1. Introduction
Malaria remains one of the biggest health problems globally, and the World Health Organization (WHO) estimated 627,000 malaria-related deaths, mostly in children aged under 5 years, in 2020 [
1]. Therefore, in addition to the expansion of existing anti-malarial control measures, it is critical to develop new tools, such as vaccines, to fight malaria. In October 2021, the first malaria vaccine, RTS,S/AS01 (RTS,S), which is a pre-erythrocytic vaccine (PEV), was recommended by WHO to prevent
Plasmodium falciparum malaria in children living in moderate to high transmission areas [
1]. PEV is designed to block sporozoite invasion to human hepatocytes and/or inhibit growth of parasites in the hepatocytes, thus it can prevent any symptoms caused by malaria infection. While this was a major milestone, further improvements in terms of the efficacy and duration of protection, and/or a combination with other intervention(s) are likely to be required for elimination of malaria [
2].
Transmission-blocking vaccines (TBVs) target a different life stage of malaria parasites, and are designed to induce antibodies in human hosts that inhibit parasite development in the mosquito bloodmeal [
3]. Using a mouse malaria model, a synergistic effect, better than an additive effect, has been shown with a combination of PEV and TBV [
4], indicating a TBV could be a strong additional tool to malaria control and eradication. Pfs230 is one of the leading TBV candidates, and the first to reach a Phase 2 clinical trial as a recombinant protein sub-fragment conjugated with
Pseudomonas aeruginosa ExoProtein A (ClinicalTrial.gov Identifier: NCT03917654). While the ultimate method to determine efficacy of a TBV is to measure reduction of malaria transmission in a field site by a cluster randomized trial, such studies are large, complex and costly, and cannot be used in early stages of TBV development. Thus, the current gold standard method to measure efficacy of vaccine-induced antibodies (either from animals or humans) in early stages of TBV development is the standard membrane-feeding assay (SMFA) or the direct membrane-feeding assay (DMFA) [
3]. In SMFA/DMFA, control or test antibodies are mixed with either cultured malaria (for SMFA) or blood from a malaria-infected human (for DMFA) and fed to mosquitoes. Approximately a week after the feed, the mosquitoes are dissected, and the reduction in parasite oocyst density in the mosquito midguts (called transmission-reducing activity (TRA)) or reduction in the proportion of infected mosquitoes (called transmission-blocking activity (TBA)) is determined by comparing the control and test groups.
During development of a subunit vaccine, the integrity of the recombinant protein must be monitored for process development and quality control of intermediate products. Furthermore, lot-to-lot consistency and stability of drug substance or drug product must be assessed. Multiple biophysical and biochemical methods, such as western blot, high-performance liquid chromatography (HPLC), circular dichroism, static light scattering, and dynamic light scattering, are commonly used for the quality control of vaccines, but a potency assay is another key evaluation [
5,
6]. In the case of a malaria TBV, based on the mode of action, an in vivo immunization study (often in mice) that assesses antisera or induced antibodies by SMFA is an acceptable potency assay. However, antibody titers in animal studies can fluctuate significantly even when the same batch of vaccine is utilized for immunization [
6]. For example, in the case of Diphtheria, Tetanus and acellular Pertussis (DTaP) vaccine, the coefficients of variance (CV) in antibody titers against pertussis antigen were reported as high as 131.7% in mouse immunization studies [
7]. In addition, precision in SMFA is poor when activity of a test antibody is low [
8]. These characteristics, coupled with the duration to conduct such testing and limited throughput, lead to the need for additional in vitro biochemical methods that may be a surrogate for such activity. Additionally, it is very challenging to detect a change in a recombinant protein by such an in vivo potency assay, especially when the level of deviation is too small to be detectable by biophysical and biochemical methods which are commonly used for vaccine quality control (QC). To avoid the issue of high variability in in vivo studies and to adhere to the “3R” principle (reduction, replacement and refinement of animals) [
9], several in vitro potency assays have been developed as alternatives [
10]. ELISA (Enzyme-linked immunosorbent assay)-based in vitro assay is one of them, and such assays have been evaluated both for licensed and developmental vaccines against multiple pathogens, such as human papillomavirus [
11], hepatitis B [
12], rabies [
13], diphtheria [
14], hepatitis E [
15] and respiratory syncytial virus (RSV) [
16].
In the present work, using a well-characterized Pfs230 recombinant protein, Pfs230D1+ [
17], and two human anti-Pfs230 monoclonal antibodies (mAbs) which have functional activity measured by SMFA [
18], we developed an in vitro sandwich ELISA, called the Antigen Integrity Assay (AIA). The expected usage of this AIA is to demonstrate integrity of functional epitopes in a new lot or a stressed (or stored) protein compared to a reference protein. We evaluated multiple validation parameters (linearity, specificity, intra- and inter-assay precision, and limit of detection) to qualify AIA following International Conference on Harmonization (ICH) Q2(R1) guidelines [
19]. To demonstrate such utility, we employed two lots of Pfs230D1+ protein (reference and an engineering lot) together with damaged antigens (reduced-and-alkylated or protease digested). The AIA coupled together with the validation parameters examined, indicate such an assay would be an additional biochemical control measure to incorporate into Pfs230 release and stability campaigns.
2. Materials and Methods
2.1. Reagents
The details of expression and characterization of the reference Pfs230D1+ recombinant protein have been described elsewhere [
17]. In brief, the recombinant protein comprising N-terminal fragment (amino acid (aa) 552–731) of Pfs230 in
P. falciparum 3D7 strain was produced in a baculovirus expression system. Two human anti-Pfs230 mAbs, Pfs230AS01-18 mAb (denoted as H18 mAb hereafter) and Pfs230AS01-73 mAb (denoted as H73 mAb) were generated based on receptor sequences of antigen-specific B cells isolated from vaccinees who had received Pfs230D1-EPA (a recombinant Pfs230 protein, which contains aa 542–736 of Pfs230 from NF54 strain, conjugated with ExoProtein A) and formulated in GlaxoSmithKline’s AS01 adjuvant (Clinicaltrials.gov # NCT02942277) according to published methods [
20]. Both mAbs recognize Pfs230D1+ in a conformation-dependent manner and have functional activities measured by SMFA [
18]. As a detection mAb, horseradish peroxidase (HRP) conjugated H18 mAb, H18-HRP mAb, was generated using a HRP Conjugation Kit-Lightning-Link (Abcam, Cambridge, UK. Cat No ab102890) following the manufacturers’ instructions. A mouse anti-Pfs230 mAb, 15A4-1B12, was isolated from mice immunized with Pfs230C1 (aa 443-731) and reacts with Pfs230D1+ protein in a conformation-dependent manner [
17], but the mAb does not have SMFA activity. HRP-conjugated 15A4-1B12 mAb was generated as H18-HRP mAb. Bovine Serum Albumin (BSA; Cat No A7906), Tween-20 (Cat No P1379), Dithiothreitol (DTT; Cat No 3483-12-3), Iodoacetamide (IAA; Cat No I1149) and 3.0 kDa Spin Concentrators (Cat No UFC800308) were purchase from Sigma (St. Louis, MO, USA). 1x Phosphate Buffered Saline (PBS; Cat No 10010-023), 2-Mercaptoethanol (BME; Cat No BP176-100) and Slide-A-Lyzer Dialysis Cassettes (Cat No 66330) were obtained from Thermo Fisher (Waltham, MA, USA); Tris buffered saline (TBS; Cat No 351-086-131) from Quality Biological (Gaithersburg, MD, USA); and KPL SureBlue TMB (3, 3’, 5, 5’—Tetramethylbenzidine) Microwell Peroxidase Substrate (TMB substrate; Cat No 5120-0075) and KPL TMB STOP Solution (stop solution; Cat No 5150-0021) from Seracare (Milford, MA, USA).
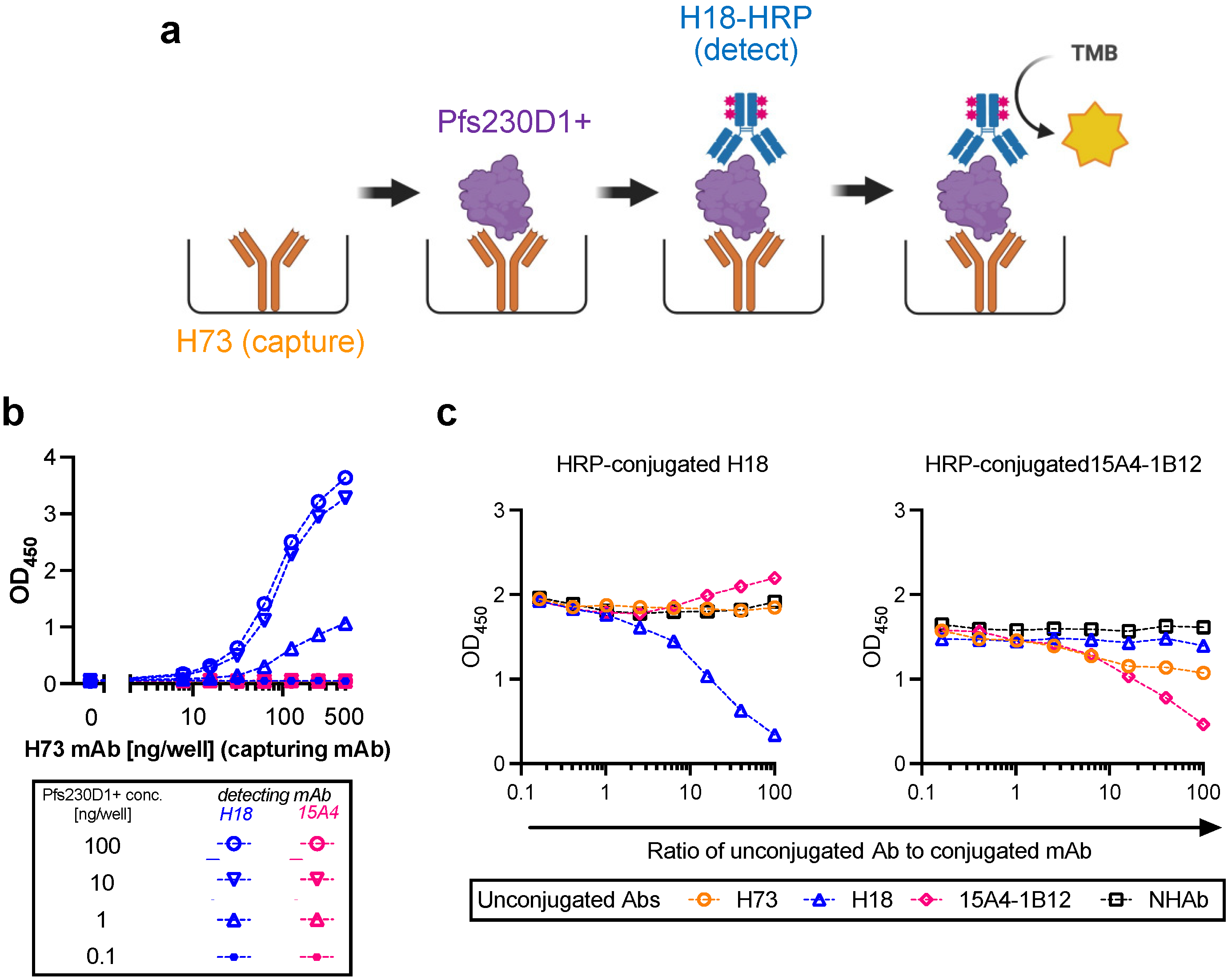
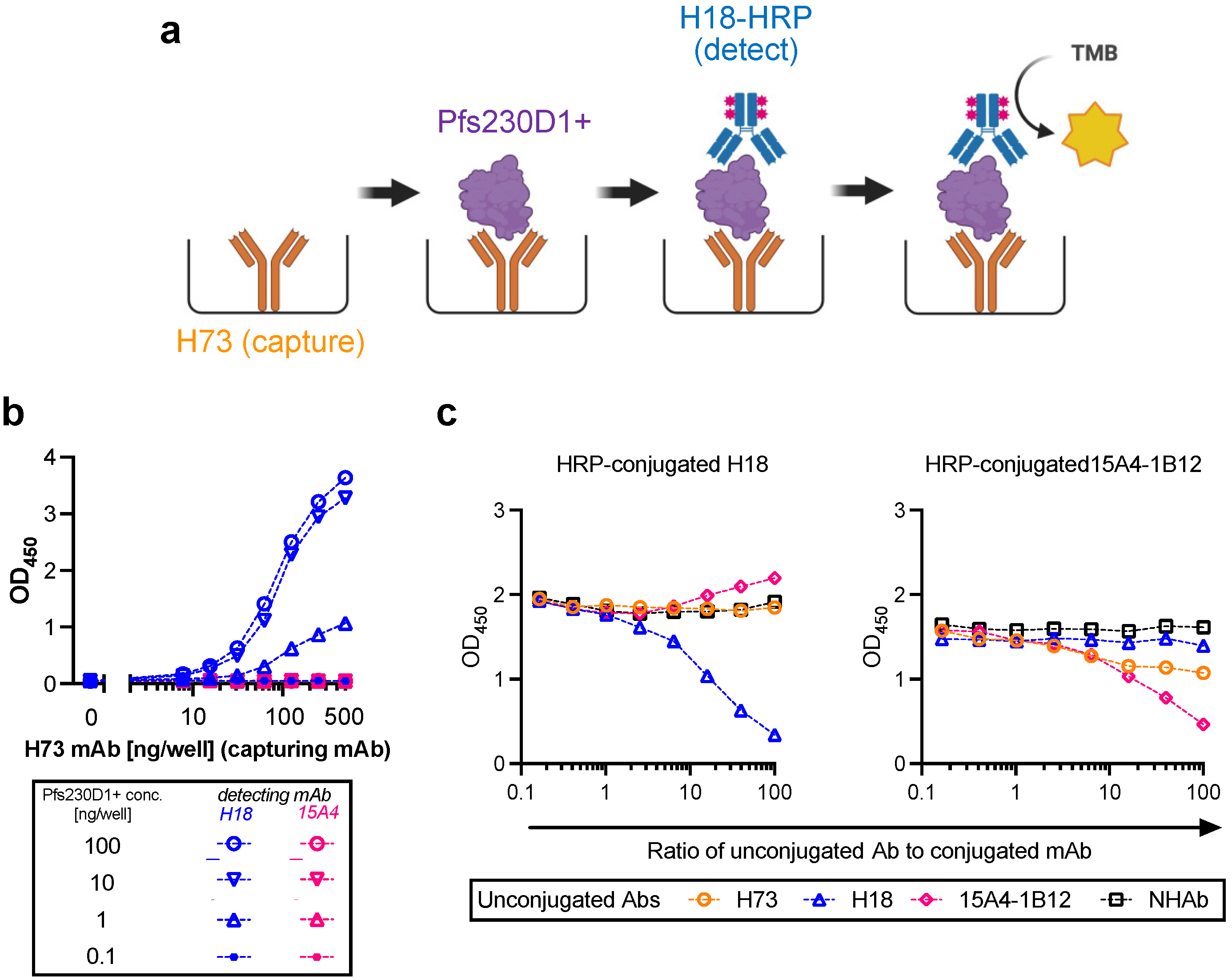
2.2. Antigen Integrity Assay (AIA)
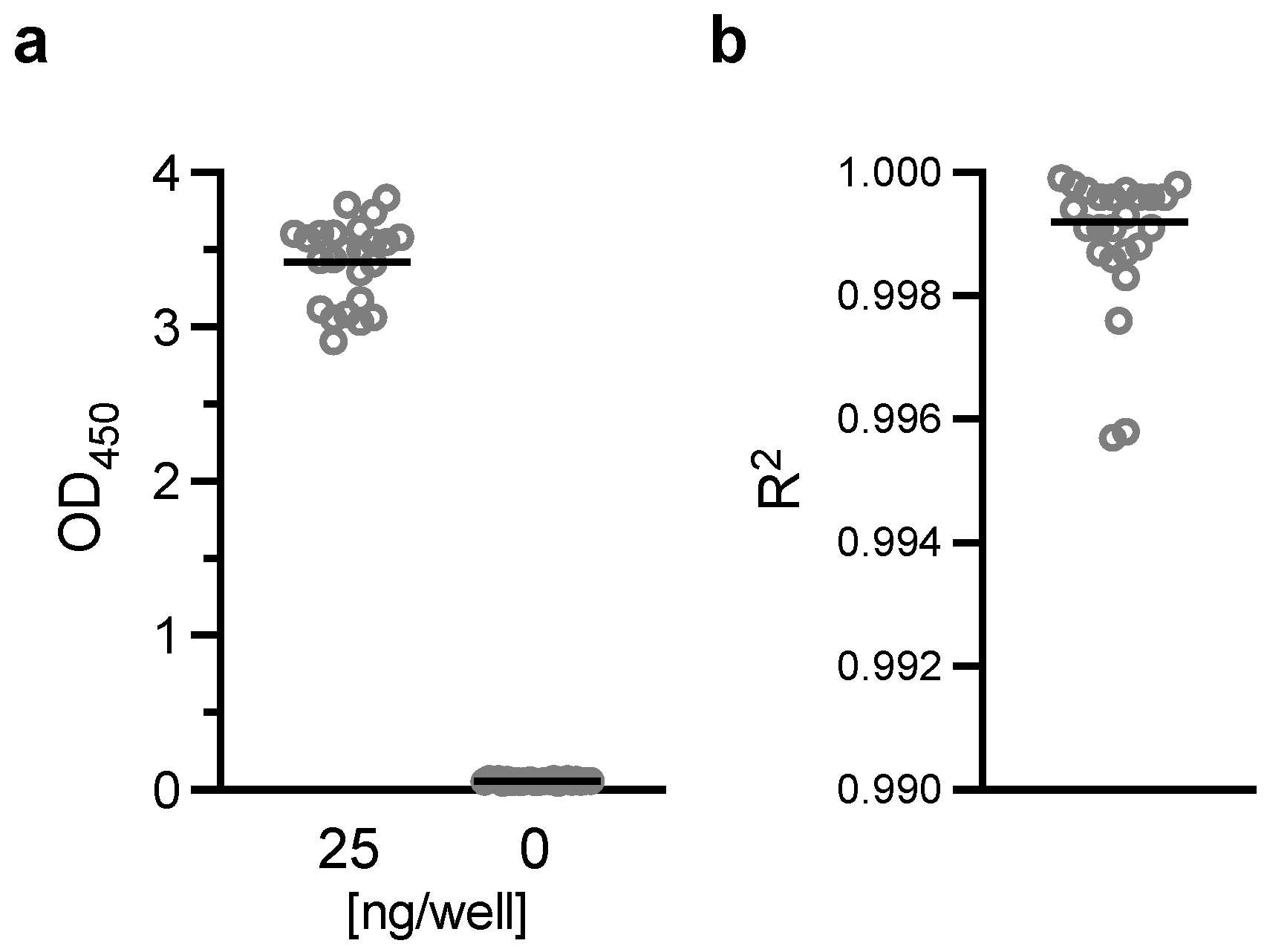
Flat-bottom 96-well ELISA plates (Immulon 4; Thermo Fisher; Cat No 3855) were coated with 200 ng/well of H73 mAb diluted in 1x PBS (pH7.4). The plates were wrapped in plastic wrap and stored at 4 °C overnight. On the next day, H73 mAb solution was discarded, and the plates were blocked with 350 µL/well of blocking/dilution buffer (1xPBS with 1% Casein; BioRad, Hercules, CA, USA; Cat No 1610783) for one hour at room temperature (RT). After extensive washing with washing buffer (0.01% Tween-20 in TBS), an indicated amount of test and reference proteins diluted in blocking/dilution buffer were added to the plates (100 µL/well). Unless otherwise stated, test samples were applied on the ELISA plates at two-fold serial dilutions (a total of six different concentrations) in triplicate wells, and a total of 11 concentrations of two-fold serial dilutions in duplicate wells for the reference protein. In each plate, two wells received only blocking/dilution buffer (blank wells). After 1.5 h of incubation at RT, the plates were washed, and further incubated for 1.5 h at RT with 27.7 ng/well (100 µL/well) of H18-HRP mAb. After washing, 100 µL/well of TMB substrate was added for 10 min at RT in the dark, and the reactions were terminated by adding 100 µL/well of stop solution. Immediately after this addition, the absorbance at 450 nm was read using a VersaMax plate reader (Molecular Devices, Sunnyvale, CA, USA).
2.3. Conversion from Absorbance Values to Protein Concentrations for Test Samples, and Calculation of Pfs230D1+ Ratio
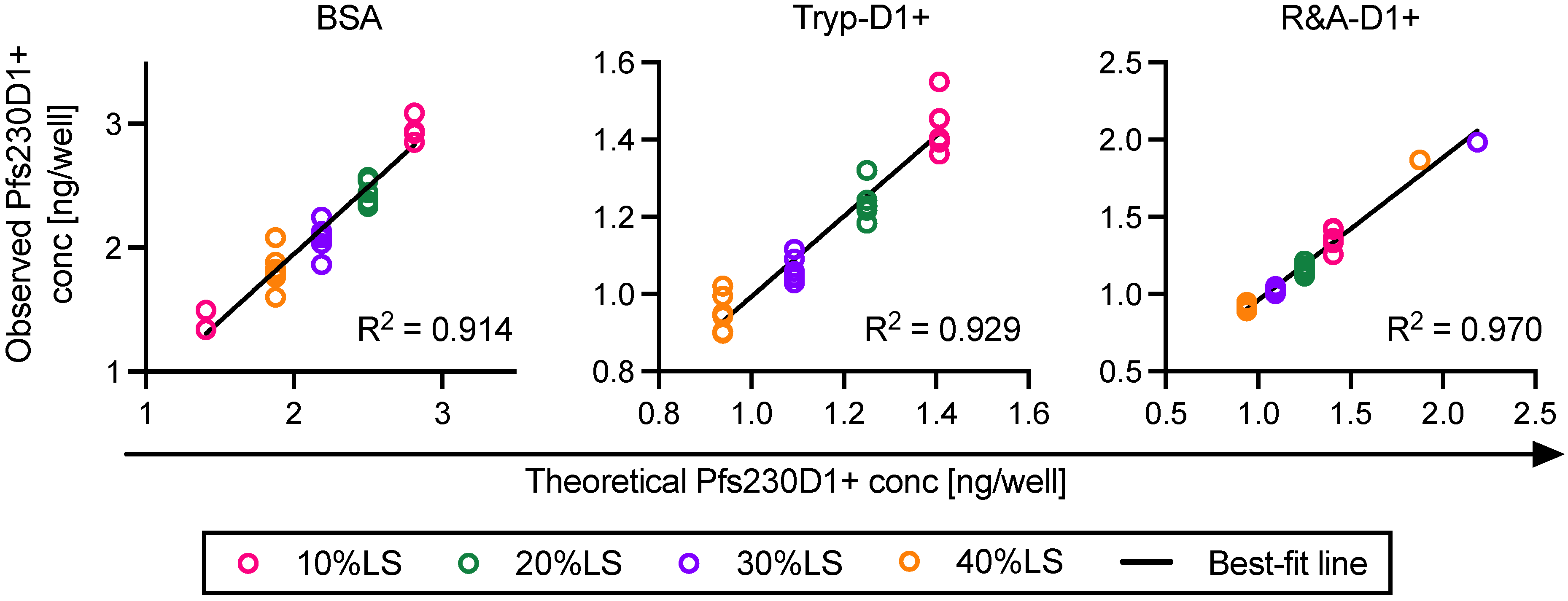
For each ELISA plate, the relation between protein concentration (log-transformed) of reference protein and optical density at 450 nm (OD450) at 11 different dilutions was approximated by a four-parameter sigmoid fit (Prism 9, GraphPad Software, La Jolla, CA, USA) and used as a standard curve. Based on the standard curve, intact Pfs230D1+ concentration in each test well in the same plate was calculated from the OD450 value (denoted as Obs-conc, i.e., observed Pfs230D1+ concentration). A theoretical Pfs230D1+ concentration (Theo-conc) was calculated based on the original intact Pfs230D1+ concentration in the undiluted test sample and the dilution factor. Finally, the Pfs230D1+ Ratio of a test sample at a given dilution was calculated as: Pfs230D1+ Ratio = (Obs − conc)/(Theo − conc).
2.4. Generation of Damaged Pfs230D1+ Proteins
Two types of damaged Pfs230D1+ proteins were generated from the reference protein [
17], one was trypsin-treated Pfs230D1+ (Tryp-D1+) and the other was reduced-and-alkylated Pfs230D1+ (R&A-D1+). Tryp-D1+ was made using Immobilized Trypsin (Promega, Madison, WI, USA; Cat No V9012) following the manufacturers’ instruction. The protein concentration of Tryp-D1+ was determined by DS-11 FX+ (DeNovix, Wilmington, DE, USA). The observed concentration of Tryp-D1+ (0.45 mg/mL) was within 5% error from a theoretical concentration (0.47 mg/mL) calculated from the original amount of protein used for the reaction and the final volume of solution. To generate R&A-D1+, 1.49 mL of 1M DDT was mixed with 1 mg of original Pfs230D1+ protein for 30 min at 60 °C, then 41 µL of 1 M IAA was added and incubated for 30 min at 37 °C. The reaction was stopped by adding 0.58 mL of BME. Using Slide-A-Lyzer Dialysis Cassettes (3.5 kDa) and 3.0 kDa Spin Concentrators, the R&A-D1+ sample was buffer exchanged to TBS (pH8.0) and concentrated to the same protein concentration of the original Pfs230D1+ protein (0.67 mg/mL). The protein concentration in the final product was determined by DS-11 FX+.
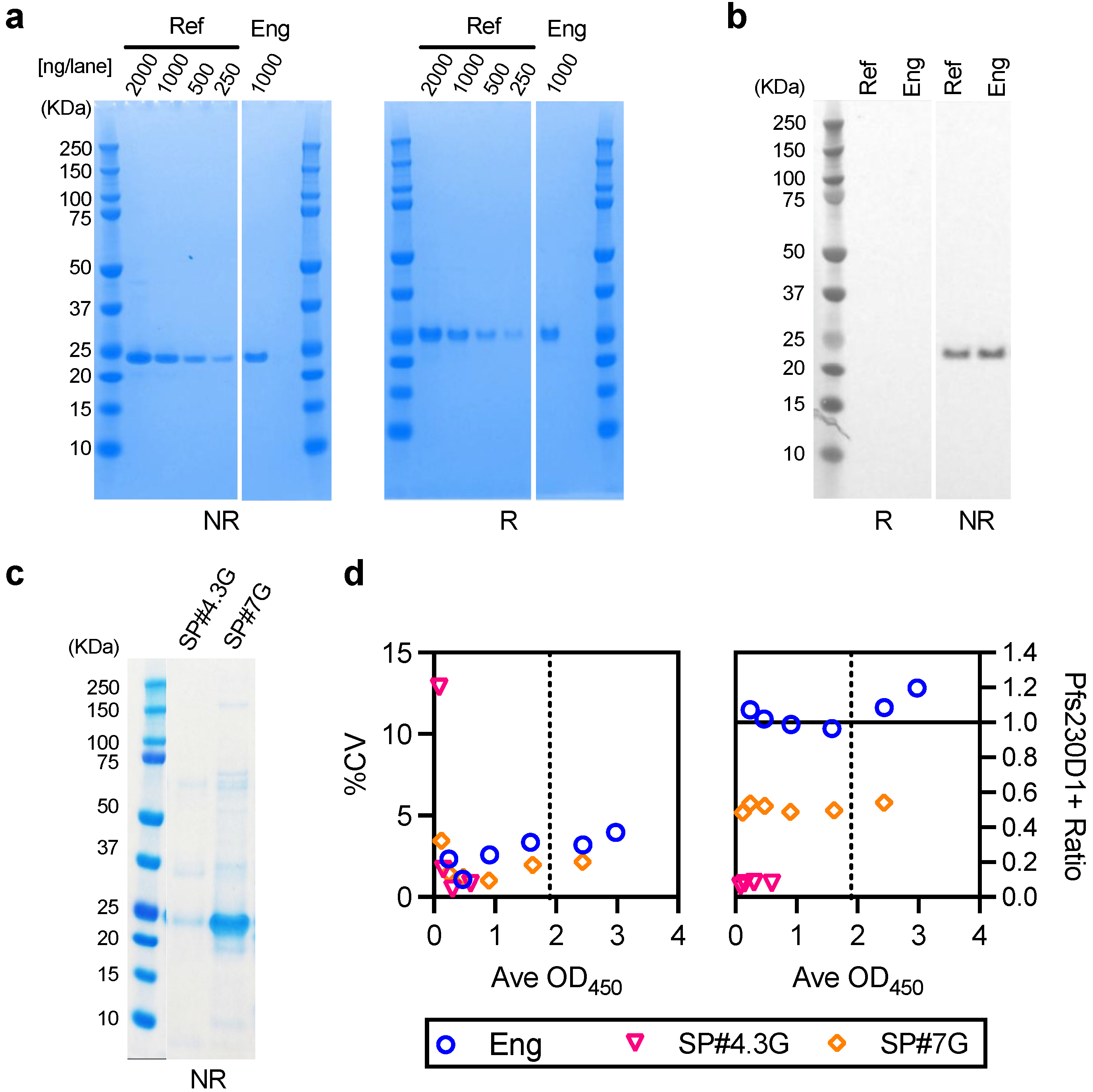
2.5. Generation of an Engineering Lot of Pfs230D1+ Protein
The methods for cloning and construction of Bacmid DNA were identical to those described for the reference Pfs230D1+ protein [
17] with the exception of removal of the histidine tag from the sequence. This Bacmid was sequence verified and used to transfect ExpiSf9 cells (Thermo Fisher, Carlsbad, CA, USA; Cat. No A35243) for the generation of recombinant baculovirus stock using BaculoFECTIN II and animal-free transfection medium Oxford reagent (Oxford Expression Technologies, Oxford, UK; Cat. No 300105) following the Bac-to-Bac manual. The baculovirus was amplified by three passages in the ExpiSf9 cells grown in ESF921 medium (Expression Systems, Davis, CA, USA; Cat. No 96-001-01) and used to infect the ExpiSf9 culture in the bioreactor. The ExpiSf9 cells were seeded at 2 × 10
6 cells/mL in ESF921 medium and MOI (multiplicity of infection) of one was used to infect a pair of 25 L ExpiSf9 wave cultures. At 3–4 days post infection and when the viability of the culture dropped to ~75%, the supernatant was harvested by centrifugation and clarified by 0.22 µm filtration. The clarified supernatant was concentrated and diafiltered using 5 kDa nominal molecular weight cut off membrane in 20 mM Tris buffer, pH 8.0 to achieve a 2.5-fold concentration, and clarified with 0.22 µm filtration. The expressed protein was captured and initially purified through Q-Sepharose FF (GE Healthcare, Uppsala Sweden; code no 17051005) column (8.1 × 9.7 cm). The column was equilibrated, loaded, washed and eluted with the same Tris buffer containing 50, 100, 200 and 1000 mM NaCl in a step gradient. The elution was monitored by UV absorbance, and the collected fractions were monitored by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). The Pfs230D1+ rich fraction (100 mM NaCl) was collected, diluted two-fold and allowed to flow through a MEP HyperCel (Sartorius Stadim, Cergy Saint Christophe, France; P/N 12035) column (5 × 11.9 cm), equilibrated with 50 mM NaCl in 20 mM Tris buffer, pH 8.0 to remove impurities. The Pfs230D1+ was then polished on a Capto DEAE (GE Healthcare; code no 17544310) column (5 × 9.6 cm) through binding and step elution with 40, 50, 60, 80, and 100 mM NaCl in 20 mM Tris buffer, pH 8.0. SDS-PAGE was used to confirm the fractions containing Pfs230D1+ (80mM NaCl). Finally, tangential flow filtration (TFF) was used for salt removal, buffer exchange, and concentration into the final buffer of 20 mM HEPES (4-(2-hydroxyethyl-1-piperazineethanesulfonic acid) buffer,150 mM NaCl, pH 7.4. The Pfs230D1+ at 0.3 mg/mL (as reported by bicinchoninic acid (BCA) assay) was stored frozen at <−60 °C.
2.6. Characterization of the Engineering lot of Pfs230D1+ Protein
Characteristics of the engineering lot of protein were evaluated as described for the reference Pfs230D1+ protein [
17]. Purity was assessed by SDS-PAGE and reverse phase HPLC (RP-HPLC), percent of monomer by Size Exclusion Chromatography HPLC (SEC-HPLC), integrity by western blot using H73 mAb, and protein concentration by BCA assay. The details of methodologies were described previously [
17].
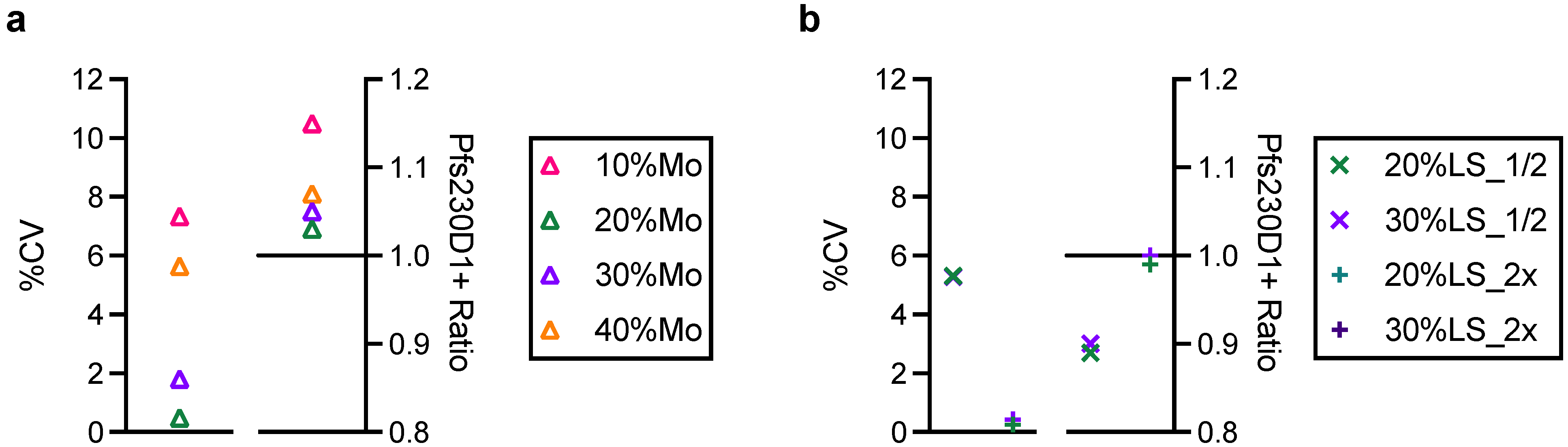
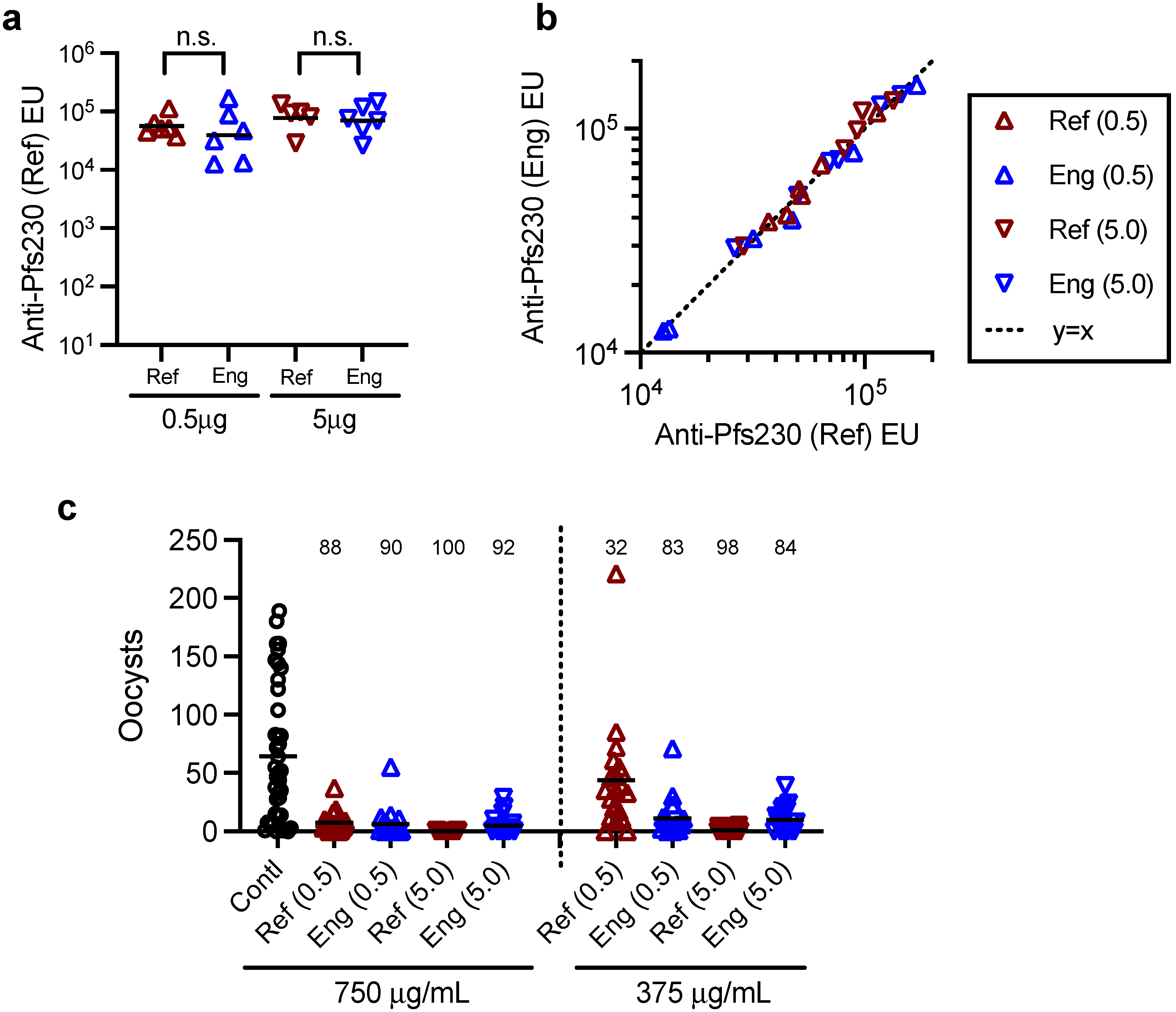
2.7. Mouse Immunization Study with Two Batches of Pfs230D1+ Proteins, ELISA and SMFA
A group of three female and three male CD-1 mice (7–9 weeks old; Charles River, Wilmington, MA, USA) were immunized with 0.5 or 5.0 µg/dose of Pfs230D1+ protein (either reference or engineering lots of proteins) formulated with Montanide ISA720 (Seppic, Fairfield, NJ, USA) on days 0 and 21 by intramuscular injection, and the serum samples were collected on day 35. The antibody titers of the collected serum samples were evaluated by ELISA [
21] using reference or engineering lots of Pfs230D1+ protein as a coating antigen, and functionality of purified IgGs from the sera was assessed by SMFA [
8] as described previously.
2.8. Statistical Analysis
To evaluate specificity and linearity of AIA, the correlation between Obs-conc and Theo-conc for each of three types of samples or for a combination of all three was assessed by a linear regression. For intra-assay precision, percent CV (%CV) of Obs-conc in triplicate wells was calculated. For inter-assay precision, Pfs230D1+ Ratio was utilized. For a normality test, the D’Agostino and Pearson test was used. To compare antibody titers (ELISA units) between two protein groups at the same immunization dose, log-transformed ELISA units were analyzed by a Student’s-
t test. To determine a concordance of log-transformed ELISA units against engineering and reference lots of Pfs230D1+ proteins, the best-fit line was compared with y = x line by F test. For SMFA, % inhibition in oocyst density (%TRA) and
p-values were calculated using a zero-inflated negative binomial (ZINB) model as described [
8].
All statistical tests were performed in Prism 9 or R (version 3.5.3, The R Foundation for Statistical Computing) and p-values < 0.05 are considered significant.
4. Discussion
In this study, using two human functional mAbs, we developed an in vitro potency assay, AIA, against the lead malaria transmission blocking vaccine candidate, Pfs230D1+. As described before, there are multiple reports for the development of in vitro ELISA-based potency assays against other pathogens. However, to the best of our knowledge, this is the first report for malaria vaccines. We qualified the assay following International Conference on Harmonization (ICH) Q2(R1) guidelines [
19], while the majority of other studies did not qualify or validate the assay, with few exceptions [
22]. While a full validation of an assay is not required until a Phase 3 trial, detailed evaluation of the assay, such as precision and limit of detection, is critical to correctly assess whether a new lot or a stressed protein (or vaccine formulation) is truly different from a reference (original) one, or a difference seen in an assay readout is within the error of assay. These results serve as a basis to incorporate the AIA into future release and stability programs related to the Pfs230D1+ protein.
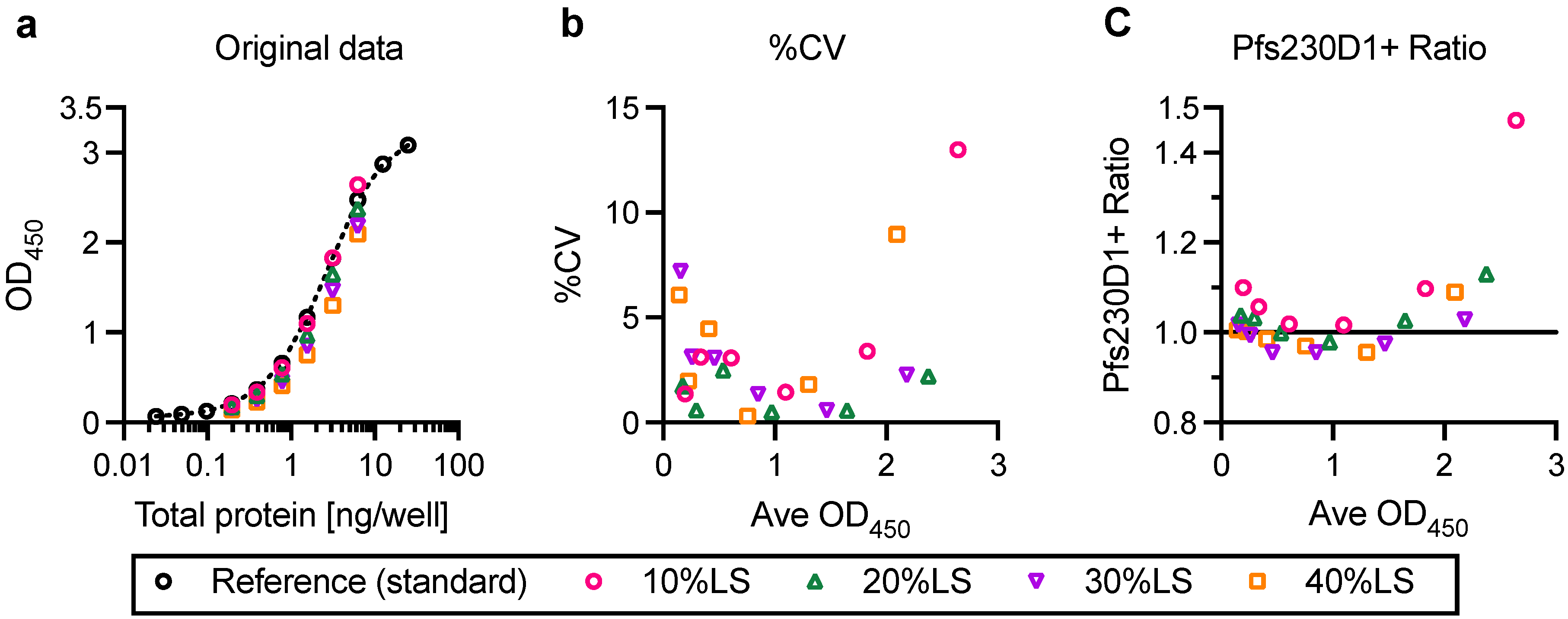
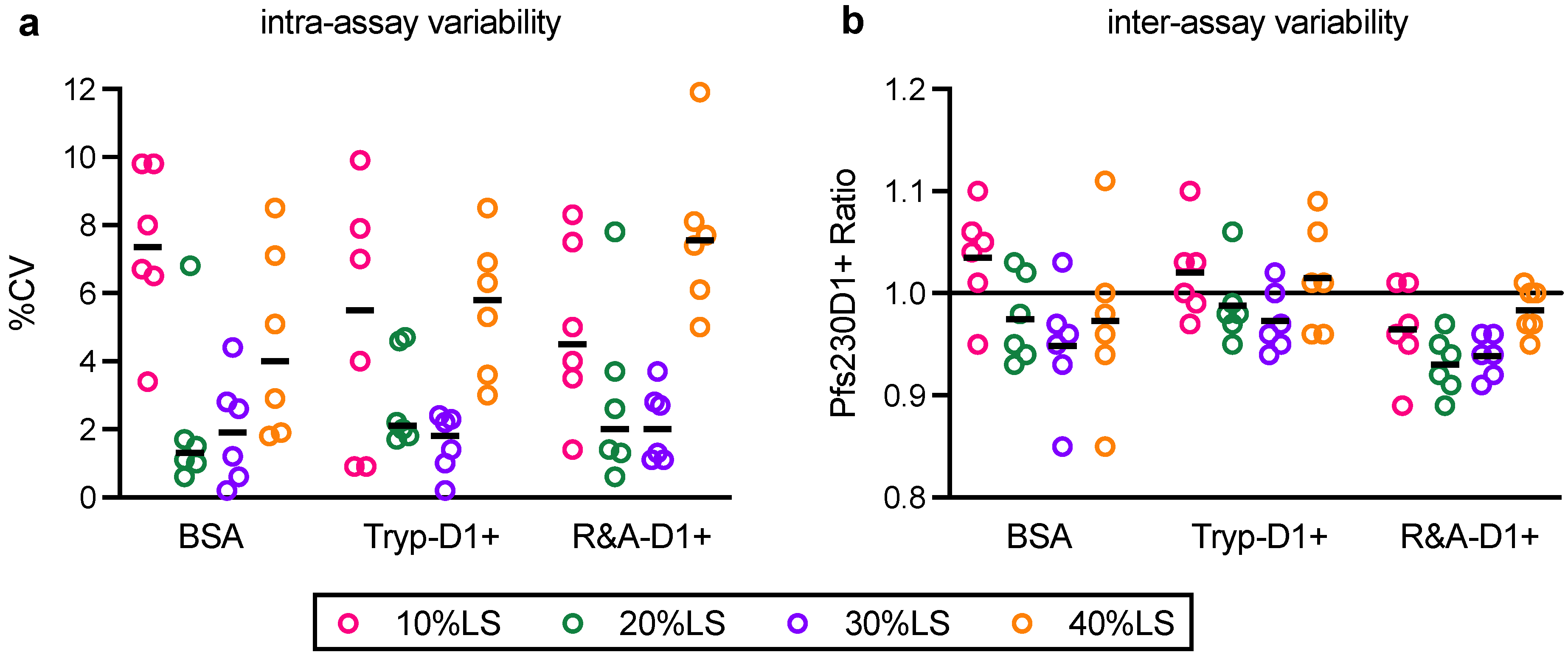
In this study, linearity, specificity, intra- and inter-assay precision, and limit of detection of AIA were evaluated. Based on the Food and Drug Administration (FDA) guidance [
23], intra- and inter-assay precision is recommended to be <20%CV, and AIA passed the criteria (%CV of intra- and inter-assay precision was determined as 3.2 and 5.4, respectively). However, several assay validation parameters were not assessed in this study (
Table 1), such as accuracy and inter-laboratory precision. The accuracy was not evaluated here because there is no field-accepted control reagent or method to determine the “true” value. For inter-laboratory precision, there is no plan to evaluate potency of the specific Pfs230 recombinant protein, which was used in this study, in multiple laboratories. Further qualification or full validation of AIA is anticipated when the Pfs230D1+ vaccine moves to phase 2 and 3 trials. In this study, a recombinant protein (drug substance) was used as a test material, but the final vaccine formulation (drug product) will likely include both the protein and an adjuvant(s) to induce strong and durable responses in humans. Thus, once an adjuvant system is selected for a human clinical trial, the utility of AIA for the drug product will be evaluated.
Practically speaking, in vivo animal immunization studies are unavoidable during novel vaccine development (e.g., testing a new antigen, new vaccine formulation and/or new delivery system). However, using such in vivo assays for quality control, process development, stability testing, or monitoring the lot-to-lot consistency of recombinant protein (or vaccine formulation) is not necessarily the best choice, because in vivo immunization assays can be highly variable and are usually time-consuming. Therefore, for those purposes, an in vitro assay is preferable to an in vivo assay where possible. US and European regulatory agencies have accepted use of ELISA-based in vitro potency assays for hepatitis B and human papillomavirus vaccines [
6].
Once a surrogate in vitro assay is developed, it is ideal to prove the correlation between in vivo and in vitro potency readouts. However, the poor precision of in vivo assays makes this difficult. When an in vitro potency assay was tested for a human papillomavirus vaccine, there was a linear correlation between in vivo and in vitro assay readouts with the correlation coefficient value of 0.75 [
11]. However, the study involved a severely damaged vaccine (the vaccine formulation was treated with DTT and it resulted in >10-fold difference in median in vivo potency between intact and damaged vaccine groups) and the analysis was done using data from >3600 mice (80 mice per assay, and >45 independent in vivo studies). In another study for respiratory syncytial virus vaccine, where a smaller number of animals (
n = 10 mice per group) was evaluated, the correlation between in vivo and in vitro readouts was weak (the correlation coefficient value or
p-value was not reported in the manuscript) [
16]. In this study, we performed an in vivo animal immunization study to test whether the Eng protein could induce functional antibodies in mice, as seen with the reference protein. Our intention was not to check whether the two proteins were equivalent in functional immunogenicity, or whether there was a correlation between in vitro and in vivo potency assay readouts. As shown in this study, the difference in geometric mean ELISA units between 0.5 and 5.0 µg dose groups of the same protein was smaller than the animal-to-animal variation within the group. In other words, the in vivo study design did not allow detection of a 10-fold difference in “intact” protein concentration. A huge study with a severely damaged protein is likely to be required if one wants to prove the correlation between the two assay readouts.
To assess the utility of AIA as a stability test, reference Pfs230D1+ protein was incubated at 37 °C for 7 days (37C-7D-D1+) and evaluated by AIA. The 37C-7D-D1+ protein was mixed with intact protein, and tested at 10, 20, 30 and 40%LS conditions, as tested with two types of damaged proteins (Tryp-D1+ and R&A-D1+). Surprisingly, all 10, 20, 30 and 40%LS samples with 37C-7D-D1+ demonstrated the same OD
450 values as reference protein alone (standard) at the same total protein concentration in AIA (
Figure S4), indicating no change after the incubation. Subsequent SDS-PAGE and western blot analysis also showed that there was no detectable difference between the original and 37C-7D-D1+ proteins (
Figure S1). While the results were encouraging for future TBV development using this recombinant protein, a further study is required before applying the AIA as a part of stability tests.
One of the limitations of this study was assay qualification was performed only up to 40% mixture of imperfect samples. Therefore, the AIA was not evaluated whether the linearity holds true at all ranges of impurity. For example, Pfs230D1+ Ratio of SP#4.3G and SP#7G were ~0.1 and ~0.5, respectively, but we cannot conclude that those samples contained ~10% and ~50% of intact Pfs230D1+ protein. Again, the expected usage of AIA is to prove the non-inferiority of integrity in a test protein compared to a reference protein, thus we believe our approach was sufficient. If a protein is severely damaged (i.e., >40% of protein is damaged), such change will be easily detected by routine SDS-PAGE, western blot or other typical protein QC analyses. On the other hand, if this AIA is to be used to determine a concentration of intact protein in a test material (at all ranges), then additional assay qualification is required. A second limitation here is that in theory, this assay only evaluates integrity of two different functional epitopes detected by the two mAbs, while it is likely that the Pfs230D1+ protein (and other Pfs230-based current vaccine candidates) contains more than two functional epitopes. As generation of human mAbs from vaccinated volunteers from phase 1 trials (or humanized mice) is becoming popular, once new functional, non-competing, mAbs are identified, such mAbs can be quickly applied to the AIA. A single operator can easily test up to 16 different protein samples in two days (including time of coating ELISA plates). Thus, even if the AIA needs to be done with 5–10 different combinations of mAbs, compared to the traditional in vivo potency assay, AIA can provide much higher throughput with excellent sensitivity and precision as described here.
Finally, the AIA could be incorporated into “point of use” stability studies, where the Pfs230 integrity (and presentation of pertinent epitopes) is monitored in the presence of adjuvants and used to support clinical pharmacy manipulations. Given sequence similarity of Pfs230D1+ to other Pfs230 vaccine candidates, such as Pfs230D1M [
24], the assay could directly be applied. General methodologies and approach to the assay validation described herein are amenable to any subunit vaccine as long as more than two functional, non-competing, mAbs are available. Thus, this study strongly supports future subunit vaccine development and use of in vitro assays for release and stability of recombinant vaccine candidates.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}