
Disruption of the NKG2A:HLA-E Immune Checkpoint Axis to Enhance NK Cell Activation against Cancer


Abstract
:1. Introduction
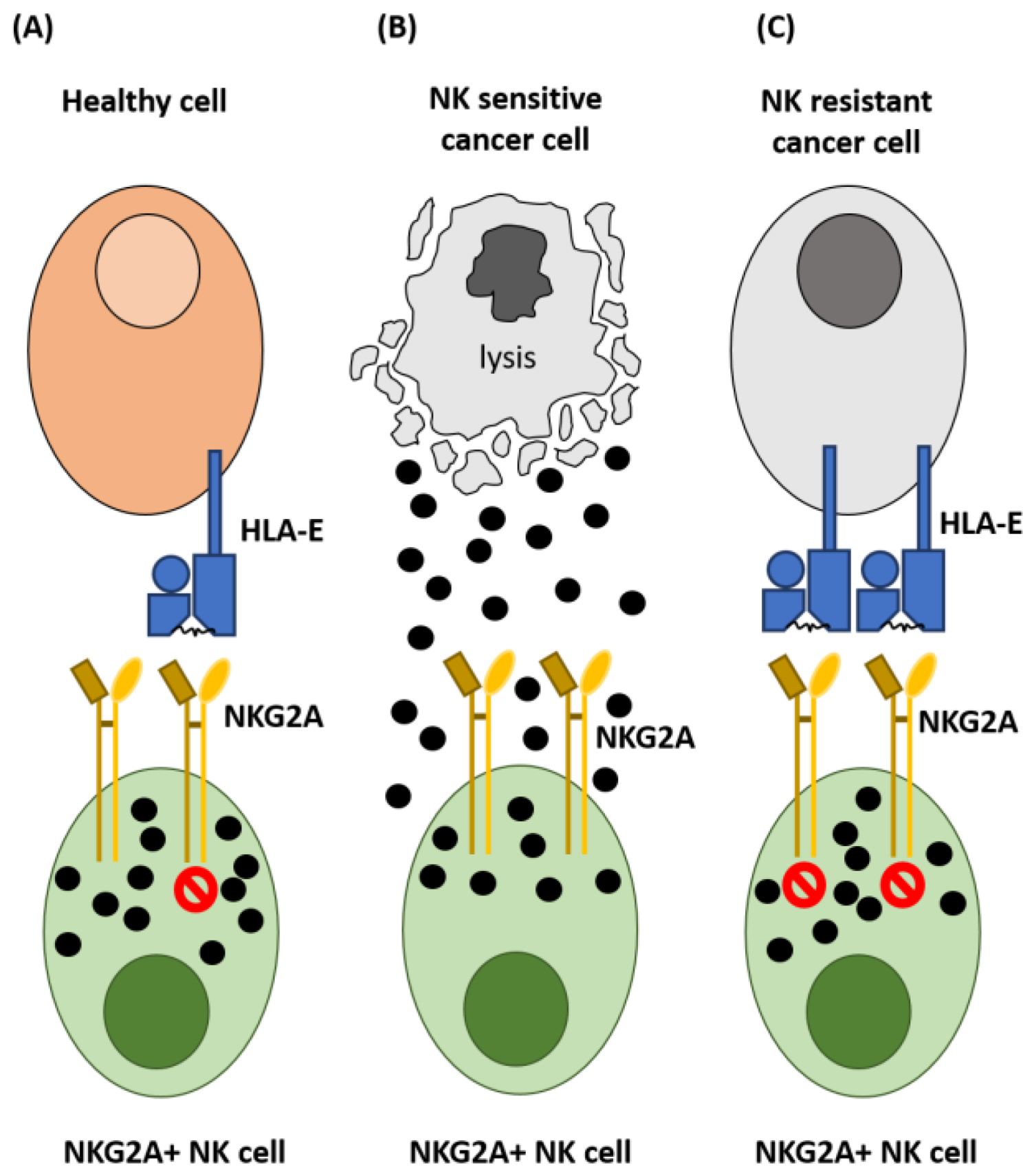
2. NKG2A and Control of NK Cell Activation
3. Expression of HLA-E in Cancer
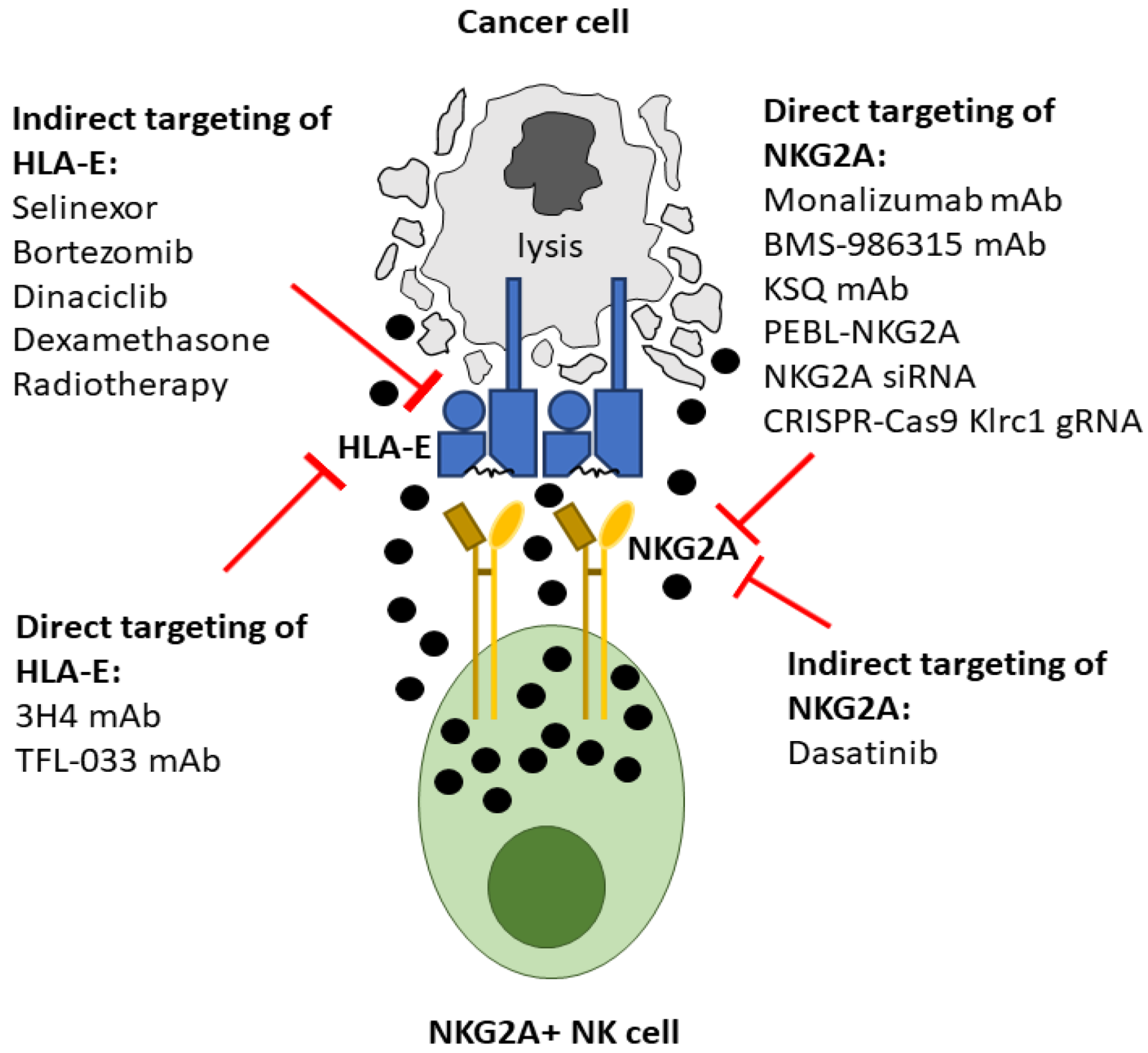
4. Direct Inhibition of the NKG2A:HLA-E Interaction
5. Antibody Mediated Targeting of NKG2A
6. NKG2A Protein Expression Blockers (PEBL)
7. NKG2A Small Interfering RNA
8. CRISPR-Cas9 Gene Targeting of NKG2A
9. Monoclonal Antibodies Targeting HLA-E

{kind=link}
{kind=link}
| Therapeutic | Mechanism for Disruption of NKG2A:HLA-E Interactions | Clinical Stage | References |
|---|---|---|---|
| Monalizumab | Binds to NKG2A | Phase 3—HNSCC, NSCLC | [62] |
| BMS-986315 | Binds to NKG2A | Pre-clinical | [71] |
| KSQ | Binds to NKG2A | Pre-clinical | [72] |
| 3H4 | Binds to HLA-E-VL9 complexes | Pre-clinical | [84] |
| TFL-033 | Binds to HLA-E | Pre-clinical | [83] |
| NKG2A PEBL | Inhibits NKG2A expression by retaining NKG2A in the ER | Pre-clinical | [25] |
| NKG2A siRNA | Inhibits NKG2A expression by degrading NKG2A mRNA | Pre-clinical | [74,75] |
| CRISPR-Cas9 Klrc1 gRNA | Targets Klrc1 gene to downregulate NKG2A expression | Pre-clinical | [79,80,81] |
10. Indirect Mechanisms for Inhibiting the NKG2A:HLA-E Interaction
11. Selective Inhibition of Nuclear Export
12. Proteasome Inhibition
13. Cyclin-Dependent Kinase Inhibition
14. Dexamethasone
15. Tyrosine Kinase Inhibitors (TKI)
16. Radiotherapy
| Therapeutic | Direct Anti-Cancer Mechanism of Action | Mechanism for Disruption of NKG2A:HLA-E Interactions | Clinical Stage | References |
|---|---|---|---|---|
| Selinexor | XPO1 inhibitor | Downregulation of HLA-E | Approved MM, DLBCL | [96] |
| Bortezomib | Proteasome inhibitor | Downregulation of HLA-E | Approved MM, MCL | [98] |
| Dinaciclib | Pan-CDK inhibitor | Downregulation of HLA-E | Phase 2—multiple cancers | [124] |
| Dasatinib | Tyrosine kinase inhibitor | Downregulation of NKG2A | Approved—CML, ALL | [137] |
| Dexamethasone | Activation of the glucocorticoid response element, upregulation of pro-apoptotic gene expression and repression of NF-κB | Downregulation of HLA-E | Approved—multiple cancers | [24] |
| Radiotherapy | DNA damage | Downregulation of HLA-E/ | Approved—multiple cancers | [142] |
| Upregulation of HLA-E | [44] |
17. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; López-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA 1998, 95, 5199–5204. [Google Scholar] [CrossRef] [Green Version]
- Cazzetta, V.; Bruni, E.; Terzoli, S.; Carenza, C.; Franzese, S.; Piazza, R.; Marzano, P.; Donadon, M.; Torzilli, G.; Cimino, M.; et al. NKG2A expression identifies a subset of human Vδ2 T cells exerting the highest antitumor effector functions. Cell Rep. 2021, 37, 109871. [Google Scholar] [CrossRef]
- Borst, L.; Sluijter, M.; Sturm, G.; Charoentong, P.; Santegoets, S.J.; Gulijk, M.; Elsas, M.J.; Groeneveldt, C.; Montfoort, N.; Finotello, F.; et al. NKG2A is a late immune checkpoint on CD8 T cells and marks repeated stimulation and cell division. Int. J. Cancer 2021, 150, 688–704. [Google Scholar] [CrossRef]
- Huntington, N.D.; Cursons, J.; Rautela, J. The cancer–natural killer cell immunity cycle. Nat. Rev. Cancer 2020, 20, 437–454. [Google Scholar] [CrossRef]
- Klanova, M.; Oestergaard, M.Z.; Trněný, M.; Hiddemann, W.; Marcus, R.; Sehn, L.H.; Vitolo, U.; Bazeos, A.; Goede, V.; Zeuner, H.; et al. Prognostic Impact of Natural Killer Cell Count in Follicular Lymphoma and Diffuse Large B-cell Lymphoma Patients Treated with Immunochemotherapy. Clin. Cancer Res. 2019, 25, 4634–4643. [Google Scholar] [CrossRef] [Green Version]
- Marshall, M.J.E.; Stopforth, R.J.; Cragg, M.S. Therapeutic Antibodies: What Have We Learnt from Targeting CD20 and Where Are We Going? Front. Immunol. 2017, 8, 1245. [Google Scholar] [CrossRef] [Green Version]
- Khlifi, H.M.; Guia, S.; Vivier, E.; Narni-Mancinelli, E. Role of the ITAM-Bearing Receptors Expressed by Natural Killer Cells in Cancer. Front. Immunol. 2022, 13, 2256. [Google Scholar] [CrossRef]
- Blunt, M.D.; Khakoo, S.I. Activating killer cell immunoglobulin-like receptors: Detection, function and therapeutic use. Int. J. Immunogenet. 2020, 47, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Manser, A.R.; Uhrberg, M. Age-related changes in natural killer cell repertoires: Impact on NK cell function and immune surveillance. Cancer Immunol. Immunother. 2015, 65, 417–426. [Google Scholar] [CrossRef]
- Masilamani, M.; Nguyen, C.; Kabat, J.; Borrego, F.; Coligan, J.E. CD94/NKG2A Inhibits NK Cell Activation by Disrupting the Actin Network at the Immunological Synapse. J. Immunol. 2006, 177, 3590–3596. [Google Scholar] [CrossRef] [Green Version]
- Le Dréan, E.; Vely, F.; Olcese, L.; Cambiaggi, A.; Guia, S.; Krystal, G.; Gervois, N.; Moretta, A.; Jotereau, F.; Vivier, E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: Association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur. J. Immunol. 1998, 28, 264–276. [Google Scholar] [CrossRef]
- Rhee, I.; Veillette, A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat. Immunol. 2012, 13, 439–447. [Google Scholar] [CrossRef]
- Stebbins, C.C.; Watzl, C.; Billadeau, D.D.; Leibson, P.J.; Burshtyn, D.N.; Long, E.O. Vav1 Dephosphorylation by the Tyrosine Phosphatase SHP-1 as a Mechanism for Inhibition of Cellular Cytotoxicity. Mol. Cell. Biol. 2003, 23, 6291–6299. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, S.A.; Cheent, K.S.; Khakoo, S. Effects of Peptide on NK Cell-Mediated MHC I Recognition. Front. Immunol. 2014, 5, 133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Feng, J.; Chen, S.; Yang, H.; Dong, Z. Synergized regulation of NK cell education by NKG2A and specific Ly49 family members. Nat. Commun. 2019, 10, 5010–5012. [Google Scholar] [CrossRef] [Green Version]
- Van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; Van Der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Bertone, S.; Schiavetti, F.; Bellomo, R.; Vitale, C.; Ponte, M.; Moretta, L.; Mingari, M.C. Transforming growth factor-β-induced expression of CD94/NKG2A inhibitory receptors in human T lymphocytes. Eur. J. Immunol. 1999, 29, 23–29. [Google Scholar] [CrossRef]
- Sheu, B.C.; Chiou, S.H.; Lin, H.H.; Chow, S.N.; Huang, S.C.; Ho, H.N.; Hsu, S.M. Up-regulation of inhibitory natural killer receptors CD94/NKG2A with suppressed intracellular perforin expression of tumor-infiltrating CD8 + T lymphocytes in human cervical carcinoma. Cancer Res. 2005, 65, 2921–2929. [Google Scholar] [CrossRef] [Green Version]
- Parry, H.M.; Stevens, T.; Oldreive, C.; Zadran, B.; McSkeane, T.; Rudzki, Z.; Paneesha, S.; Chadwick, C.; Stankovic, T.; Pratt, G.; et al. NK cell function is markedly impaired in patients with chronic lymphocytic leukaemia but is preserved in patients with small lymphocytic lymphoma. Oncotarget 2016, 7, 68513–68526. [Google Scholar] [CrossRef]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1226720. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, A.W.; Jillab, M.; Smith, M.R.; Alpaugh, R.K.; Cole, M.E.; Litwin, S.; Millenson, M.M.; Al-Saleem, T.; Cohen, A.D.; Campbell, K.S. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology 2017, 6, e1330235. [Google Scholar] [CrossRef] [Green Version]
- Cooley, S.; McCullar, V.; Wangen, R.; Bergemann, T.; Spellman, S.; Weisdorf, D.J.; Miller, J.S. KIR reconstitution is altered by T cells in the graft and correlates with clinical outcomes after unrelated donor transplantation. Blood 2005, 106, 4370–4376. [Google Scholar] [CrossRef] [Green Version]
- Thangaraj, J.L.; Ahn, S.-Y.; Jung, S.-H.; Vo, M.-C.; Chu, T.-H.; Phan, M.-T.T.; Kwon, M.; Lee, K.-H.; Kim, M.; Song, G.-Y.; et al. Expanded natural killer cells augment the antimyeloma effect of daratumumab, bortezomib, and dexamethasone in a mouse model. Cell. Mol. Immunol. 2021, 18, 1652–1661. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. OncoImmunology 2016, 6, e1264562. [Google Scholar] [CrossRef] [Green Version]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van, Ham, V.J.; Ehsan, I.; Charoentong, P.; Ander, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755.e15. [Google Scholar] [CrossRef] [Green Version]
- Sandoval-Borrego, D.; Moreno-Lafont, M.C.; Vazquez-Sanchez, E.A.; Gutierrez-Hoya, A.; López-Santiago, R.; Montiel-Cervantes, L.A.; Ramírez-Saldaña, M.; Vela-Ojeda, J. Overexpression of CD158 and NKG2A Inhibitory Receptors and Underexpression of NKG2D and NKp46 Activating Receptors on NK Cells in Acute Myeloid Leukemia. Arch. Med. Res. 2016, 47, 55–64. [Google Scholar] [CrossRef]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Azoulay, T.; Slouzky, I.; Karmona, M.; Filatov, M.; Hayun, M.; Ofran, Y.; Sarig, G.; Ringelstein-Harlev, S. Compromised activity of natural killer cells in diffuse large b-cell lymphoma is related to lymphoma-induced modification of their surface receptor expression. Cancer Immunol. Immunother. 2022, 1–12. [Google Scholar] [CrossRef]
- Orr, M.T.; Wu, J.; Fang, M.; Sigal, L.J.; Spee, P.; Egebjerg, T.; Dissen, E.; Fossum, S.; Phillips, J.H.; Lanier, L.L. Development and Function of CD94-Deficient Natural Killer Cells. PLoS ONE 2010, 5, e15184. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Goodlett, D.R.; Ishitani, A.; Marquardt, H.; Geraghty, D.E. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J. Immunol. 1998, 160, 4951–4960. [Google Scholar]
- Petrie, E.; Clements, C.S.; Lin, J.; Sullivan, L.; Johnson, D.; Huyton, T.; Heroux, A.; Hoare, H.L.; Beddoe, T.; Reid, H.H.; et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J. Exp. Med. 2008, 205, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, A.B.; Carretero, J.; Aptsiauri, N.; Garrido, F. Targeting HLA class I expression to increase tumor immunogenicity. Tissue Antigens 2012, 79, 147–154. [Google Scholar] [CrossRef]
- Raulet, D.H.; Guerra, N. Oncogenic stress sensed by the immune system: Role of natural killer cell receptors. Nat. Rev. Immunol. 2009, 9, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Palmisano, G.L.; Contardi, E.; Morabito, A.; Gargaglione, V.; Ferrara, G.B.; Pistillo, M.P. HLA-E surface expression is independent of the availability of HLA class I signal sequence-derived peptides in human tumor cell lines. Hum. Immunol. 2005, 66, 1–12. [Google Scholar] [CrossRef]
- de Kruijf, E.M.; Sajet, A.; van Nes, J.G.; Natanov, R.; Putter, H.; Smit, V.T.; Liefers, J.G.; van den Elsen, P.J.; van de Velde, C.J.H.; Kuppen, P.J.K. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef] [Green Version]
- Hannoun, Z.; Lin, Z.; Brackenridge, S.; Kuse, N.; Akahoshi, T.; Borthwick, N.; McMichael, A.; Murakoshi, H.; Takiguchi, M.; Hanke, T. Identification of novel HIV-1-derived HLA-E-binding peptides. Immunol. Lett. 2018, 202, 65–72. [Google Scholar] [CrossRef]
- Cheent, K.S.; Jamil, K.M.; Cassidy, S.; Liu, M.; Mbiribindi, B.; Mulder, A.; Claas, F.H.J.; Purbhoo, M.A.; Khakoo, S.I. Synergistic inhibition of natural killer cells by the nonsignaling molecule CD94. Proc. Natl. Acad. Sci. USA 2013, 110, 16981–16986. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.G.; Wu, H.L.; Burwitz, B.J.; Hughes, C.M.; Hammond, K.B.; Ventura, A.B.; Reed, J.S.; Gilbride, R.M.; Ainslie, E.; Morrow, D.W.; et al. Broadly targeted CD8+ T cell responses restricted by major histocompatibility complex E. Science 2016, 351, 714–720. [Google Scholar] [CrossRef]
- Battin, C.; Kaufmann, G.; Leitner, J.; Tobias, J.; Wiedermann, U.; Rölle, A.; Meyer, M.; Momburg, F.; Steinberger, P. NKG2A -checkpoint inhibition and its blockade critically depends on peptides presented by its ligand HLA-E. Immunology 2022, 166, 507–521. [Google Scholar] [CrossRef]
- Morinaga, T.; Iwatsuki, M.; Yamashita, K.; Matsumoto, C.; Harada, K.; Kurashige, J.; Iwagami, S.; Baba, Y.; Yoshida, N.; Komohara, Y.; et al. Evaluation of HLA-E Expression Combined with Natural Killer Cell Status as a Prognostic Factor for Advanced Gastric Cancer. Ann. Surg. Oncol. 2022, 29, 4951–4960. [Google Scholar] [CrossRef]
- Hrbac, T.; Kopkova, A.; Siegl, F.; Vecera, M.; Ruckova, M.; Kazda, T.; Jancalek, R.; Hendrych, M.; Hermanova, M.; Vybihal, V.; et al. HLA-E and HLA-F Are Overexpressed in Glioblastoma and HLA-E Increased After Exposure to Ionizing Radiation. Cancer Genom.—Proteom. 2022, 19, 151–162. [Google Scholar] [CrossRef]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Lagana, A.; Ruan, D.F.; Melnekoff, D.; Leshchenko, V.; Perumal, D.; Rahman, A.; Kim-Schultze, S.; Song, Y.; Keats, J.J.; Yesil, J.; et al. Increased HLA-E Expression Correlates with Early Relapse in Multiple Myeloma. Blood 2018, 132, 59. [Google Scholar] [CrossRef]
- Hamid, M.A.; Wang, R.Z.; Yao, X.; Fan, P.; Li, X.; Chang, X.M.; Feng, Y.; Jones, S.; Maldonado-Perez, D.; Waugh, C.; et al. Enriched HLA-E and CD94/NKG2A Interaction Limits Antitumor CD8 + Tumor-Infiltrating T Lymphocyte Responses. Cancer Immunol. Res. 2019, 7, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Yazdi, M.T.; van Riet, S.; van Schadewijk, A.; Fiocco, M.; van Hall, T.; Taube, C.; Hiemstra, P.S.; van der Burg, S.H. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget 2015, 7, 3477–3488. [Google Scholar] [CrossRef] [Green Version]
- Gooden, M.; Lampen, M.; Jordanova, E.S.; Leffers, N.; Trimbos, J.B.; van der Burg, S.H.; Nijman, H.; van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10656–10661. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, S.; Dufva, O.; Huuhtanen, J.; Dashevsky, O.; Klievink, J.; Bouhlal, J.; Scheffer, M.; Kankainen, M.; Simoes, R.D.M.; Mitsiades, C.; et al. OAB-019: CRISPR screens with single-cell transcriptome readout reveal potential mechanisms of response to natural killer cell treatment in multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, S12–S13. [Google Scholar] [CrossRef]
- Sheffer, M.; Lowry, E.; Beelen, N.; Borah, M.; Amara, S.N.-A.; Mader, C.C.; Roth, J.A.; Tsherniak, A.; Freeman, S.S.; Dashevsky, O.; et al. Genome-scale screens identify factors regulating tumor cell responses to natural killer cells. Nat. Genet. 2021, 53, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, K.-J.; Levitsky, V.; Norell, H.; De Matos, C.T.; Carlsten, M.; Schedvins, K.; Rabbain, H.; Moretta, A.; Söderström, K.; Levitskaya, J.; et al. IFN-γ protects short-term ovarian carcinoma cell lines from CTL lysis via a CD94/NKG2A-dependent mechanism. J. Clin. Investig. 2002, 110, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Beziat, V.; Dhedin, N.; Kuentz, M.; Vernant, J.P.; Debre, P.; Vieillard, V. HLA-E upregulation on IFN-gamma-activated AML blasts impairs CD94/NKG2A-dependent NK cytolysis after haplo-mismatched hematopoietic SCT. Bone Marrow Transplant. 2009, 43, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoerster, K.; Uhrberg, M.; Wiek, C.; Horn, P.A.; Hanenberg, H.; Heinrichs, S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for “Off-the-Shelf” Immunotherapy. Front. Immunol. 2021, 11, 586168. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, B.; Wu, Z.; Bo, J.; Tong, C.; Chen, D.; Wang, J.; Wang, H.; Wang, Y.; Han, W. Mutant B2M-HLA-E and B2M-HLA-G fusion proteins protects universal chimeric antigen receptor-modified T cells from allogeneic NK cell-mediated lysis. Eur. J. Immunol. 2021, 51, 2513–2521. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.B.; Ha, S.-J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 1074. [Google Scholar] [CrossRef]
- Sun, H.; Sun, C. The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front. Immunol. 2019, 10, 2354. [Google Scholar] [CrossRef] [Green Version]
- Borrego, F.; Ulbrecht, M.; Weiss, E.H.; Coligan, J.E.; Brooks, A.G. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J. Exp. Med. 1998, 187, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Tognarelli, S.; Wirsching, S.; Von Metzler, I.; Rais, B.; Jacobs, B.; Serve, H.; Bader, P.; Ullrich, E. Enhancing the Activation and Releasing the Brakes: A Double Hit Strategy to Improve NK Cell Cytotoxicity Against Multiple Myeloma. Front. Immunol. 2018, 9, 2743. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinker, A.V.; Hirte, H.W.; Provencher, D.; Butler, M.; Ritter, H.; Tu, D.; Azim, H.A.; Paralejas, P.; Grenier, N.; Hahn, S.-A.; et al. Dose-Ranging and Cohort-Expansion Study of Monalizumab (IPH2201) in Patients with Advanced Gynecologic Malignancies: A Trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res. 2019, 25, 6052–6060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galot, R.; Le Tourneau, C.; Saada-Bouzid, E.; Daste, A.; Even, C.; Debruyne, P.; Henry, S.; Zanetta, S.; Rutten, A.; Licitra, L.; et al. A phase II study of monalizumab in patients with recurrent/metastatic squamous cell carcinoma of the head and neck: The I1 cohort of the EORTC-HNCG-1559 UPSTREAM trial. Eur. J. Cancer 2021, 158, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Majem, M.; Barlesi, F.; Carcereny, E.; Chu, Q.; Monnet, I.; Sanchez-Hernandez, A.; Dakhil, S.; Camidge, D.R.; Winzer, L.; et al. COAST: An Open-Label, Phase II, Multidrug Platform Study of Durvalumab Alone or in Combination With Oleclumab or Monalizumab in Patients With Unresectable, Stage III Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 3383–3393. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Salomé, B.; Sfakianos, J.P.; Ranti, D.; Daza, J.; Bieber, C.; Charap, A.; Hammer, C.; Banchereau, R.; Farkas, A.M.; Ruan, D.F.; et al. NKG2A and HLA-E define an alternative immune checkpoint axis in bladder cancer. Cancer Cell 2022, 40, 1027–1043.e9. [Google Scholar] [CrossRef]
- Ruggeri, L.; Urbani, E.; André, P.; Mancusi, A.; Tosti, A.; Topini, F.; Bléry, M.; Animobono, L.; Romagné, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2015, 101, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xin, Z.; Huang, L.; Zhao, L.; Wang, S.; Cheng, J.; Wu, P.; Chai, Y. CD8+ T Cells Form the Predominant Subset of NKG2A+ Cells in Human Lung Cancer. Front. Immunol. 2020, 10, 3002. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Definition of Anti-NKG2A Monoclonal Antibody BMS-986315—NCI Drug Dictionary—NCI. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/anti-nkg2a-monoclonal-antibody-bms-986315 (accessed on 15 October 2022).
- Spinosa, P.; Musial-Siwek, M.; Presler, M.; Betts, A.; Rosentrater, E.; Villali, J.; Wille, L.; Zhao, Y.; McCaughtry, T.; Subramanian, K.; et al. Quantitative modeling predicts competitive advantages of a next generation anti-NKG2A monoclonal antibody over monalizumab for the treatment of cancer. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Jiang, Q.; Jiang, H.; Hu, L.J.; Zhao, T.; Yu, X.X.; Huang, X.J. Expanded clinical-grade membrane-bound IL-21/4-1BBL NK cell products exhibit activity against acute myeloid leukemia in vivo. Eur. J. Immunol. 2020, 50, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.; Seltsam, A.; Blasczyk, R. Permanent silencing of NKG2A expression for cell-based therapeutics. J. Mol. Med. 2008, 87, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Furutani, E.; Su, S.; Smith, A.; Berg, M.; Childs, R. siRNA Inactivation of the Inhibitory Receptor NKG2A Augments the Anti-Tumor Effects of Adoptively Transferred NK Cells In Tumor-Bearing Hosts. Blood 2010, 116, 1015. [Google Scholar] [CrossRef]
- Biber, G.; Sabag, B.; Raiff, A.; Ben-Shmuel, A.; Puthenveetil, A.; Benichou, J.I.C.; Jubany, T.; Levy, M.; Killner, S.; Barda-Saad, M. Modulation of intrinsic inhibitory checkpoints using nano-carriers to unleash NK cell activity. EMBO Mol. Med. 2021, 14, e14073. [Google Scholar] [CrossRef]
- Wood, H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 2018, 14, 570. [Google Scholar] [CrossRef]
- Ledford, H. Gene-silencing technology gets first drug approval after 20-year wait. Nature. 2018, 560, 291–292. [Google Scholar] [CrossRef]
- Huang, R.-S.; Shih, H.-A.; Lai, M.-C.; Chang, Y.-J.; Lin, S. Enhanced NK-92 Cytotoxicity by CRISPR Genome Engineering Using Cas9 Ribonucleoproteins. Front. Immunol. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Huang, R.-S.; Lai, M.-C.; Shih, H.-A.; Lin, S. A robust platform for expansion and genome editing of primary human natural killer cells. J. Exp. Med. 2021, 218, e20201529. [Google Scholar] [CrossRef]
- Bexte, T.; Alzubi, J.; Reindl, L.M.; Wendel, P.; Schubert, R.; Salzmann-Manrique, E.; von Metzler, I.; Cathomen, T.; Ullrich, E. CRISPR-Cas9 based gene editing of the immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma. OncoImmunology 2022, 11, 2081415. [Google Scholar] [CrossRef]
- Borrego, F.; Kabat, J.; Sanni, T.B.; Coligan, J.E. NK Cell CD94/NKG2A Inhibitory Receptors Are Internalized and Recycle Independently of Inhibitory Signaling Processes. J. Immunol. 2002, 169, 6102–6111. [Google Scholar] [CrossRef]
- Ravindranath, M.H.; Filippone, E.J.; Devarajan, A.; Asgharzadeh, S. Enhancing Natural Killer and CD8+T Cell-Mediated Anticancer Cytotoxicity and Proliferation of CD8+T Cells with HLA-E Monospecific Monoclonal Antibodies. Monoclon. Antibodies Immunodiagn. Immunother. 2019, 38, 38–59. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Brackenridge, S.; Walters, L.C.; Swanson, O.; Harlos, K.; Rozbesky, D.; Cain, D.W.; Wiehe, K.; Scearce, R.M.; Barr, M.; et al. Mouse and human antibodies bind HLA-E-leader peptide complexes and enhance NK cell cytotoxicity. Commun. Biol. 2022, 5, 1–17. [Google Scholar] [CrossRef]
- Braud, V.; Jones, E.Y.; McMichael, A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur. J. Immunol. 1997, 27, 1164–1169. [Google Scholar] [CrossRef]
- Michaëlsson, J.; Teixeira de Matos, C.; Achour, A.; Lanier, L.L.; Kärre, K.; Söderström, K. A signal peptide derived from hsp60 binds HLA-E and interferes with CD94/NKG2A recognition. J. Exp. Med. 2002, 196, 1403–1414. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, T.; Celik, A.A.; Huyton, T.; Kunze-Schumacher, H.; Blasczyk, R.; Bade-Döding, C. HLA-E: Presentation of a Broader Peptide Repertoire Impacts the Cellular Immune Response—Implications on HSCT Outcome. Stem Cells Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-Dependent Cellular Cytotoxicity Activity of a Novel Anti–PD-L1 Antibody Avelumab (MSB0010718C) on Human Tumor Cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-E.; Kim, S.-E.; Keam, B.; Park, H.-R.; Kim, S.; Kim, M.; Kim, T.M.; Doh, J.; Kim, D.-W.; Heo, D.S. Anti-tumor effects of NK cells and anti-PD-L1 antibody with antibody-dependent cellular cytotoxicity in PD-L1-positive cancer cell lines. J. Immunother. Cancer 2020, 8, e000873. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; Wilson, D.; McMichael, A.J. TAP- and tapasin-dependent HLA-E surface expression correlates with the binding of an MHC class I leader peptide. Curr. Biol. 1998, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Brooks, C.R.; Elliott, T.; Parham, P.; Khakoo, S.I. The Inhibitory Receptor NKG2A Determines Lysis of Vaccinia Virus-Infected Autologous Targets by NK Cells. J. Immunol. 2006, 176, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Kojima, K.; Yamamoto, S.; Sekihara, K.; Matsushita, H.; Davis, R.E.; Wang, Z.; Ma, W.; Ishizawa, J.; Kazuno, S.; et al. Ribosomal Biogenesis and Translational Flux Inhibition by the Selective Inhibitor of Nuclear Export (SINE) XPO1 Antagonist KPT-185. PLoS ONE 2015, 10, e0137210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sendino, M.; Omaetxebarria, M.J.; Rodríguez, J.A. Hitting a moving target: Inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer. Cancer Drug Resist. 2018, 1, 139–163. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.Y.; Liu, H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 2019, 6, 6. [Google Scholar] [CrossRef]
- Fisher, J.G.; Walker, C.J.; Doyle, A.D.; Johnson, P.W.; Forconi, F.; Cragg, M.S.; Landesman, Y.; Khakoo, S.I.; Blunt, M.D. Selinexor Enhances NK Cell Activation Against Malignant B Cells via Downregulation of HLA-E. Front. Oncol. 2021, 11, 5088. [Google Scholar] [CrossRef]
- Kambayashi, T.; Kraft-Leavy, J.R.; Dauner, J.G.; Sullivan, B.A.; Laur, O.; Jensen, P.E. The Nonclassical MHC Class I Molecule Qa-1 Forms Unstable Peptide Complexes. J. Immunol. 2004, 172, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Carlsten, M.; Namazi, A.; Reger, R.; Levy, E.; Berg, M.; Hilaire, C.S.; Childs, R.W. Bortezomib sensitizes multiple myeloma to NK cells via ER-stress-induced suppression of HLA-E and upregulation of DR5. OncoImmunology 2018, 8, e1534664. [Google Scholar] [CrossRef] [Green Version]
- Tyler, P.M.; Servos, M.M.; de Vries, R.C.; Klebanov, B.; Kashyap, T.; Sacham, S.; Landesman, Y.; Dougan, M.; Dougan, S.K. Clinical Dosing Regimen of Selinexor Maintains Normal Immune Homeostasis and T-cell Effector Function in Mice: Implications for Combination with Immunotherapy. Mol. Cancer Ther. 2017, 16, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Farren, M.R.; Hennessey, R.C.; Shakya, R.; Elnaggar, O.; Young, G.; Kendra, K.; Landesman, Y.; Elloul, S.; Crochiere, M.; Klebanov, B.; et al. The Exportin-1 Inhibitor Selinexor Exerts Superior Antitumor Activity when Combined with T-Cell Checkpoint Inhibitors. Mol. Cancer Ther. 2017, 16, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Lapalombella, R.; Sun, Q.; Williams, K.; Tangeman, L.; Jha, S.; Zhong, Y.; Goett, V.; Mahoney, E.; Berglund, C.; Gupta, S.; et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012, 120, 4621–4634. [Google Scholar] [CrossRef] [Green Version]
- Stephens, D.M.; Huang, Y.; Ruppert, A.S.; Walker, J.S.; Canfield, D.; Cempre, C.B.; Fu, Q.; Baker, S.; Hu, B.; Shah, H.; et al. Selinexor Combined with Ibrutinib Demonstrates Tolerability and Safety in Advanced B-Cell Malignancies: A Phase I Study. Clin. Cancer Res. 2022, 28, 3242–3247. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, C.; Lentzsch, S.; Schiller, G.; Callander, N.; Tuchman, S.; Chen, C.; White, D.; Kotb, R.; Sutherland, H.; Sebag, M.; et al. Selinexor, daratumumab, and dexamethasone in patients with relapsed or refractory multiple myeloma. eJHaem 2021, 2, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Chen, X.-Q.; Li, X.-P.; Lu, Y.-X.; Chen, S.-L.; Wang, D.-W.; Liang, Y.; Dai, Y.-J. Drug resistance biomarker ABCC4 of selinexor and immune feature in multiple myeloma. Int. Immunopharmacol. 2022, 108, 108722. [Google Scholar] [CrossRef]
- Restrepo, P.; Bhalla, S.; Ghodke-Puranik, Y.; Aleman, A.; Leshchenko, V.; Melnekoff, D.T.; Agte, S.; Jiang, J.; Madduri, D.; Richter, J.; et al. A Three-Gene Signature Predicts Response to Selinexor in Multiple Myeloma. JCO Precis. Oncol. 2022, 6, e2200147. [Google Scholar] [CrossRef]
- Xu, J.; Niu, T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 1–20. [Google Scholar] [CrossRef]
- Bachanova, V.; Cooley, S.; DeFor, T.E.; Verneris, M.R.; Zhang, B.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef]
- Wang, S.; Sellner, L.; Wang, L.; Sauer, T.; Neuber, B.; Gong, W.; Stock, S.; Ni, M.; Yao, H.; Kleist, C.; et al. Combining selective inhibitors of nuclear export (SINEs) with chimeric antigen receptor (CAR) T cells for CD19-positive malignancies. Oncol. Rep. 2021, 46, 1–12. [Google Scholar] [CrossRef]
- Martini, S.; Figini, M.; Croce, A.; Frigerio, B.; Pennati, M.; Gianni, A.M.; De Marco, C.; Daidone, M.G.; Argueta, C.; Landesman, Y.; et al. Selinexor Sensitizes TRAIL-R2-Positive TNBC Cells to the Activity of TRAIL-R2xCD3 Bispecific Antibody. Cells 2020, 9, 2231. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Bartsch, T.; Schanz, F.; Oesch, F.; Wieser, R.J. p53-dependent cell cycle arrest induced by N-acetyl-L-leucinyl-L-leucinyl-L-norleucinal in platelet-derived growth factor-stimulated human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 10815–10819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagosklonny, M.V.; Wu, G.S.; Omura, S.; El-Deiry, W.S. Proteasome-dependent regulation of p21WAF1/CIP1 expression. Biochem. Biophys. Res. Commun. 1996, 227, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome Inhibitors Induce a Terminal Unfolded Protein Response in Multiple Myeloma Cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Gelman, J.S.; Sironi, J.; Berezniuk, I.; Dasgupta, S.; Castro, L.M.; Gozzo, F.C.; Ferro, E.S.; Fricker, L.D. Alterations of the Intracellular Peptidome in Response to the Proteasome Inhibitor Bortezomib. PLoS ONE 2013, 8, e53263. [Google Scholar] [CrossRef] [Green Version]
- Ulbrecht, M.; Modrow, S.; Srivastava, R.; Peterson, P.A.; Weiss, E.H. Interaction of HLA-E with peptides and the peptide transporter in vitro: Implications for its function in antigen presentation. J. Immunol. 1998, 160, 4375–4385. [Google Scholar]
- Hallett, W.H.; Ames, E.; Motarjemi, M.; Barao, I.; Shanker, A.; Tamang, D.L.; Sayers, T.J.; Hudig, D.; Murphy, W.J. Sensitization of Tumor Cells to NK Cell-Mediated Killing by Proteasome Inhibition. J. Immunol. 2007, 180, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: A role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913. [Google Scholar] [PubMed]
- Yun, H.D.; Schirm, D.K.; Felices, M.; Miller, J.S.; Eckfeldt, C.E. Dinaciclib enhances natural killer cell cytotoxicity against acute myelogenous leukemia. Blood Adv. 2019, 3, 2448–2452. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.-C.; Ho, Y.-J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell–mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kα Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Chauhan, D.; Auclair, D.; Robinson, E.K.; Hideshima, T.; Li, G.; Podar, K.; Gupta, D.; Richardson, P.; Schlossman, R.L.; Krett, N.; et al. Identification of genes regulated by Dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene 2002, 21, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Liechtenstein, A. Dexamethasone-induced apoptotic mechanisms in myeloma cells investigated by analysis of mutant glucocorticoid receptors. Blood 2008, 112, 1338. [Google Scholar] [CrossRef] [Green Version]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2018, 98, 19–28. [Google Scholar] [CrossRef]
- Boyle, E.M.; Petillon, M.-O.; Herbaux, C.; Mimouni, J.; Leleu, X.; Karlin, L.; Doyen, C.; Wetterwald, M.; Roussel, M.; Hulin, C.; et al. Daratumumab in Combination with Dexamethasone in Resistant or Refractory Multiple Myeloma: Primary Results of the IFM2014-04 Trial. Blood 2016, 128, 2138. [Google Scholar] [CrossRef]
- Keating, G.M. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs 2016, 77, 85–96. [Google Scholar] [CrossRef]
- Snead, J.L.; O’Hare, T.; Adrian, L.T.; Eide, C.A.; Lange, T.; Druker, B.J.; Deininger, M.W. Acute dasatinib exposure commits Bcr-Abl–dependent cells to apoptosis. Blood 2009, 114, 3459–3463. [Google Scholar] [CrossRef]
- Shah, N.P.; Kasap, C.; Weier, C.; Balbas, M.; Nicoll, J.M.; Bleickardt, E.; Nicaise, C.; Sawyers, C.L. Transient Potent BCR-ABL Inhibition Is Sufficient to Commit Chronic Myeloid Leukemia Cells Irreversibly to Apoptosis. Cancer Cell 2008, 14, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Hassold, N.; Seystahl, K.; Kempf, K.; Urlaub, D.; Zekl, M.; Einsele, H.; Watzl, C.; Wischhusen, J.; Seggewiss-Bernhardt, R. Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int. J. Cancer 2012, 131, E916–E927. [Google Scholar] [CrossRef]
- Uchiyama, T.; Sato, N.; Narita, M.; Yamahira, A.; Iwabuchi, M.; Furukawa, T.; Sone, H.; Takahashi, M. Direct effect of dasatinib on proliferation and cytotoxicity of natural killer cells in in vitro study. Hematol. Oncol. 2013, 31, 156–163. [Google Scholar] [CrossRef]
- Chang, M.-C.; Cheng, H.-I.; Hsu, K.; Hsu, Y.-N.; Kao, C.-W.; Chang, Y.-F.; Lim, K.-H.; Chen, C.G. NKG2A Down-Regulation by Dasatinib Enhances Natural Killer Cytotoxicity and Accelerates Effective Treatment Responses in Patients With Chronic Myeloid Leukemia. Front. Immunol. 2019, 9, 3152. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Nakamae, H.; Katayama, T.; Nakane, T.; Koh, H.; Nakamae, M.; Hirose, A.; Hagihara, K.; Terada, Y.; Nakao, Y.; et al. Different immunoprofiles in patients with chronic myeloid leukemia treated with imatinib, nilotinib or dasatinib. Leuk. Lymphoma 2012, 53, 1084–1089. [Google Scholar] [CrossRef]
- Marusina, A.I.; Kim, D.-K.; Lieto, L.D.; Borrego, F.; Coligan, J.E. GATA-3 Is an Important Transcription Factor for Regulating Human NKG2A Gene Expression. J. Immunol. 2005, 174, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Mondini, M.; Levy, A.; Meziani, L.; Milliat, F.; Deutsch, E. Radiotherapy-immunotherapy combinations—Perspectives and challenges. Mol. Oncol. 2020, 14, 1529–1537. [Google Scholar] [CrossRef]
- Patin, E.C.; Dillon, M.T.; Nenclares, P.; Grove, L.; Soliman, H.; Leslie, I.; Northcote, D.; Bozhanova, G.; Crespo-Rodriguez, E.; Baldock, H.; et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage–response inhibition and immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004306. [Google Scholar] [CrossRef]
- Michelin, S.; Gallegos, C.E.; Dubner, D.; Favier, B.; Carosella, E.D. Ionizing radiation modulates the surface expression of human leukocyte antigen-G in a human melanoma cell line. Hum. Immunol. 2009, 70, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, N.G.; Murphy, J.D.; Uccello, T.P.; Hughson, A.; Gavras, N.W.; Caldon, J.J.; Gerber, S.A.; Lord, E.M. Combination of NKG2A and PD-1 Blockade Improves Radiotherapy Response in Radioresistant Tumors. J. Immunol. 2022, 209, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, K.; Mukherjee, D.; Ambrose, A.R.; Harikrishnan, A.; Mole, H.; Hurlstone, A.; Önfelt, B.; Honeychurch, J.; Davis, D.M. Radiotherapy transiently reduces the sensitivity of cancer cells to lymphocyte cytotoxicity. Proc. Natl. Acad. Sci. USA 2022, 119, e2111900119. [Google Scholar] [CrossRef]
- Reger, R.N.; Berg, M.; Lundqvist, A.; Donohue, T.; Carlsten, M.; Betters, D.M.; Cook, L.; Ramos, C.; Grasmeder, S.; Su, S.; et al. A Phase I Trial of Adoptively Transferred Ex-Vivo Expanded Autologous Natural Killer (NK) Cells Following Treatment with Bortezomib to Sensitize Tumors to NK Cell Cytotoxicity. Blood 2011, 118, 1001. [Google Scholar] [CrossRef]
- Berg, M.; Lundqvist, A.; McCoy, P.; Samsel, L.; Fan, Y.; Tawab, A.; Childs, R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy 2009, 11, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Ciurea, S.O.; Schafer, J.R.; Bassett, R.; Denman, C.J.; Cao, K.; Willis, D.; Rondon, G.; Chen, J.; Soebbing, D.; Kaur, I.; et al. Phase 1 clinical trial using mbIL21 ex vivo–expanded donor-derived NK cells after haploidentical transplantation. Blood 2017, 130, 1857–1868. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-C.; Shimasaki, N.; Lim, J.S.; Wong, A.; Yadav, K.; Yong, W.P.; Tan, L.K.; Koh, L.P.; Poon, M.L.; Tan, S.H.; et al. Phase I Trial of Expanded, Activated Autologous NK-cell Infusions with Trastuzumab in Patients with HER2-positive Cancers. Clin. Cancer Res. 2020, 26, 4494–4502. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, J.G.; Doyle, A.D.P.; Graham, L.V.; Khakoo, S.I.; Blunt, M.D. Disruption of the NKG2A:HLA-E Immune Checkpoint Axis to Enhance NK Cell Activation against Cancer. Vaccines 2022, 10, 1993. https://doi.org/10.3390/vaccines10121993
Fisher JG, Doyle ADP, Graham LV, Khakoo SI, Blunt MD. Disruption of the NKG2A:HLA-E Immune Checkpoint Axis to Enhance NK Cell Activation against Cancer. Vaccines. 2022; 10(12):1993. https://doi.org/10.3390/vaccines10121993
Chicago/Turabian StyleFisher, Jack G., Amber D. P. Doyle, Lara V. Graham, Salim I. Khakoo, and Matthew D. Blunt. 2022. "Disruption of the NKG2A:HLA-E Immune Checkpoint Axis to Enhance NK Cell Activation against Cancer" Vaccines 10, no. 12: 1993. https://doi.org/10.3390/vaccines10121993
APA StyleFisher, J. G., Doyle, A. D. P., Graham, L. V., Khakoo, S. I., & Blunt, M. D. (2022). Disruption of the NKG2A:HLA-E Immune Checkpoint Axis to Enhance NK Cell Activation against Cancer. Vaccines, 10(12), 1993. https://doi.org/10.3390/vaccines10121993