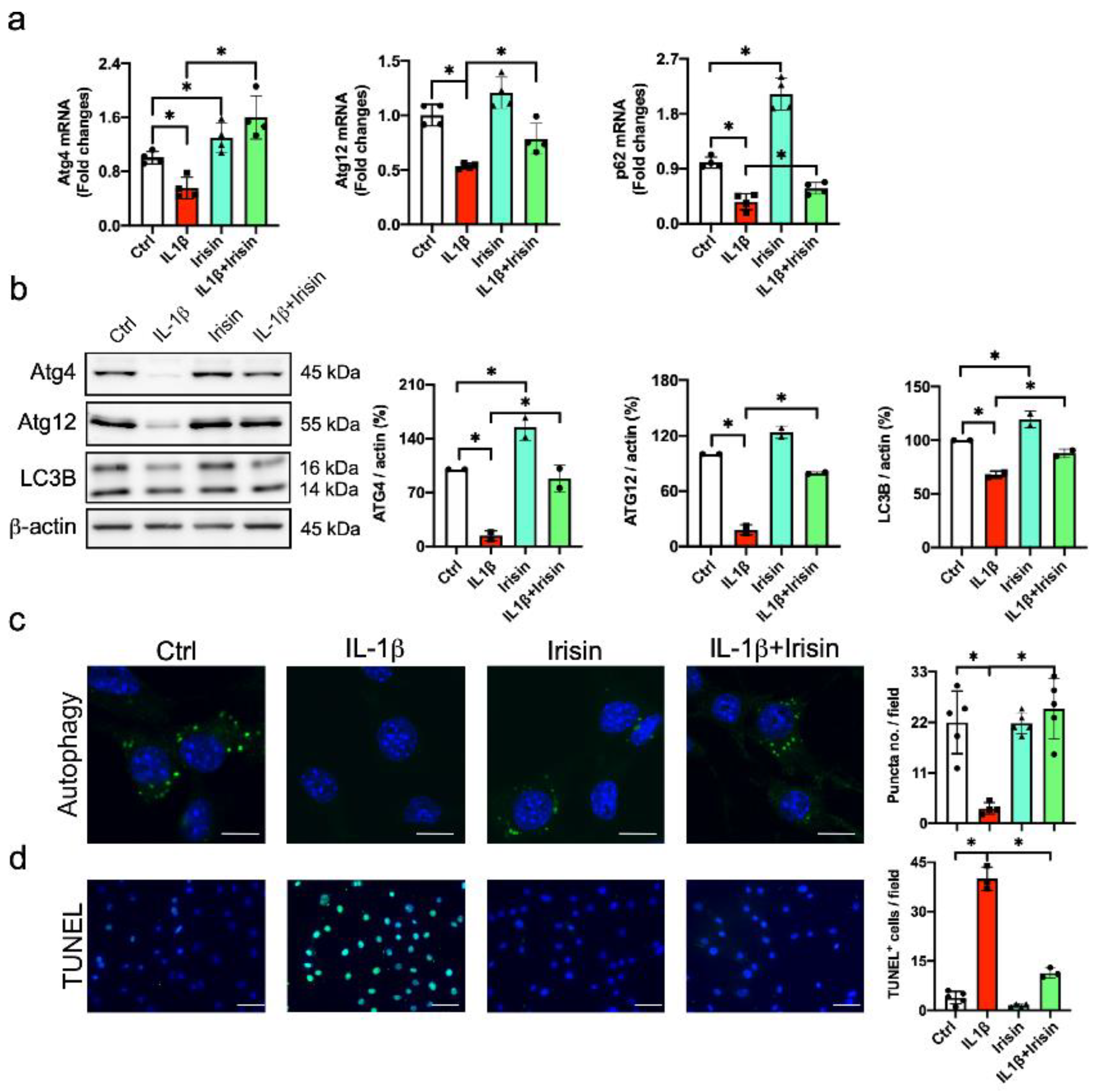
3.5. Irisin Improved Autophagy and Apoptosis in Inflamed Chondrocytes
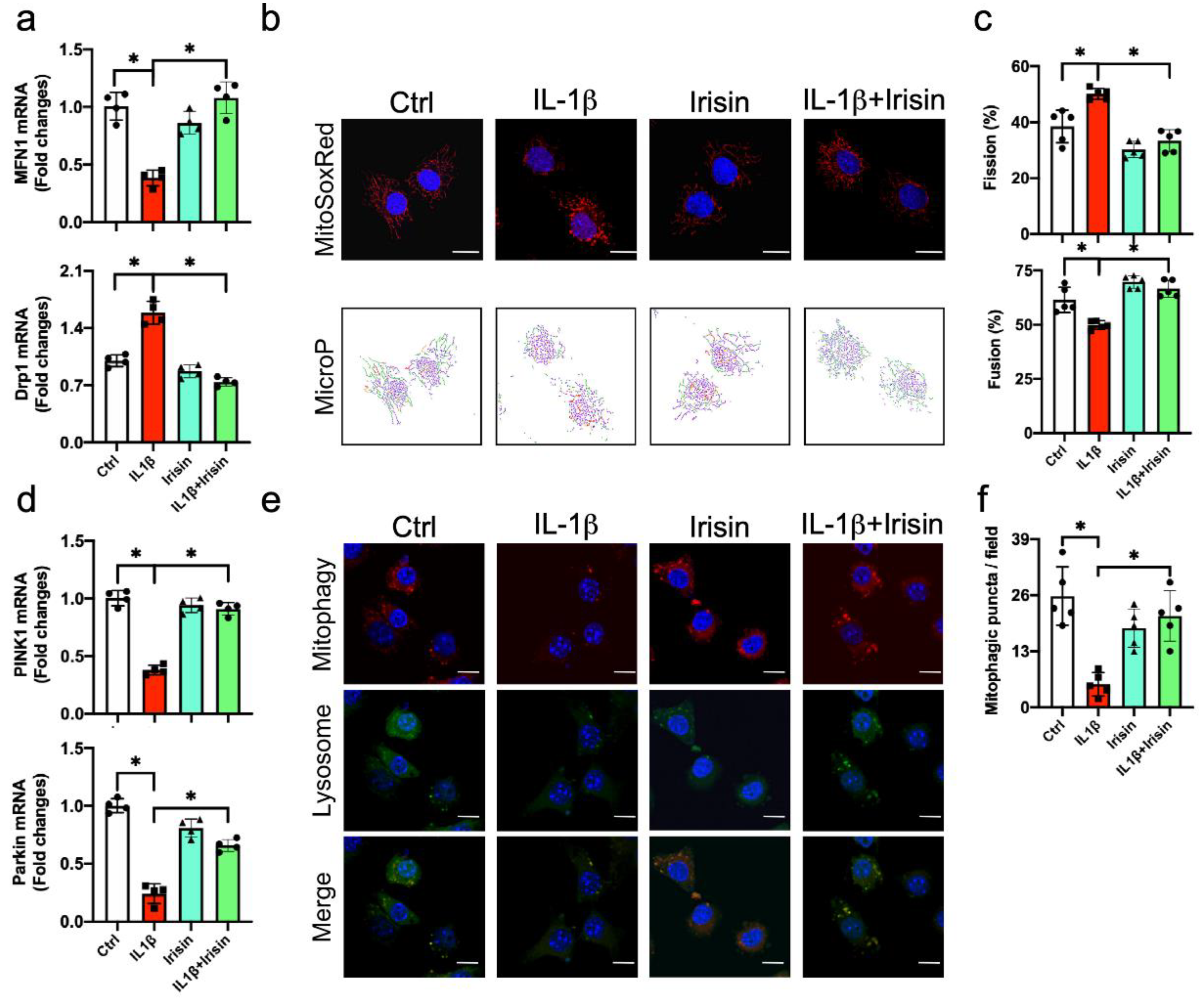
3.6. Irisin Preserved Mitochondrial Fusion and Mitophagy in Inflamed Chondrocytes
3.7. Irisin Protected from UCP-1, Sirt3, Catalase Loss and Mitochondrial Dysfunction
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Kim, J.H.; Jeon, J.; Shin, M.; Won, Y.; Lee, M.; Kwak, J.S.; Lee, G.; Rhee, J.; Ryu, J.H.; Chun, C.H.; et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 2014, 156, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.J.; Beier, F.; Young, D.A.; Loughlin, J. Interplay between genetics and epigenetics in osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 268–281. [Google Scholar] [CrossRef]
- Blanco, F.J.; Fernández-Moreno, M. Mitochondrial biogenesis: A potential therapeutic target for osteoarthritis. Osteoarthr. Cartil. 2020, 28, 1003–1006. [Google Scholar] [CrossRef]
- Coleman, M.C.; Goetz, J.E.; Brouillette, M.J.; Seol, D.; Willey, M.C.; Petersen, E.B.; Anderson, H.D.; Hendrickson, N.R.; Compton, J.; Khorsand, B.; et al. Targeting mitochondrial responses to intra-articular fracture to prevent posttraumatic osteoarthritis. Sci. Transl. Med. 2018, 10, eaan5372. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Moreno, M.; Soto-Hermida, A.; Vázquez-Mosquera, M.E.; Cortés-Pereira, E.; Relaño, S.; Hermida-Gómez, T.; Pértega, S.; Oreiro-Villar, N.; Fernández-López, C.; Garesse, R.; et al. Mitochondrial DNA haplogroups influence the risk of incident knee osteoarthritis in OAI and CHECK cohorts. A meta-analysis and functional study. Ann. Rheum. Dis. 2017, 76, 1114–1122. [Google Scholar] [CrossRef]
- Rellmann, Y.; Dreier, R. Different forms of ER stress in chondrocytes result in short stature disorders and degenerative cartilage diseases: New insights by cartilage-Specific ERp57 knockout mice. Oxid. Med. Cell Longev. 2018, 2018, 8421394. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, H.; Wan, X.; Lu, L.; Zhang, J.; Zhang, H.; Ye, T.; Liu, Q.; Xie, M.; Liu, X.; et al. Prevention of injury-induced osteoarthritis in rodent temporomandibular joint by targeting chondrocyte CaSR. J. Bone Miner. Res. 2019, 34, 726–738. [Google Scholar] [CrossRef]
- Zheng, W.; Li, X.; Liu, D.; Li, J.; Yang, S.; Gao, Z.; Wang, Z.; Yokota, H.; Zhang, P. Mechanical loading mitigates osteoarthritis symptoms by regulating endoplasmic reticulum stress and autophagy. FASEB J. 2019, 33, 4077–4088. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2016, 75, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-Specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Xiong, Y.; Wu, Z.; Zhang, B.; Wang, C.; Mao, F.; Liu, X.; Hu, K.; Sun, X.; Jin, W.; Kuang, S. Fndc5 loss-of-function attenuates exercise-induced browning of white adipose tissue in mice. FASEB J. 2019, 33, 5876–5886. [Google Scholar] [CrossRef]
- Reza, M.M.; Subramaniyam, N.; Sim, C.M.; Ge, X.; Sathiakumar, D.; McFarlane, C.; Sharma, M.; Kambadur, R. Irisin is a pro-myogenic factor that induces skeletal muscle hypertrophy and rescues denervation-induced atrophy. Nat. Commun. 2017, 8, 1104. [Google Scholar] [CrossRef]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.A.; Novick, S.J.; et al. Irisin mediates effects on bone and fat via αV integrin receptors. Cell 2018, 175, 1756–1768. [Google Scholar] [CrossRef] [Green Version]
- Storlino, G.; Colaianni, G.; Sanesi, L.; Lippo, L.; Brunetti, G.; Errede, M.; Colucci, S.; Passeri, G.; Grano, M. Irisin prevents disuse-Induced osteocyte apoptosis. J. Bone Miner. Res. 2020, 35, 766–775. [Google Scholar] [CrossRef]
- Vadalà, G.; Di Giacomo, G.; Ambrosio, L.; Cannata, F.; Cicione, C.; Papalia, R.; Denaro, V. Irisin recovers osteoarthritic chondrocytes in vitro. Cells 2020, 9, 1478. [Google Scholar] [CrossRef]
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919. [Google Scholar] [CrossRef]
- Gosset, M.; Berenbaum, F.; Thirion, S.; Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Wang, X.; Cornelis, F.M.F.; Lories, R.J.; Monteagudo, S. Exostosin-1 enhances canonical Wnt signaling activity during chondrogenic differentiation. Osteoarthr. Cartil. 2019, 27, 1702–1710. [Google Scholar] [CrossRef]
- Ko, J.Y.; Sun, Y.C.; Li, W.C.; Wang, F.S. Chaperonin 60 regulation of SOX9 ubiquitination mitigates the development of knee osteoarthritis. J. Mol. Med. 2016, 94, 755–769. [Google Scholar] [CrossRef]
- Lian, W.S.; Ko, J.Y.; Chen, Y.S.; Ke, H.C.; Wu, S.L.; Kuo, C.W.; Wang, F.S. Chaperonin 60 sustains osteoblast autophagy and counteracts glucocorticoid aggravation of osteoporosis by chaperoning RPTOR. Cell Death Dis. 2018, 9, 938. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.Y.; Lee, M.S.; Lian, W.S.; Weng, W.T.; Sun, Y.C.; Chen, Y.S.; Wang, F.S. MicroRNA-29a counteracts synovitis in knee osteoarthritis pathogenesis by targeting VEGF. Sci. Rep. 2017, 7, 3584. [Google Scholar] [CrossRef]
- Peng, J.Y.; Lin, C.C.; Chen, Y.J.; Kao, L.S.; Liu, Y.C.; Chou, C.C.; Huang, Y.H.; Chang, F.R.; Wu, Y.C.; Tsai, Y.S.; et al. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PLoS Comput. Biol. 2011, 7, e1002212. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.K.; Chen, S.D.; Chuang, Y.C.; Lan, M.Y.; Chuang, J.H.; Wang, P.W.; Hsu, T.Y.; Wang, F.S.; Tsai, M.H.; Huang, S.T.; et al. Mitochondrial transfer of Whaton’s jelly mesenchymal stem cells eliminates mutation burden and rescues mitochondrial bioenergetics in rotenone-stressed MELAS fibroblasts. Oxid. Med. Cell Longev. 2019, 2019, 9537504. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Yang, L.; Wang, T.; Zhang, J.; Li, T.; Ren, Y.; Wang, M.; Chen, X.; Lv, Y.; Wu, R. Irisin improves autophagy of aged hepatocytes via increasing telomerase activity in liver injury. Oxid. Med. Cell Longev. 2020, 2020, 6946037. [Google Scholar] [CrossRef]
- Sophocleous, A.; Huesa, C. Osteoarthritis mouse model of destabilization of the medial meniscus. Methods Mol. Biol. 2019, 1914, 281–293. [Google Scholar]
- Bian, Q.; Wang, Y.J.; Liu, S.F.; Li, Y.P. Osteoarthritis: Genetic factors, animal models, mechanisms, and therapies. Front. Biosci. 2012, 4, 74–100. [Google Scholar] [CrossRef]
- D’Adamo, S.; Cetrullo, S.; Guidotti, S.; Silvestri, Y.; Minguzzi, M.; Santi, S.; Cattini, L.; Filardo, G.; Flamigni, F.; Borzì, R.M. Spermidine rescues the deregulated autophagic response to oxidative stress of osteoarthritic chondrocytes. Free Radic. Biol. Med. 2020, 153, 159–172. [Google Scholar] [CrossRef]
- Chen, L.Y.; Wang, Y.; Terkeltaub, R.; Liu-Bryan, R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr. Cartil. 2018, 26, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Li, X.; Wang, X.; Chen, T.; Tao, F.; Liu, C.; Tu, Q.; Shen, G.; Chen, J.J. Irisin deficiency disturbs bone metabolism. J. Cell Physiol. 2020. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Q.; Lou, T.; Qin, J.; Jung, S.; Shetty, V.; Li, F.; Wang, Y.; Feng, X.H.; Mitch, W.E.; et al. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat. Commun. 2017, 8, 1493. [Google Scholar] [CrossRef] [Green Version]
- Carlson, A.K.; Rawle, R.A.; Wallace, C.W.; Brooks, E.G.; Adams, E.; Greenwood, M.C.; Olmer, M.; Lotz, M.K.; Bothner, B.; June, R.K. Characterization of synovial fluid metabolomic phenotypes of cartilage morphological changes associated with osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1174–1184. [Google Scholar] [CrossRef]
- Lee, K.I.; Choi, S.; Matsuzaki, T.; Alvarez-Garcia, O.; Olmer, M.; Grogan, S.P.; D’Lima, D.D.; Lotz, M.K. FOXO1 and FOXO3 transcription factors have unique functions in meniscus development and homeostasis during aging and osteoarthritis. Proc. Natl. Acad. Sci. USA 2020, 117, 3135–3143. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Xiong, X.Q.; Ren, X.S.; Zhao, M.X.; Shi, C.X.; Wang, J.J.; Zhou, Y.B.; Zhang, F.; Han, Y.; Gao, X.Y.; et al. FNDC5 alleviates hepatosteatosis by restoring AMPK/mTOR-mediated autophagy, fatty acid oxidation, and iipogenesis in mice. Diabetes 2016, 65, 3262–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.L.; Wu, S.S.; Wu, Y.; Wang, X.X.; Chen, H.Y.; Xin, J.J.; Li, H.; Lan, J.; Xue, K.Y.; Li, X.; et al. Irisin alleviates pressure overload-induced cardiac hypertrophy by inducing protective autophagy via mTOR-independent activation of the AMPK-ULK1 pathway. J. Mol. Cell Cardiol. 2018, 121, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef]
- Chen, K.; Xu, Z.; Liu, Y.; Wang, Z.; Li, Y.; Xu, X.; Chen, C.; Xia, T.; Liao, Q.; Yao, Y.; et al. Irisin protects mitochondria function during pulmonary ischemia/reperfusion injury. Sci. Transl. Med. 2017, 9, eaao6298. [Google Scholar] [CrossRef] [Green Version]
- Nazem, S.; Rabiee, F.; Ghaedi, K.; Babashah, S.; Sadeghizadeh, M.; Nasr-Esfahani, M.H. Fndc5 knockdown induced suppression of mitochondrial integrity and significantly decreased cardiac differentiation of mouse embryonic stem cells. J. Cell Biochem. 2018, 119, 4528–4539. [Google Scholar] [CrossRef]
- Huang, M.L.; Chiang, S.; Kalinowski, D.S.; Bae, D.H.; Sahni, S.; Richardson, D.R. The role of the antioxidant response in mitochondrial dysfunction in degenerative diseases: Cross-Talk between Aantioxidant defense, autophagy, and apoptosis. Oxid. Med. Cell Longev. 2019, 2019, 6392763. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wu, Z.; He, Y.; Chen, Z.; Xu, K.; Yu, W.; Fang, W.; Ma, C.; Moqbel, S.A.A.; Ran, J.; et al. MFN2 contributes to metabolic disorders and inflammation in the aging of rat chondrocytes and osteoarthritis. Osteoarthr. Cartil. 2020, 28, 1079–1091. [Google Scholar] [CrossRef]
- Shin, H.J.; Park, H.; Shin, N.; Kwon, H.H.; Yin, Y.; Hwang, J.A.; Song, H.J.; Kim, J.; Kim, D.W.; Beom, J. Pink1-Mediated chondrocytic mitophagy contributes to cartilage degeneration in osteoarthritis. J. Clin. Med. 2019, 8, 1849. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.Y.; Khan, N.M.; Ahmad, I.; Haqqi, T.M. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr. Cartil. 2018, 26, 1087–1097. [Google Scholar] [CrossRef]
- Castro, C.M.; Corciulo, C.; Solesio, M.E.; Liang, F.; Pavlov, E.V.; Cronstein, B.N. Adenosine A2A receptor (A2AR) stimulation enhances mitochondrial metabolism and mitigates reactive oxygen species-mediated mitochondrial injury. FASEB J. 2020, 34, 5027–5045. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, Z.; Xu, L.; Xu, K.; Chen, Z.; Ran, J.; Wu, L. The role of SIRT3-mediated mitochondrial homeostasis in osteoarthritis. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 2019, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Bobba, N.; Richelsen, B.; Lund, S.; Pedersen, S.B. Inflammation downregulates UCP1 expression in brown adipocytes potentially via SIRT1 and DBC1 interaction. Int. J. Mol. Sci. 2017, 18, 1006. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.-S.; Kuo, C.-W.; Ko, J.-Y.; Chen, Y.-S.; Wang, S.-Y.; Ke, H.-J.; Kuo, P.-C.; Lee, C.-H.; Wu, J.-C.; Lu, W.-B.; et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants 2020, 9, 810. https://doi.org/10.3390/antiox9090810
Wang F-S, Kuo C-W, Ko J-Y, Chen Y-S, Wang S-Y, Ke H-J, Kuo P-C, Lee C-H, Wu J-C, Lu W-B, et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants. 2020; 9(9):810. https://doi.org/10.3390/antiox9090810
Chicago/Turabian StyleWang, Feng-Sheng, Chung-Wen Kuo, Jih-Yang Ko, Yu-Shan Chen, Shao-Yu Wang, Huei-Jing Ke, Pei-Chen Kuo, Chin-Huei Lee, Jian-Ching Wu, Wen-Bin Lu, and et al. 2020. "Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy" Antioxidants 9, no. 9: 810. https://doi.org/10.3390/antiox9090810