Mitochondrial Oxidative and Nitrosative Stress and Alzheimer Disease


Abstract
1. Introduction
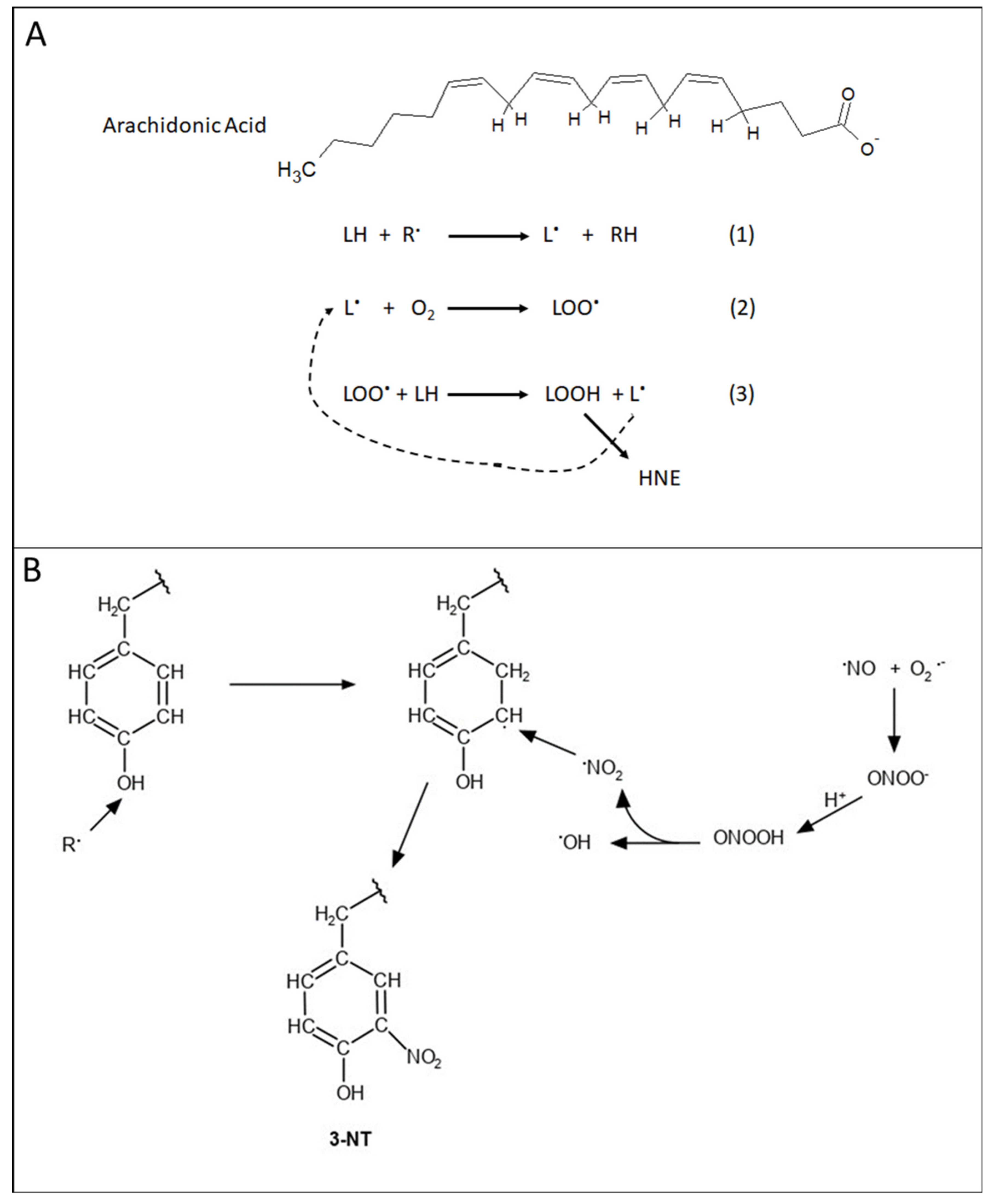
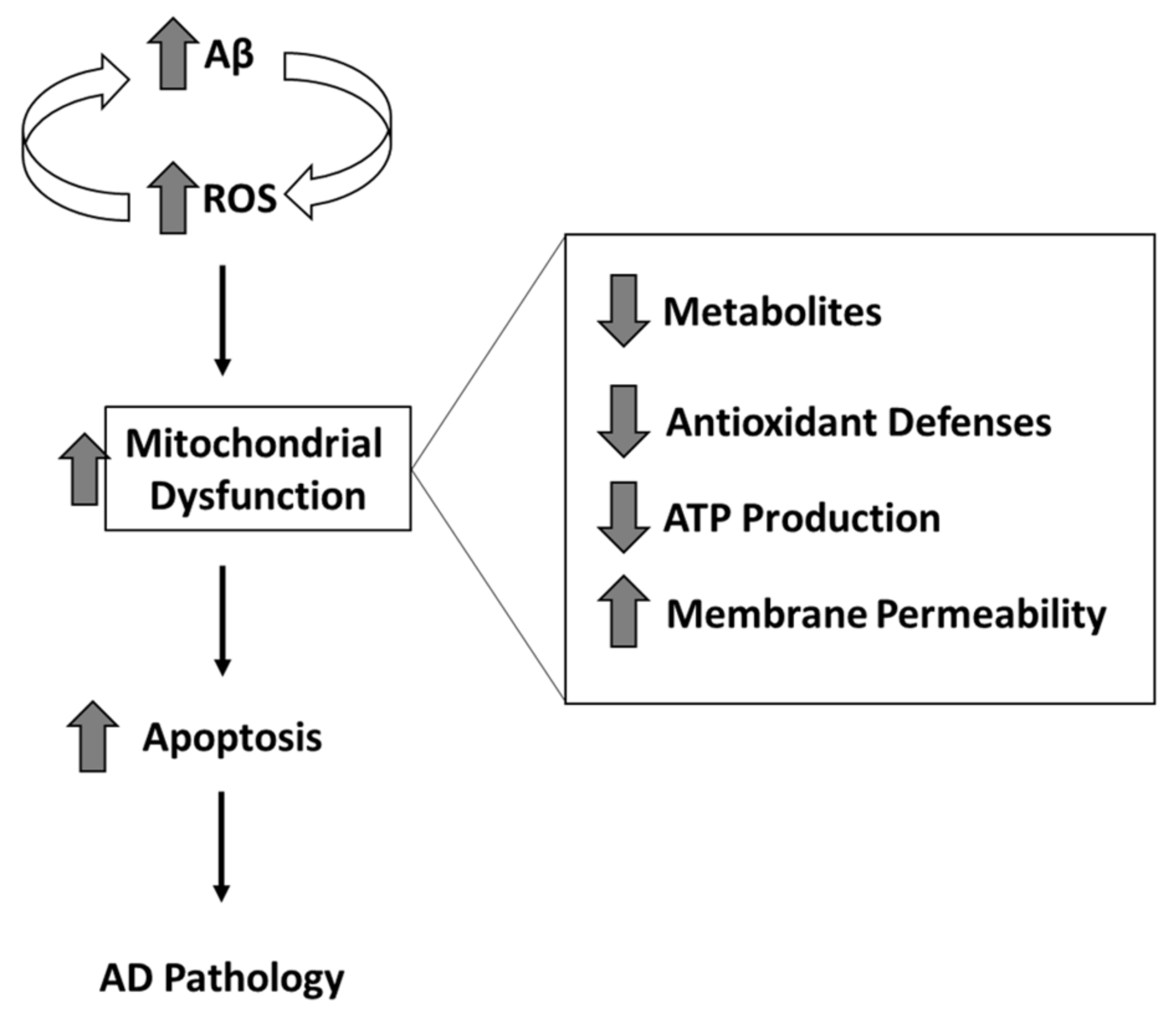
2. Mitochondria and Oxidative and Nitrosative Stress
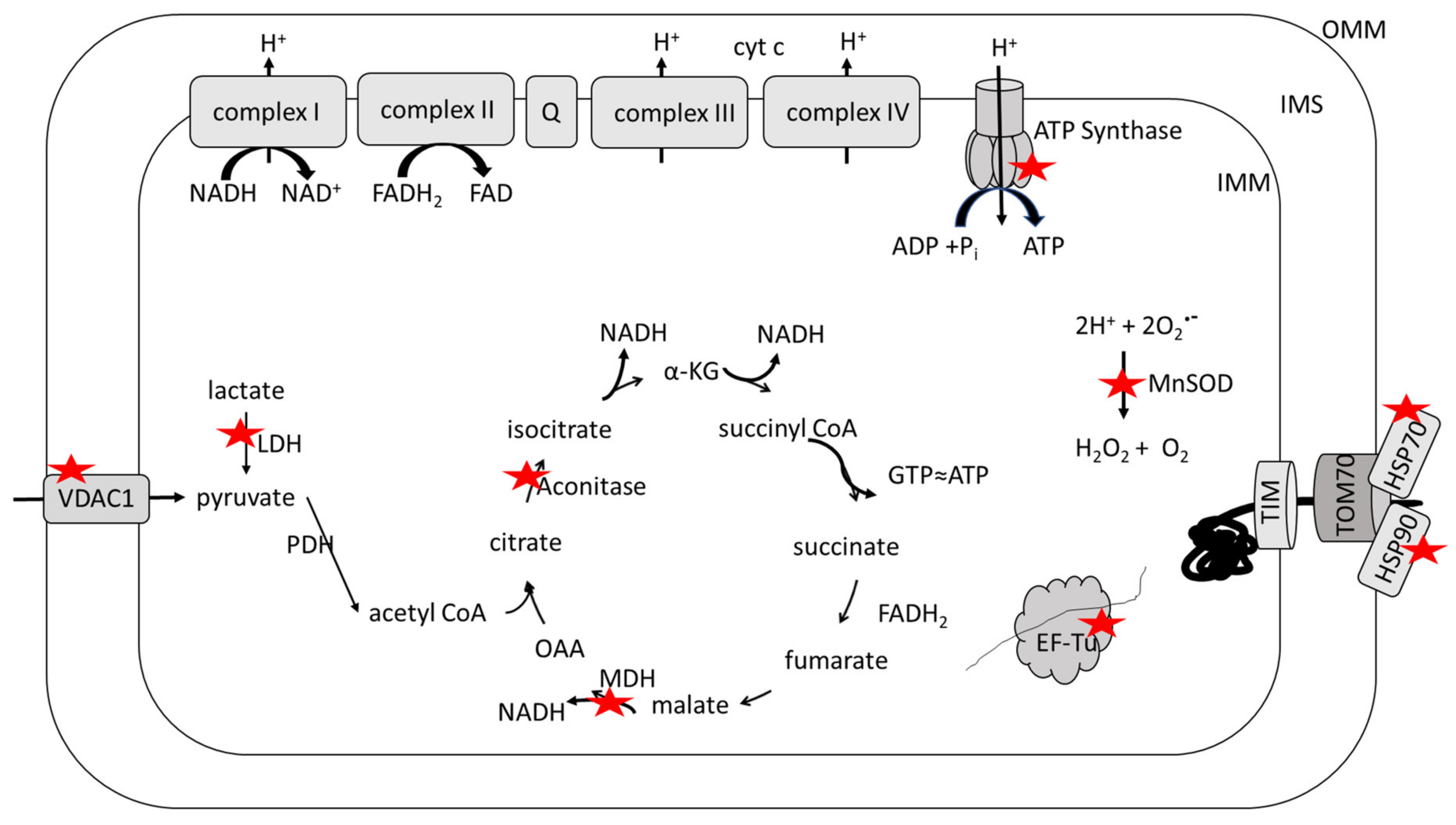
3. Selected Biological Alterations in AD Mitochondria
4. Redox Proteomics
5. Oxidative and Nitrosative Modifications of Mitochondrial Proteins in AD and MCI and in Systems of Relevance Thereof
6. Implications of Mitochondrial Oxidative and Nitrosative Stress in MCI and AD: Correlations with Imaging Studies and Targeted Therapies
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Katzman, R.; Saitoh, T. Advances in Alzheimer’s disease. FASEB J. 1991, 5, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, in press. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A. Alzheimer’s disease-related alterations in synaptic density: Neocortex and hippocampus. J. Alzheimers Dis. 2006, 9 (Suppl. 3), 101–115. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment: Current research and clinical implications. Semin. Neurol. 2007, 27, 22–31. [Google Scholar] [CrossRef]
- Markesbery, W.R. Neuropathologic alterations in mild cognitive impairment: A review. J. Alzheimers Dis. 2010, 19, 221–228. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, glucose dysmetabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Hyoung-gon Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. The 2013 SFRBM Discovery Award: Selected discoveries from the Butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radic. Biol. Med. 2014, 74, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology & Medicine, 5th ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- van der Bliek, A.M.; Margaret, M.; Sedensky, M.M.; Morgan, P.G. Cell biology of the mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Keeney, J.T.R.; Miriyala, S.; Noel, T.; Powell, D.K.; Chaiswing, L.; Bondada, S.; St. Clair, D.K.; Butterfield, D.A. Bottom of Form the triangle of death of neurons: Oxidative damage, mitochondrial dysfunction, and loss of choline-containing biomolecules in brains of mice treated with doxorubicin. Advanced insights into mechanisms of chemotherapy induced cognitive impairment (“chemobrain”) involving TNF-α. Free Radic. Biol. Med. 2019, 134, 1–8. [Google Scholar] [PubMed]
- Dhar, S.K.; St. Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef]
- Nelson, S.K.; Bose, S.K.; Grunwald, G.K.; Myhill, P.; McCord, J.M. The induction of human superoxide dismutase and catalase in vivo: A fundamentally new approach to antioxidant therapy. Free Radic. Biol. Med. 2006, 40, 341–347. [Google Scholar] [CrossRef]
- Sies, H.; Brendt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kil, I.S. Multiple functions and regulation of mammalian peroxiredoxin. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, lipid peroxidation, and cell death: Discoveries, rediscoveries, and open issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Jiang, B.; Moskovitz, J. The functions of the mammalian methionine reductase systems and related diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Stadtman, E.R. Protein oxidation processes in aging brain. Adv. Cell Aging Gerontol. 1997, 2, 161–191. [Google Scholar]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Hinchie, A.; Swomley, A.; Powell, D.K.; Butterfield, D.A. Profiles of brain oxidative damage, ventricular alterations, and neurochemical metabolites in the striatum of PINK1 Kkockout rats as functions of age and gender: Relevance to parkinson disease. Free Radic. Biol. Med. 2019, 143, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.N.; Landar, A.; Barnes, S.; Darley-Usmar, V.M. The electrophile responsive proteome: Integrating proteomics and lipidomics with cellular function. Antioxid. Redox Signal. 2012, 17, 1580–1589. [Google Scholar] [CrossRef]
- Lauderback, C.M.; Hackett, J.M.; Huang, F.F.; Keller, J.N.; Szweda, L.I.; Markesbery, W.R.; Butterfield, D.A. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-Hydroxy-2-nonenal in the Alzheimer’s disease brain: Role of Aβ1-42. J. Neurochem. 2001, 78, 413–416. [Google Scholar] [CrossRef]
- Hardas, S.S.; Sultana, R.; Clark, A.M.; Bracket, C.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef]
- Subramaniam, R.; Roediger, F.; Jordan, B.; Mattson, M.P.; Keller, J.N.; Waeg, G.; Butterfield, D.A. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortial synaptosomal membrane proteins. J. Neurochem. 1997, 69, 1161–1169. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Batthyány, C.; Bartesaghi, S.; Mastrogiovanni, M.; Lima, A.; Demicheli, V.; Radi, R. Tyrosine-nitrated proteins: Proteomic and bioanalytical aspects. Antioxid. Redox Signal. 2017, 26, 313–328. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Wisniewski, T.; Drummond, E. APOE-amyloid interaction: Therapeutic targets. Neurobiol. Dis. 2020, 138, 104784. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Redox proteomics and amyloid β-peptide: Insights into Alzheimer disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Sultana, R.; Mecocci, P.; Mangialasche, F.; Cecchetti, R.; Butterfield, D.A. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer disease: Insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J. Alzheimers Dis. 2011, 24, 77–84. [Google Scholar]
- Sultana, R.; Baglioni, M.; Cecchetti, R.; Cai, J.; Klein, J.B.; Bastiani, P.; Ruggiero, C.; Mecocci, P.; Butterfield, D.A. Lymphocyte mitochondria: Toward identification of peripheral biomarkers in progression of Alzheimer disease. Free Radic. Biol. Med. 2013, 65, 595–606. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Molecular biology of Alzheimers disease: Focus on mitochondria. J. Alzheimers Dis. 2019, 72, S95–S116. [Google Scholar] [CrossRef]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 ameliorates synaptic depression, Aβ deposition, and cognitive impairment in an Alzheimer’s disease model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondrial-division inhibitor 1 protects against amyloid-b induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Scaloni, A.; Butterfield, D.A. Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases; Wiley Press: New York, NY, USA, 2006. [Google Scholar]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Perluigi, M.; Reed, T.; Muharib, T.; Hughes, C.P.; Robinson, R.A.; Sultana, R. Redox proteomics in selected neurodegenerative disorders. From its infancy to future applications. Antioxidant. Redox Signal. 2012, 17, 1610–1655. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Gu, L.; Di Domenico, F.; Robinson, R.A.S. Mass spectrometry and redox proteomics: Applications in disease. Mass Spectrom. Rev. 2014, 33, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: Insights into mechanism of neurodegeneration from redox proteomics. Antioxidant. Redox Signal. 2006, 8, 2021–2037. [Google Scholar] [CrossRef]
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: A toxic combination illuminated by redox proteomics Studies. Antioxidant. Redox Signal. 2012, 17, 590–1609. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers Neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Perluigi, M.; Swomley, A.M.; Butterfield, D.A. Redox proteomics and the dynamic molecular landscape of the aging brain. Ageing Res. Rev. 2014, 13, 75–89. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Swomley, A.M.; Head, E.; Perluigi, M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: Overlaps in Down syndrome and Alzheimer disease brain. Biochem. J. 2014, 463, 177–189. [Google Scholar] [CrossRef]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. The triangle of death in Alzheimer disease brain: The aberrant cross talk among energy metabolism, mTOR signaling and protein homeostasis revealed by redox proteomics. Antioxidant. Redox Signal. 2017, 26, 364–387. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteom. Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of HNE-bound proteins in early Alzheimer’s disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2008, 30, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Reed, T.; Perluigi, M.; Coccia, R.; Pierce, W.M.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: A regional study. J. Cell. Molec. Med. 2007, 11, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Newman, S.F.; Pierce, W.M.; Cini, C.; Coccia, R.; Butterfield, D.A. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease. Antioxid. Redox Signal. 2010, 12, 327–336. [Google Scholar] [CrossRef]
- Aluise, C.D.; Robinson, R.A.S.; Cai, J.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: Insights into memory loss in MCI. J. Alzheimers Dis. 2011, 23, 257–269. [Google Scholar] [CrossRef]
- Terni, B.; Boada, J.; Portero-Otin, M.; Ramplona, R.; Ferrer, I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Path. 2010, 20, 222–233. [Google Scholar] [CrossRef]
- Lushchak, O.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014, 19, 8–15. [Google Scholar] [CrossRef]
- Shen, L.; Chen, C.; Yang, A.; Chen, Y.; Liu, Q.; Ni, J. Redox proteomics identification of specifically carbonylated proteins in the hippocampi of triple transgenic Alzheimer’s disease mice at its earliest pathological stage. J. Proteomics 2015, 123, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Minárik, P.; Tomášková, N.; Kollárová, M.; Antalík, M. Malate Dehydrogenases—Structure and Function. Gen. Physiol. Biophys. 2002, 21, 257–265. [Google Scholar] [PubMed]
- Sultana, R.; Newman, S.F.; Mohmmad-Abdul, H.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Butterfield, D.A. Protective Effect of D609 Against Amyloid-Beta1-42-Induced Oxidative Modification of Neuronal Proteins: Redox Proteomics Study. J. Neurosci. Res. 2006, 84, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Boyd-Kimball, D.; Poon, H.F.; Lynn, B.C.; Cai, J.; Pierce, W.M., Jr.; Klein, J.B.; Ferguson, J.; Link, C.D.; Butterfield, D.A. Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Aβ(1-42): Implications for Alzheimer’s Disease. Neurobiol. Aging 2006, 27, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Hussien, R.; Cho, H.-S.; Kaufer, D.; Brooks, G.A. Evidence for the mitochondrial lactate oxidation complex in rat neurons: Demonstration of an essential component of brain lactate shuttles. PLoS ONE 2008, 3, e2915. [Google Scholar] [CrossRef] [PubMed]
- Bittar, P.G.; Charnay, Y.; Pellerin, L.; Bouras, C.; Magistretti, P.J. Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Met. 1996, 16, 1079–1089. [Google Scholar] [CrossRef]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011, 31, 7477–7485. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Wallace, D.C. Bioenergetic origins of complexity of disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef]
- Yellen, G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 2018, 217, 2235–2246. [Google Scholar] [CrossRef]
- Sultana, R.; Robinson, R.A.S.; Di Domenico, F.; Abdul, H.M.; St. Clair, D.K.; Markesbery, W.R.; Cai, J.; Pierce, W.M.; Butterfield, D.A. Proteomics identification of specifically carbonylated brain proteins in APPNLh/ APPNLh x PS-1P264L/ PS-1P264L human double mutant knock-in mice model of Alzheimer disease as a function of age. J. Proteom. 2011, 74, 2430–2440. [Google Scholar]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.S.; Huang, S.-C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Wardelmann, K.; Grune, T.; Kleinridders, A. Mitochondrial chaperones in brain: Safeguarding brain health and metabolism? Front. Endrocrinol. 2018, 9, 196. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Maracci, C.; Rodnina, M.V. Review: Translational GTPases. Biopolymers 2016, 105, 463–475. [Google Scholar] [CrossRef]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F.U. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St. Clair, D.K. Manganese Superoxide Dismutase: Guardian of the Powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Holley, A.K.; Dhar, S.K.; Xu, Y.; St. Clair, D.K. Manganese superoxide dismutase: Beyond life and death. Amino Acids 2012, 42, 139–158. [Google Scholar] [CrossRef]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Rad. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Marcus, D.L.; Strafaci, J.A.; Freedman, M.L. Differential neuronal expression of manganese superoxide dismutase in Alzheimer’s disease. Med. Sci. Monit. 2006, 12, BR8–BR14. [Google Scholar] [PubMed]
- Zhao, Y.; Chaiswing, L.; Velez, J.M.; Batinic-Haberle, I.; Colburn, N.H.; Oberley, T.D.; St. Clair, D.K. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005, 65, 3745–3750. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.-Q.; Luo, T.-T.; Luo, S.-C.; Wang, J.-Q.; Wang, S.-M.; Bai, Y.-H.; Yang, Y.-L.; Wang, Y.-Y. p53 mitochondrial dysfunction: Novel insight of neurodegenerative disease. J. Bioenerg. Biomembr. 2016, 48, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer’s disease. Free Rad. Biol. Med. 2008, 45, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 987–994. [Google Scholar] [CrossRef]
- Fiorini, A.; Sultana, R.; Barone, E.; Cenini, G.; Perluigi, M.; Mancuso, C.; Cai, J.; Klein, J.B.; St. Clair, D.K.; Butterfield, D.A. Lack of p53 affects the expression of several brain mitochondrial proteins: Insights from proteomics in important pathways regulated by p53. PLoS ONE 2012, 7, e49846. [Google Scholar] [CrossRef]
- Drane, P.; Bravard, A.; Bouvard, V.; May, E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene 2001, 20, 430–439. [Google Scholar] [CrossRef]
- Dhar, S.K.; Tangpong, J.; Chaiswing, L.; Oberley, T.D.; St. Clair, D.K. Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 2011, 71, 6684–6695. [Google Scholar] [CrossRef]
- Barone, E.; Cenini, G.; Di Domenico, F.; Noel, T.; Wang, C.; Perluigi, M.; St. Clair, D.K.; Butterfield, D.A. Basal brain oxidative and nitrosative stress levels are finely regulated by the interplay between superoxide dismutase 2 and p53. J. Neurosci. Res. 2015, 93, 1728–1739. [Google Scholar] [CrossRef]
- Tramutola, A.; Pupo, G.; Di Domenico, F.; Barone, E.; Arena, A.; Lanzillotta, C.; Brokeaart, D.; Blarzino, C.; Head, E.; Butterfield, D.A.; et al. Activation of p53 in Down Syndrome and in the Ts65Dn mouse brain is associated with a pro-apoptotic phenotype. J. Alzheimers Dis. 2016, 52, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Naveed, H.; Liang, J.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1 protein: Defining oligomer contact sites. J. Biol. Chem. 2012, 287, 2179–2190. [Google Scholar] [CrossRef]
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, J.; Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell. Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef]
- Arbel, N.; Shoshnan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, S. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Shoshan-Barmtz, V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim. Biophys. Acta 2016, 1863, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Krelin, Y.; Chen, Q. VDAC1 as a player in mitochondria-mediated apoptosis and target for modulating apoptosis. Curr. Med. Chem. 2017, 24, 4435–4446. [Google Scholar] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmtz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The voltage-dependent anion channel 1 mediates amyloid β toxicity and represents a potential target for Alzheimer disease therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef]
- Feldhaus, P.; Fraga, D.B.; Ghedim, F.V.; De Luca, R.D.; Bruna, T.D.; Heluany, M.; Matos, M.P.; Ferreira, G.K.; Jeremias, I.C.; Heluany, C.; et al. Evaluation of respiratory chain activity in lymphocytes of patients with Alzheimer disease. Metab. Brain Dis. 2011, 26, 229–236. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Proenςa, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Mangialasche, F.; Baglioni, M.; Cecchetti, R.; Kivipelto, M.; Ruggiero, C.; Piobbico, D.; Kussmaul, L.; Monastero, R.; Brancorsini, S.; Mecocci, P. Lymphocytic mitochondrial aconitase activity is reduced in Alzheimer’s disease and mild cognitive impairment. J. Alzheimers Dis. 2015, 44, 649–660. [Google Scholar] [CrossRef]
- Zahid, S.; Khan, R.; Oellerid, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Zareba-Koziol, M.; Swajda, A.; Dadlez, M.; Wyslouch-Cieszynsha, A. Global analysis of S-nitrosylation sites in the wild type (APP) transgenic mouse brain: Clues for synaptic pathology. Mol. Cell. Proteom. 2014, 13, 2288–2305. [Google Scholar] [CrossRef] [PubMed]
- Wijasa, T.S.; Sylveser, M.; Broche-Ahmadinejad, N.; Schwartz, S.; Santarelli, F.; Geiselmann, V.; Klockgether, F.; Heneka, M.T. Quantitative proteomics of synaptosome S-nitrosylation in Alzheimer disease. J. Neurochem. 2020, 152, 710–726. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.-K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.-I.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Rolfe, D.F.S.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; de Freitas, A.E.; et al. The energetic brain – A review from students to students. J. Neurochem. 2020, 151, 139–165. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Bawanowska-Basiacka, I. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment, and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.-A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, E.E.; Bocti, C.; et al. Ketongenic medium chain triglycerides increase brain energy metabolism in Alzheimer’s disease. J. Alzheimers Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Yip, J.; Geng, X.; Shen, J.; Ding, Y. Cerebral gluconeogenesis and diseases. Front Pharmacol. 2017, 7, 521. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, G.K.; Cruz, N.F.; Ball, K.K.; Dienel, G.A. Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J. Neurochem. 2009, 111, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Mullarky, E.; Cantley, L.C. Diverting glycolysis to combat oxidative stress. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 3–23. [Google Scholar]
- Williams, H.C.; Farmer, B.C.; Piron, M.A.; Walsh, A.E.; Bruntz, R.C.; Gentry, M.S.; Sun, R.C.; Johnson, L.A. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol. Dis. 2020, 136, 104742. [Google Scholar] [CrossRef]
- Griffin, J.W.D.; Bradshaw, P.C. Amino acid catabolism in Alzheimer’s disease brain: Friend or foe? Oxid. Med. Cell. Longev. 2017, 2017, 5472792. [Google Scholar] [CrossRef]
- Cooper, A.J.; Jeitner, T.M. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef]
- Polis, B.; Samson, A.O. Role of the metabolism of branched-chain amino acids in the development of Alzheimer’s disease and other metabolic disorders. Neural Regen. Res. 2020, 15, 1460–1470. [Google Scholar] [CrossRef]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A. Excess brain protein oxidation and enzyme dysfunction in normal and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1991, 87, 10540–10543. [Google Scholar] [CrossRef]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, R.; Harris, M.; Aksenov, M.; Aksenova, A.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brian regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Arnold, M.; Kastenmüller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. The Alzheimer’s Disease Neuroimaging Initiative and the Alzheimer’s Disease Metabolomics Consortium. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Metabolite profiling for the identification of altered metabolic pathways in Alzheimer’s disease. J. Pharm. Biomed. Anal. 2015, 107, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Lowe, V.J.; Weigand, S.D.; Wiste, H.J.; Senjem, M.L.; Knopman, D.S.; Shiung, M.M.; Gunter, J.L.; Boeve, B.F.; Kemp, B.J.; et al. The Alzheimer’s Disease Neuroimaging Initiative. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: Implications for sequence of pathological events in Alzheimer’s disease. Brain 2009, 132, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Zuiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012, 11, 1048–1056. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvador, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individual from families with autosomal dominant Alheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid β-peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef]
- Oda, T.; Wals, P.; Osterburg, H.H.; Johnson, S.A.; Pasinetti, G.M.; Morgan, T.E.; Rozovsky, I.; Stine, W.B.; Snyder, S.W.; Holzman, T.F.; et al. Clusterin (apoJ) alters the aggregation of amyloid β-peptide (Aβ1-42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp. Neurol. 1995, 136, 22–31. [Google Scholar] [CrossRef]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid beta-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Mann, D.M.; Harris, J.M.; Chartier-Harlin, M.C.; Cumming, A.; Coates, J.; Lemmon, H.; St. Clair, D.K.; Iwatsubo, T.; Lendon, C. The -48 C/T polymorphism in the presenilin 1 promoter is associated with increased Abeta load in brain. J. Med. Genet. 2001, 38, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.; Link, C.D.; Butterfield, D.A. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol. Aging 2003, 24, 415–420. [Google Scholar] [CrossRef]
- Tobore, T.O. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer’s disease. Neurol. Sci. 2019, 40, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting Mitochondria in Alzheimer disease: Rationale and perspectives. CNS Drugs 2019, 33, 957–969. [Google Scholar] [CrossRef]
- Opii, W.O.; Joshi, G.; Head, E.; Milgram, N.W.; Muggenburg, B.A.; Klein, J.B.; Pierce, W.M.; Cotman, C.W.; Butterfield, D.A. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and program of behavioral enrichment: Relevance to Alzheimer’s disease. Neurobiol. Aging 2008, 29, 51–70. [Google Scholar] [CrossRef]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Moreley, J.E. The antioxidants α-Lipoic Acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef]
- Hager, K.; Marahrens, A.; Kenklies, M.; Riederer, P.; Munich, G. Alpha-lipoic acid as a new treatment option for Alzheimer type dementia. Arch. Gerontol. Geriat. 2001, 32, 275–282. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Li, G.; Wang, J.; Yang, E.S. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J. Mol. Neurosci. 2008, 34, 165–171. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Cherbuin, N.; Budge, M.; Young, J. Body mass index in midlife and late-life as a risk factor for dementia: A meta-analysis of prospective studies. Obes. Rev. 2011, 12, e426–e437. [Google Scholar] [CrossRef] [PubMed]
- Albanese, E.; Launer, L.J.; Egger, M.; Prince, M.J.; Giannakopoulos, P.; Wolters, F.J.; Egan, K. Body mass index in midlife and dementia: Systemic review and meta-regression analysis of 589,649 men and women following in longitudinal studies. Alzheimers Dement. (Amst.) 2017, 8, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Soloman, A.; Kivipelto, M.; Wolozin, B.; Zhou, J.; Whitmer, R.A. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 2009, 28, 75–80. [Google Scholar] [CrossRef]
- Meng, X.F.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Wang, C.; Tan, C.C.; Tan, L. Midlife vascular risk factors and the risk of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2014, 42, 1295–1310. [Google Scholar] [CrossRef]
- Debette, S.; Seshadri, S.; Beiser, A.; Au, R.; Himali, J.J.; Palumbo, C.; Wolf, P.A.; DeCarli, C. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology 2011, 77, 461–468. [Google Scholar] [CrossRef]
- Ninomiya, T.; Ohara, T.; Hirakawa, Y.; Yoshida, D.; Hata, J.; Kanba, S.; Iwaki, T.; Kiyohara, Y. Midlife and late-life blood pressure and dementia in Japanese elderly: The Hisayama study. Hypertension 2011, 58, 22–28. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Albert, M.S.; Alonso, A.; Coker, L.H.; Coresh, J.; Davis, S.M.; Deal, J.A.; McKhann, G.M.; Mosley, T.H.; Sharrett, A.R.; et al. Associations between midlife vascular risk factors and 25-year incident dementia in atherosclerosis risk in communities (ARIC) cohort. JAMA Neurol. 2017, 74, 1246–1254. [Google Scholar] [CrossRef]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef]
- Wu, W.; Brickman, A.M.; Luchsinger, J.; Ferrazzano, P.; Pichiule, P.; Yoshita, M.; DeCarli, C.; Barnes, C.A.; Mayeux, R.; Vannucci, S.J.; et al. The brain in the age of old: The hippocampal formation is targeted differentially by disease of late life. Ann. Neurol. 2008, 64, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer’s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef]
- Santos-Lozano, A.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Quindós-Rubial, M.; Fiuza-Luces, C.; Cristi-Montero, C.; Emanuele, E.; Garatachea, N.; Lucia, A. Physical activity and Alzheimer disease: A protective association. Mayo Clin. Proc. 2016, 91, 999–1020. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brain harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Ratnaike, T.E.; De Gruyter, H.L.M.; Jaros, E.; Turnbull, D.M. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 2210–2214. [Google Scholar] [CrossRef]
- Bargerger-Gateau, P.; Raffaitin, C.; Letenneur, L.; Tzourio, C.; Dartigues, J.F.; Alpérovitch, A. Dietary patterns and risk of dementia: The Three-City cohort study. Neurology 2007, 69, 1921–1930. [Google Scholar] [CrossRef]
- Lourida, I.; Soni, M.; Thompson-Coon, J.; Purandare, N.; Lang, I.A.; Ukoumunne, O.C.; Llewellyn, D.J. Mediterranean diet, cognitive function, and dementia: A systematic review. Epidemiology 2013, 24, 479–489. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Barnes, L.L.; Bennett, D.A.; Aggarwal, N.T. MIND diet slows cognitive decline with aging. Alzheimers Dement. 2015, 11, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Hardman, R.J.; Kennedy, G.; Macpherson, H.; Scholey, A.B.; Pipingas, A. Adherence to a Mediterranean-Style Diet and Effects on Cognition in Adults: A Qualitative Evaluation and Systematic Review of Longitudinal and Prospective Trials. Front. Nutr. 2016, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Stephen, R.; Hongisto, K.; Solomon, A.; Lönnroos, E. Physical activity and Alzheimer’s disease: A Systematic Review. J. Gerontol. A Biol. Med. Sci. 2017, 72, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.S.; Spartano, N.L.; Beiser, A.S.; DeCarli, C.; Auerbach, S.H.; Vasan, R.S.; Seshadri, S. Physical activity, brain volume, and dementia risk: The Framingham Study. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Ogino, E.; Manly, J.J.; Schupf, N.; Mayeuz, R.; Gu, Y. Current and past leisure time physical activity in relation to risk of Alzheimer’s disease in older adults. Alzheimers Dement. 2019, 15, 1603–1611. [Google Scholar] [CrossRef]
- Najar, J.; Östling, S.; Gudmundsson, P.; Sundh, V.; Johansson, L.; Kern, S.; Guo, X.; Hällström, T.; Skoog, I. Cognitive and physical activity and dementia: A 44-year longitudinal population study of women. Neurology 2019, 92, e1322–e1330. [Google Scholar] [CrossRef]
- Rosenberg, A.; Ngandu, T.; Rusanen, M.; Antikainen, R.; Bäckman, L.; Havulinna, S.; Hänninen, T.; Laatikainen, T.; Lehtisalo, J.; Levälahti, E.; et al. Multidomain lifestyle intervention benefits a large population at risk for cognitive decline and dementia regardless of baseline characteristics: The FINGER trial. Alzheimers Dement. 2018, 14, 263–270. [Google Scholar] [CrossRef]
- Solomon, A.; Turunen, H.; Ngandu, T.; Peltonen, M.; Levälahti, E.; Seppo, H.; Antikainen, R.; Backman, L.; Hanninen, T.; Jula, A.; et al. Effect of the apolipoprotein E genotype on cognitive change during a multidomain lifestyle intervention: A subgroup analysis of a randomized clinical trial. JAMA Neurol. 2018, 75, 462–470. [Google Scholar] [CrossRef]
- Stephen, R.; Liu, Y.; Ngandu, T.; Antikainen, R.; Hulkkonen, J.; Koikkalainen, J.; Kemppainen, N.; Lötjönen, J.; Levälahti, E.; Parkkola, R.; et al. Brain volumes and cortical thickness on MRI in the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER). Alzheimers Res. Ther. 2019, 11, 53. [Google Scholar] [CrossRef]
- Bernardo, T.C.; Marques-Aleixo, I.; Beleza, J.; Oliveira, P.J.; Ascensão, A.; Magalhães, J. Physical exercise and brain mitochondrial fitness: The possible role against Alzheimer’s disease. Brain Pathol. 2016, 26, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Cavalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Silzer, T.K.; Phillips, N.R. Etiology of type 2 diabetes and Alzheimer’s disease: Exploring the mitochondria. Mitochondrion 2018, 43, 16–24. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Oxidative Modification | Disease State | Brain Region | Activity | Reference |
|---|---|---|---|---|---|
| Aconitase | H | Late AD | Hippocampus | ↓ | [54] |
| MDH | H | Early AD | IPL | ↑ | [55] |
| LDH | H | MCI | Hippocampus | ↓ | [56] |
| ATP Synthase | H | MCI | Hippocampus | ↓ | [56] |
| H | MCI | IPL | ↓ | [56] | |
| H | Early AD | IPL | [55] | ||
| H | Late AD | IPL | ↓ | [54] | |
| N | Late AD | Hippocampus | [57] | ||
| MnSOD | H | Early AD | IPL | ↓ | [55] |
| H | Late AD | IPL | [54] | ||
| EF-Tu | H | MCI | IPL | [56] | |
| HSP70 | H | MCI | Hippocampus | [56] | |
| N | MCI | Hippocampus | [58] | ||
| C | MCI | IPL | [59] | ||
| HSP90 | C | MCI | IPL | [60] | |
| VDAC1 | N | Late AD | Hippocampus | [57] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butterfield, D.A.; Boyd-Kimball, D. Mitochondrial Oxidative and Nitrosative Stress and Alzheimer Disease. Antioxidants 2020, 9, 818. https://doi.org/10.3390/antiox9090818
Butterfield DA, Boyd-Kimball D. Mitochondrial Oxidative and Nitrosative Stress and Alzheimer Disease. Antioxidants. 2020; 9(9):818. https://doi.org/10.3390/antiox9090818
Chicago/Turabian StyleButterfield, D. Allan, and Debra Boyd-Kimball. 2020. "Mitochondrial Oxidative and Nitrosative Stress and Alzheimer Disease" Antioxidants 9, no. 9: 818. https://doi.org/10.3390/antiox9090818
APA StyleButterfield, D. A., & Boyd-Kimball, D. (2020). Mitochondrial Oxidative and Nitrosative Stress and Alzheimer Disease. Antioxidants, 9(9), 818. https://doi.org/10.3390/antiox9090818