
Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach


 ,
,  ,
,  , , , and
, , , and 
Abstract
:1. Introduction
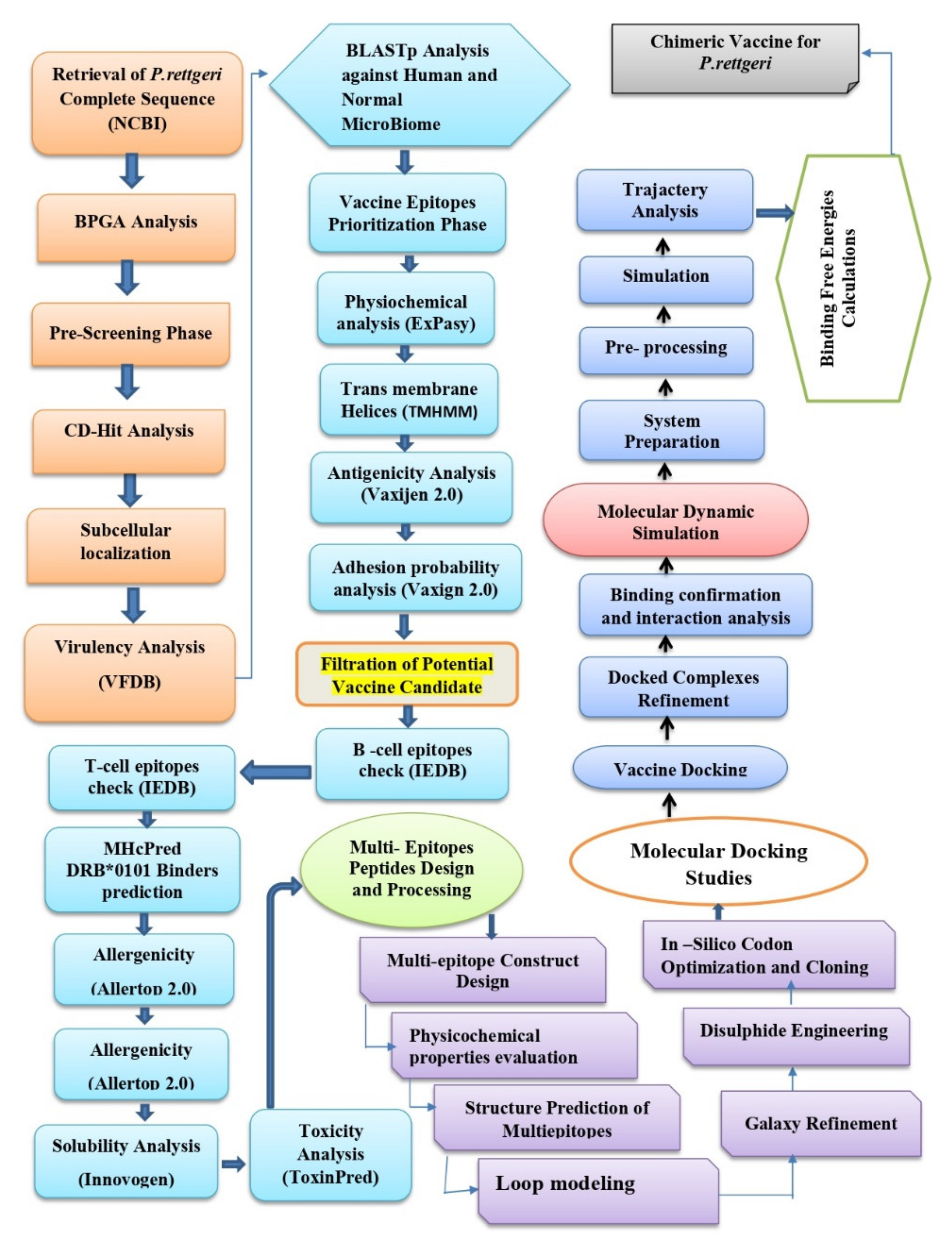
2. Research Methodology
2.1. P. rettgeri Complete Proteome Retrieval
2.2. BPGA Analysis
2.3. Pre-Screening Phase
2.4. CD-Hit Analysis (Cluster Data with High Identity and Tolerance)
2.5. Subcellular Localization Analysis
2.6. Virulent Protein Analysis
2.7. BLASTp Analysis against Humans and MicroBiome
2.8. Vaccine Epitopes Prioritization Phase
2.9. Physiochemical Analysis
2.10. Transmembrane Helices
2.11. Antigenicity Prediction
2.12. Adhesion Probability Analysis
2.13. Epitopes Prediction Phase
2.14. Multi-Epitope Peptide Designing and Processing
2.15. Physiochemical Properties of the Final Vaccine Construct
2.16. Structure Modeling of the Final Vaccine Construct
2.17. Loops Modeling
2.18. Galaxy Refinement
2.19. Disulfide Engineering
2.20. Codon Optimization
2.21. Molecular Docking
2.22. Molecular Dynamic Simulation
2.23. Binding Free Energies Calculation
2.24. C-Immune Simulation
3. Results
3.1. Providencia rettgeri Complete Proteome Retrieval
3.2. Bacterial Pan Genome Analysis
3.3. CD-HIT Analysis
3.4. Subcellular Localization
3.5. VFDB Analysis
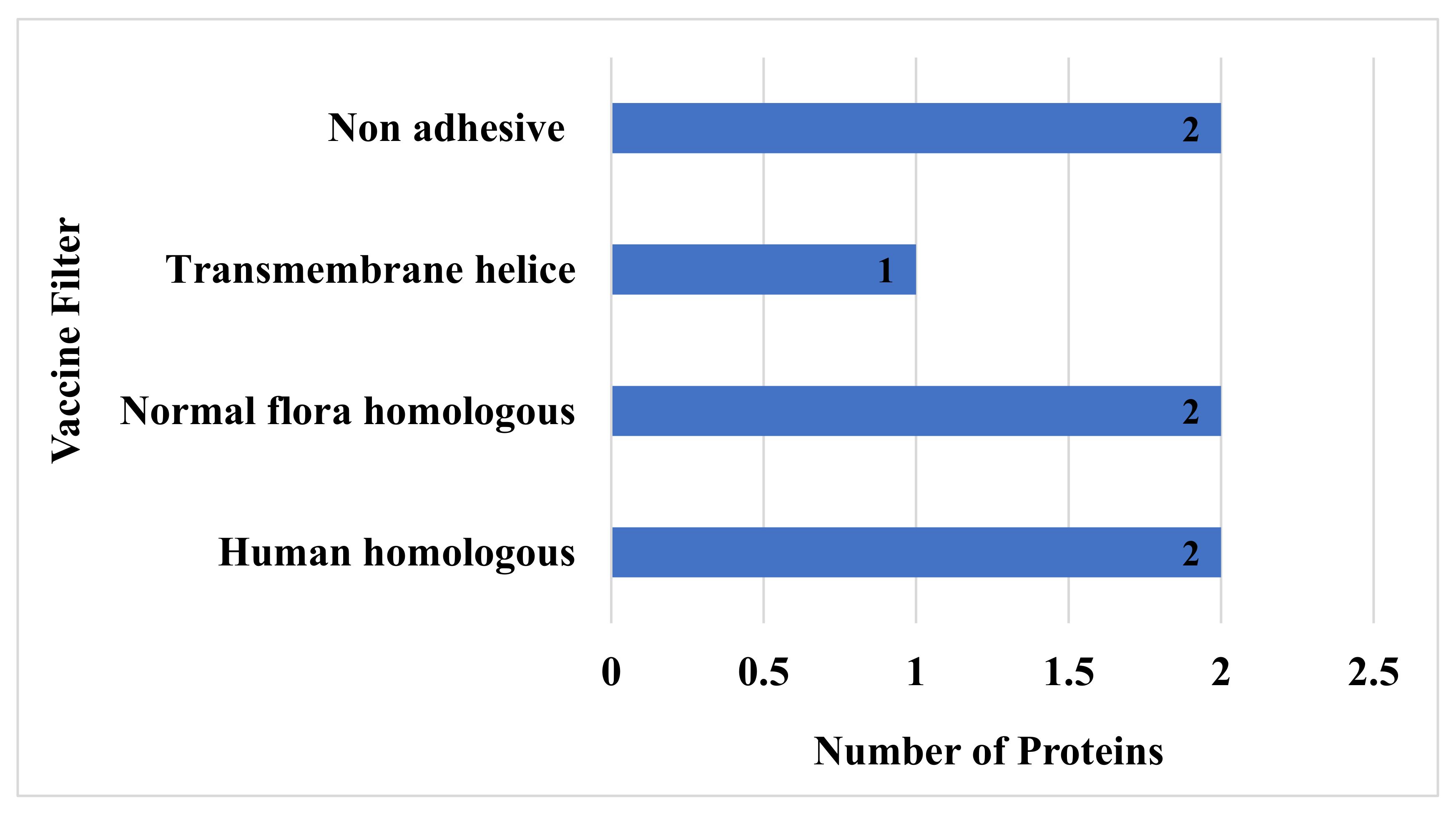
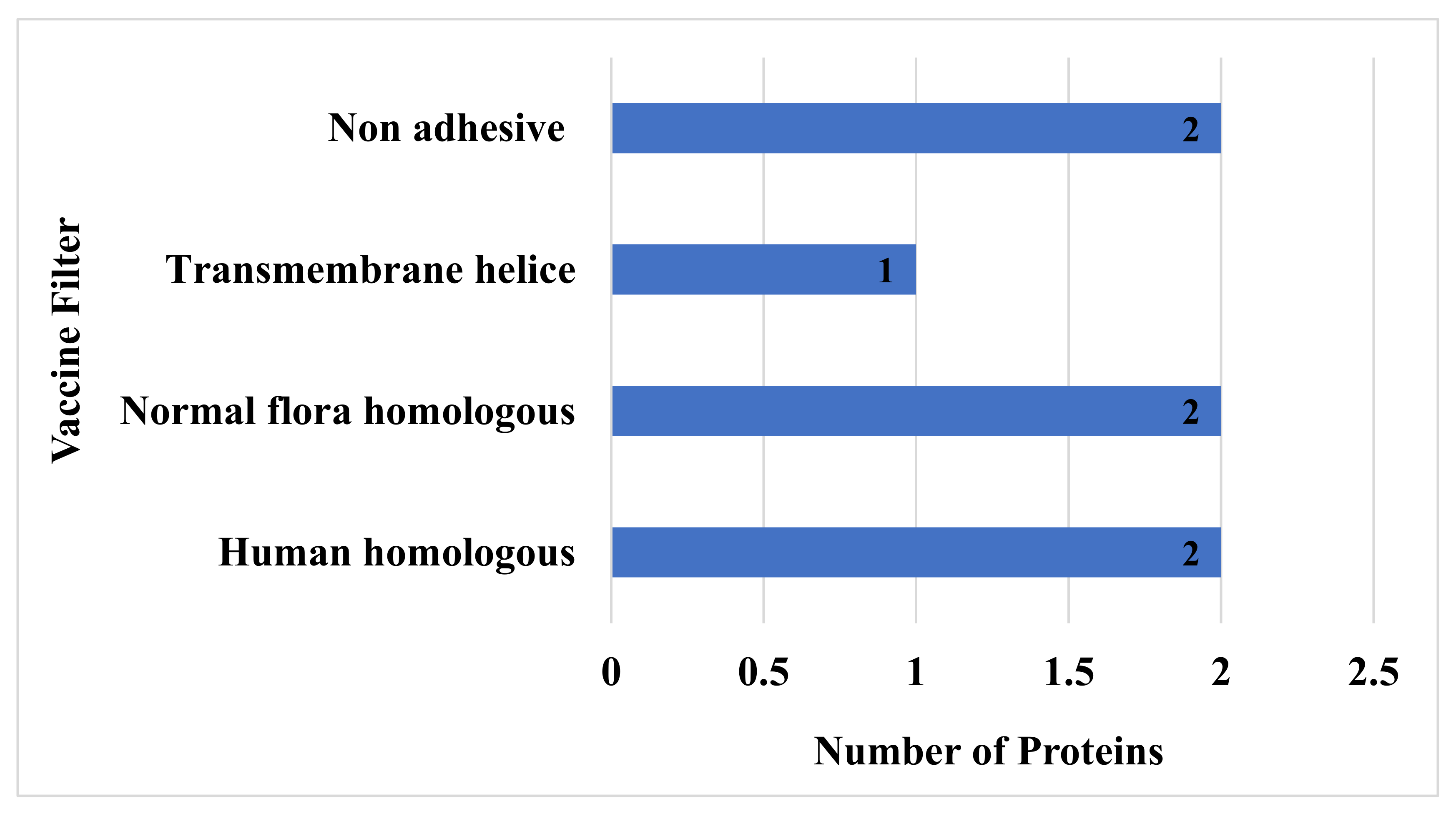
3.6. Human and Normal Flora, Adhesion Probability, Physiochemical Property Analysis
3.7. Vaccine Epitopes Prioritization Phase
3.8. B-Cell Epitopes Prediction
3.9. T-Cell Epitopes Prediction
3.10. Epitope Prioritization Phase
3.11. DRB*0101 Binding Analysis
3.12. Antigenicity, Allergenicity, Solubility, and Toxicity Analysis of Selected Epitopes
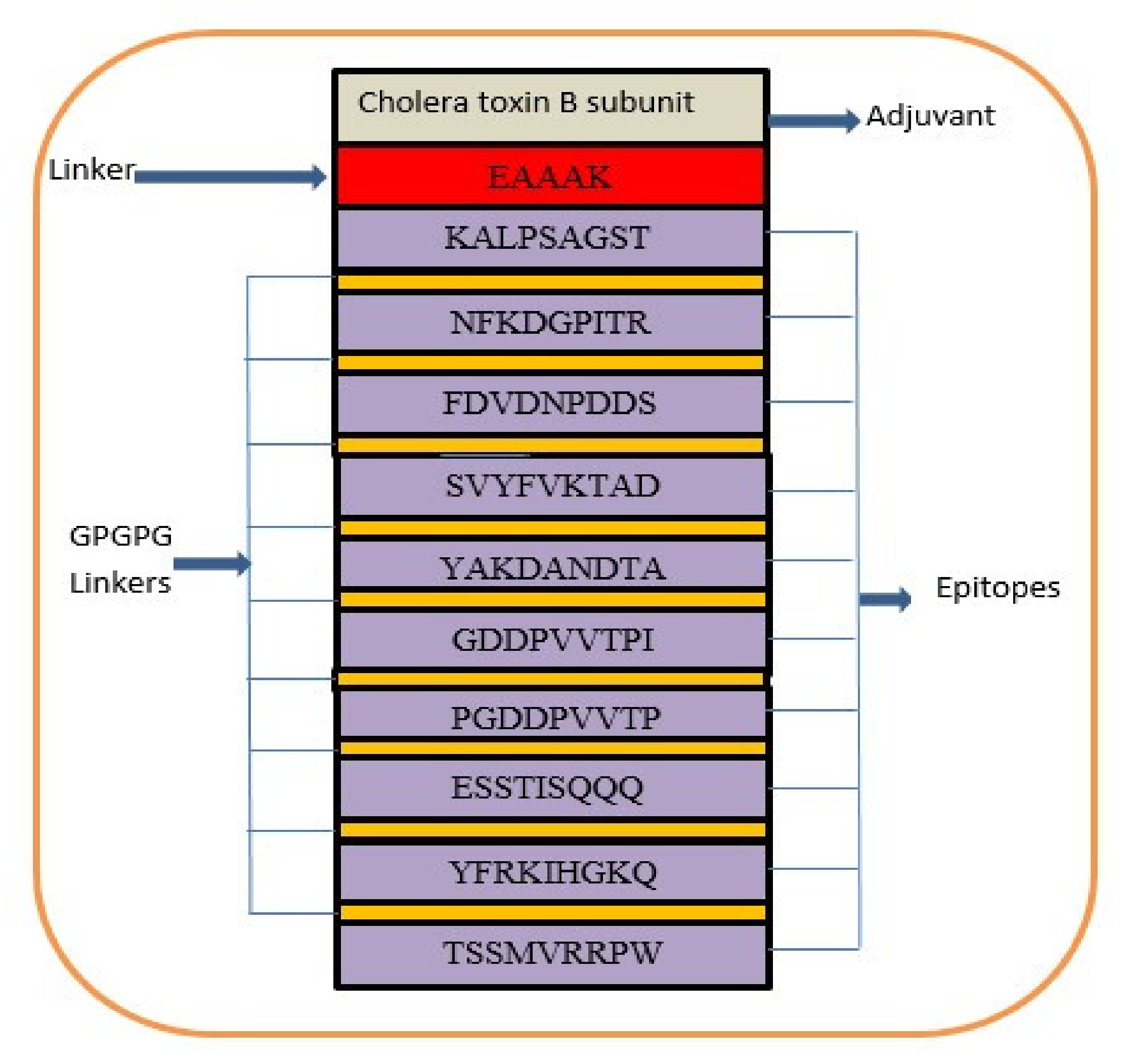
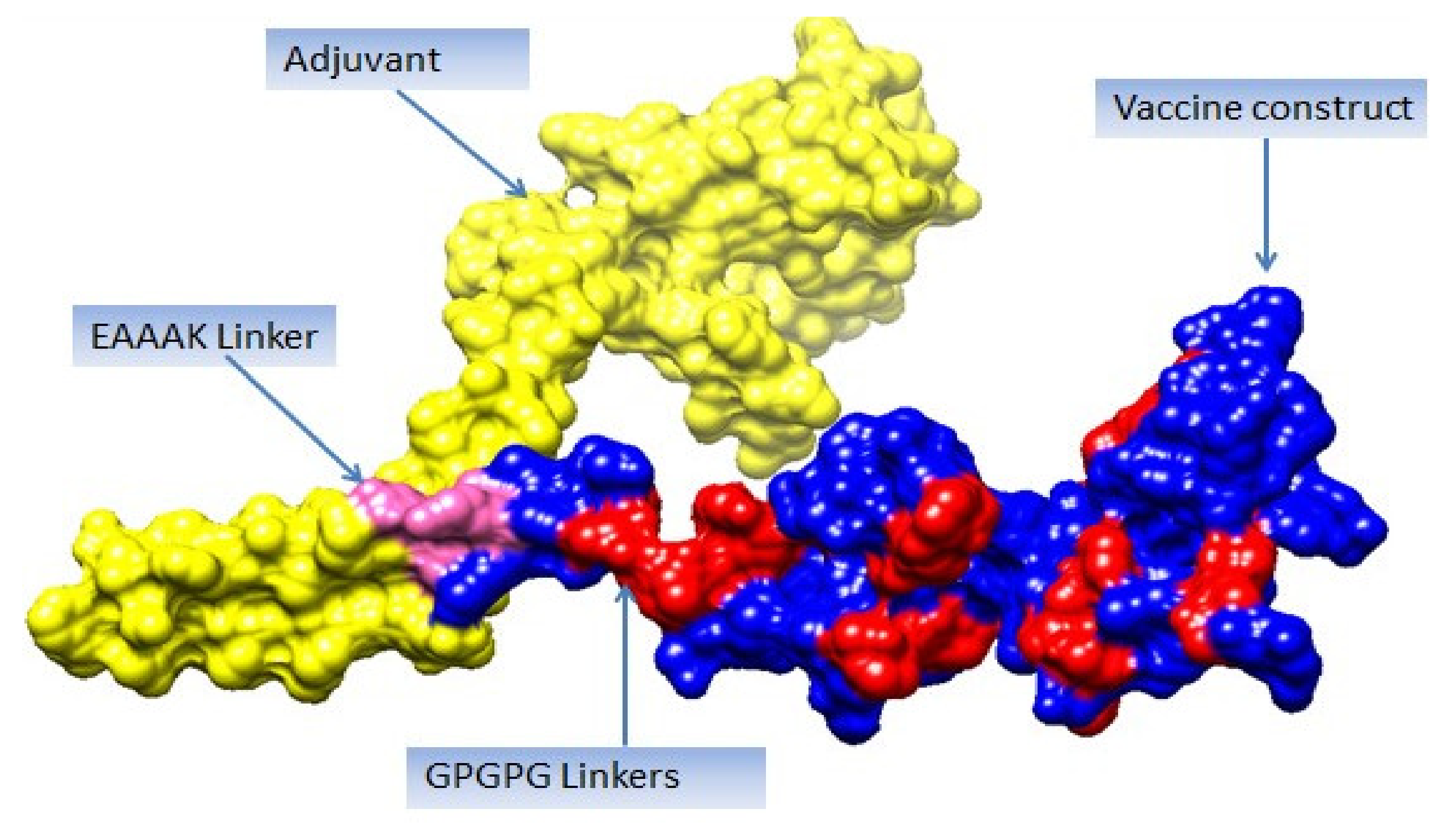
3.13. Multi-Epitope Vaccine Construction and Processing
3.14. Structure Modeling
3.15. Loops Modeling and Refinement
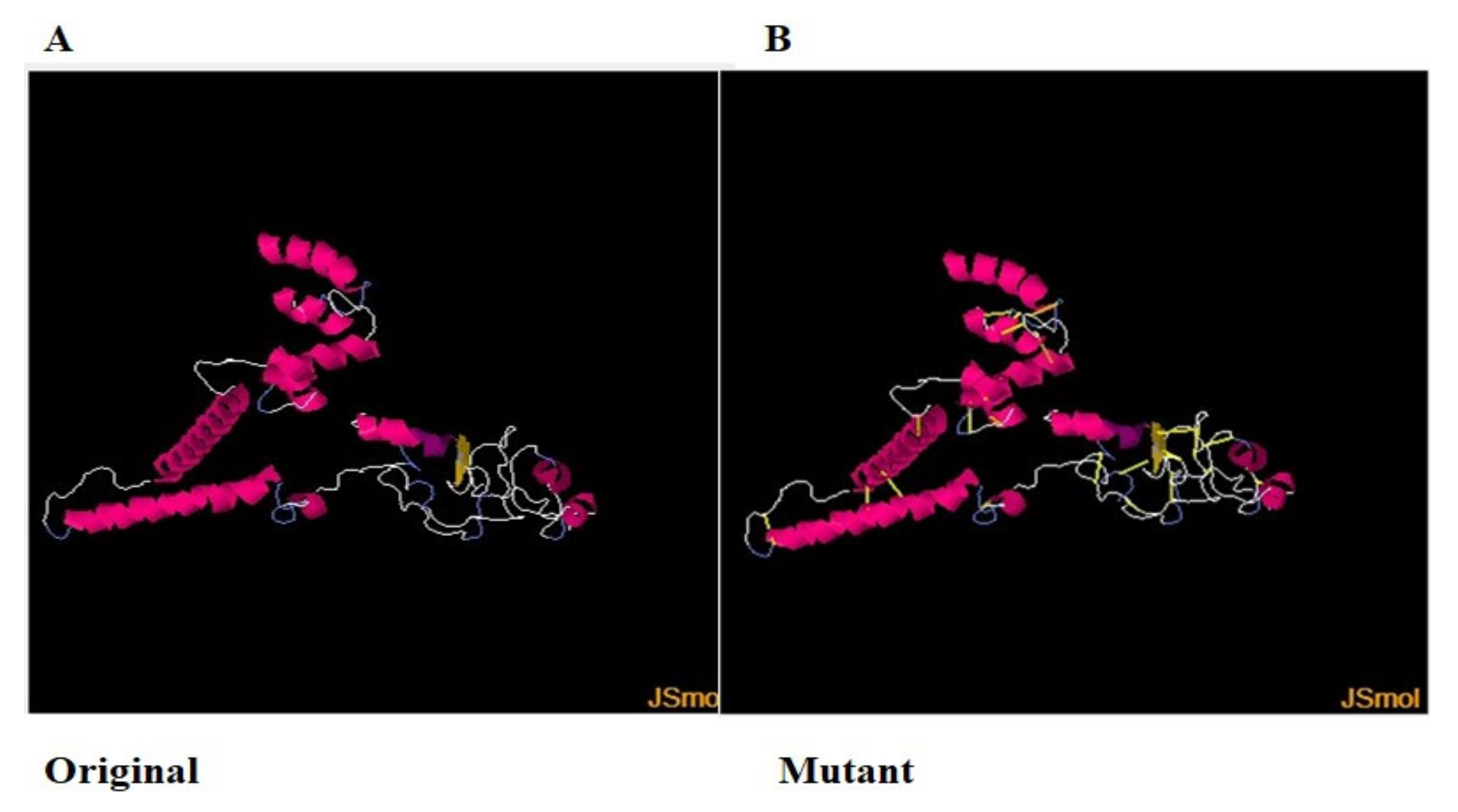
3.16. Disulfide Engineering
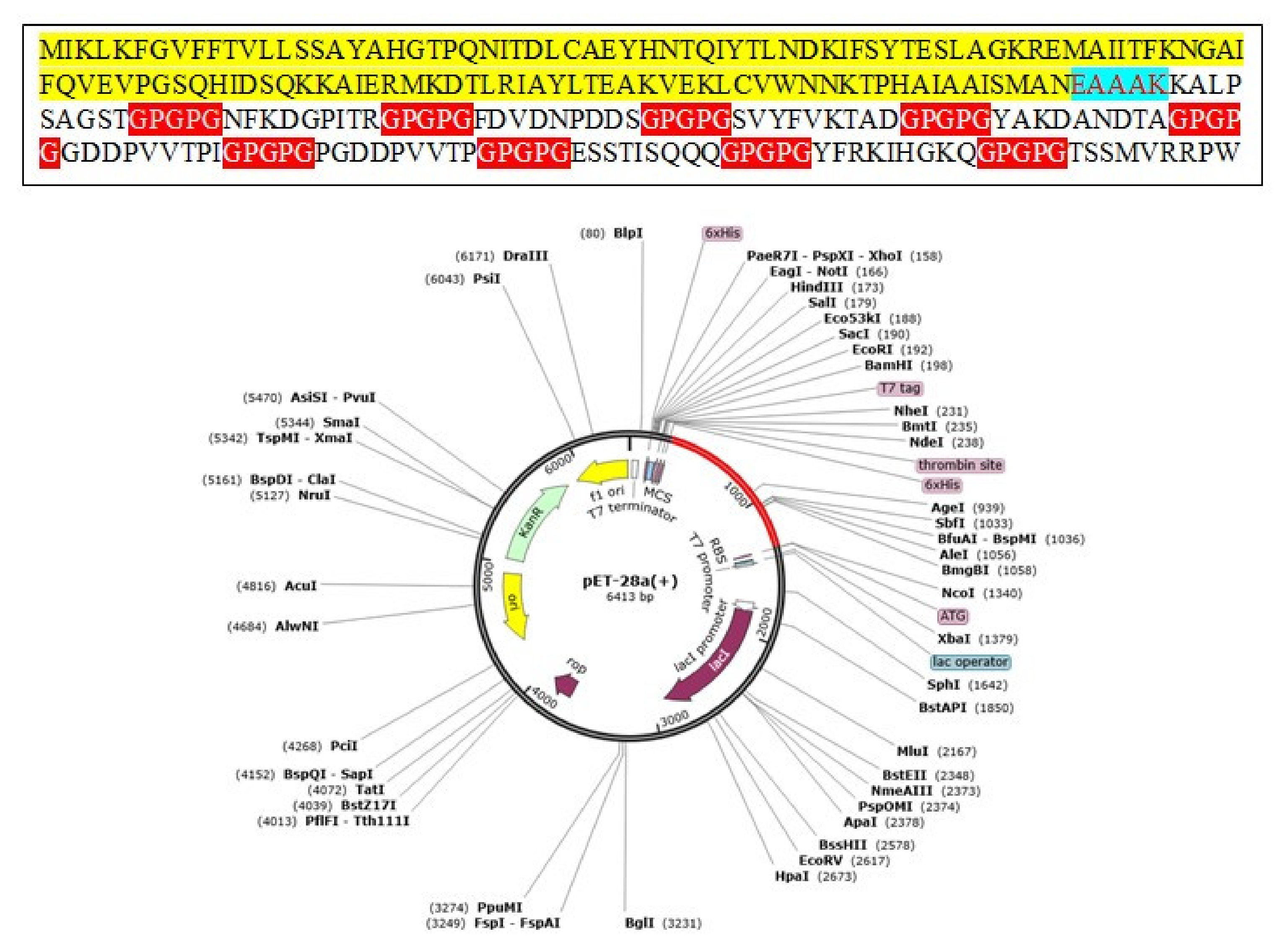
3.17. Codon Optimization
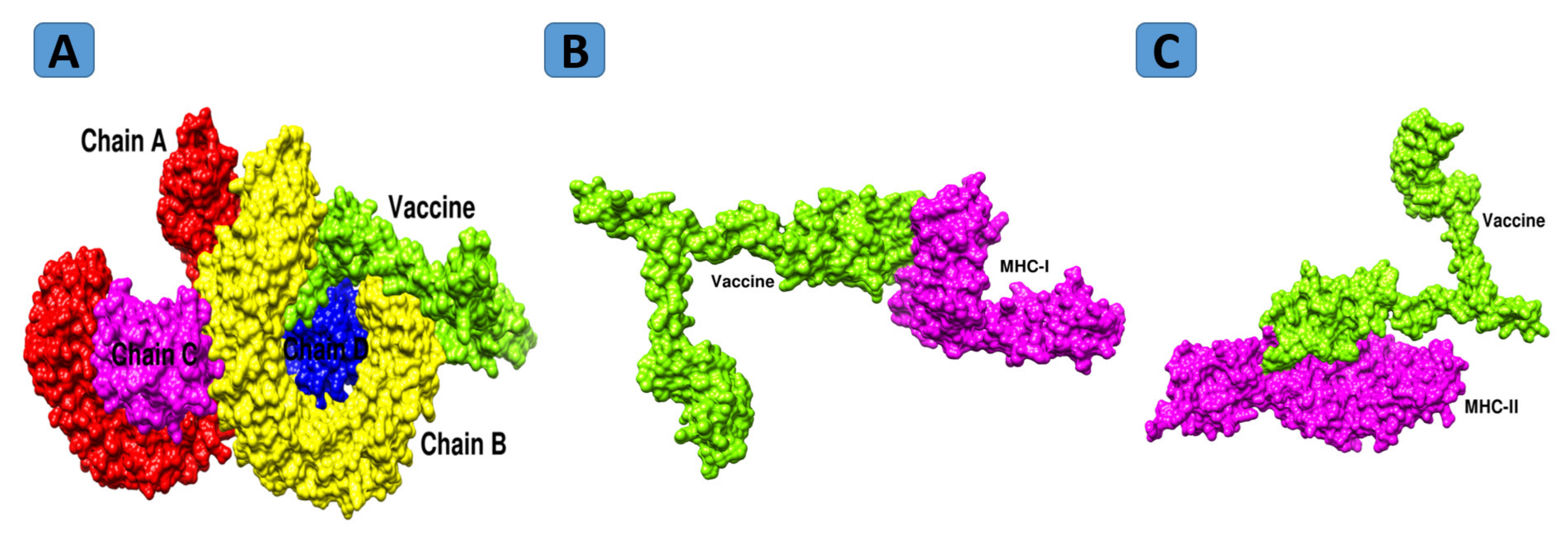
3.18. Molecular Docking
3.19. Refinement of Docked Complexes
3.20. Chemical Interactions of Vaccine to Immune Cells Receptors
3.21. Molecular Dynamic Simulation
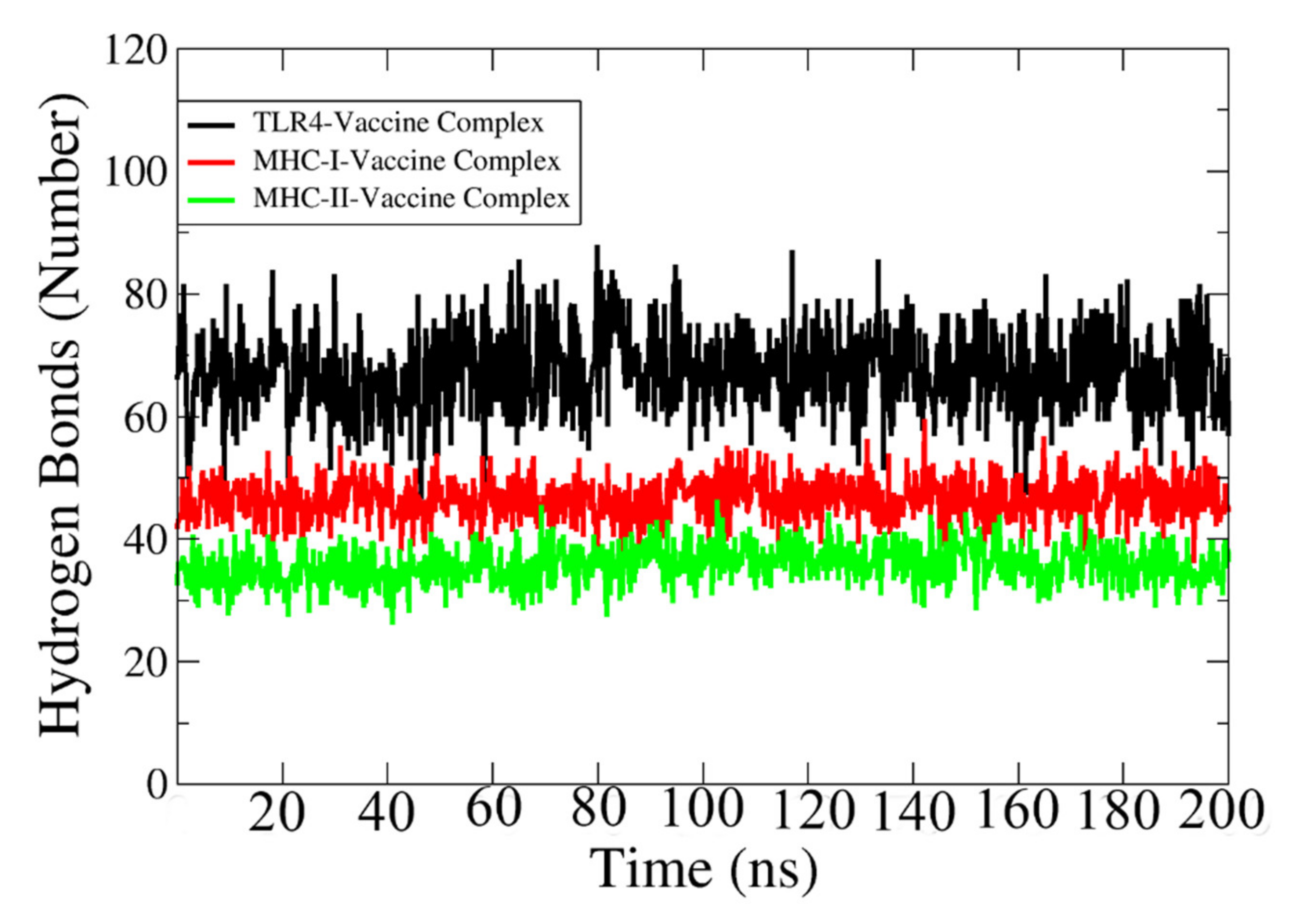
3.22. Hydrogen Bonding
3.23. Binding Free Energies Calculation
3.24. In Silico Immune Simulation
4. Discussion
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowler, P.; Murphy, C.; Wolcott, R. Biofilm exacerbates antibiotic resistance: Is this a current oversight in antimicrobial stewardship? Antimicrob. Resist. Infect. Control 2020, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Roth, N.; Käsbohrer, A.; Mayrhofer, S.; Zitz, U.; Hofacre, C.; Domig, K.J. The application of antibiotics in broiler production and the resulting antibiotic resistance in Escherichia coli: A global overview. Poult. Sci. 2019, 98, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, C.B.; Cohen, A.; Engelhard, D.; Hays, J.P.; Karlsson, R.; Moore, E.; Fernández, D.; Kreisberg, R.; Collins, L.V.; de Waal, W. Antibiotic misuse in respiratory tract infections in children and adults—A prospective, multicentre study (TAILORED Treatment). Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, R.; Halder, U.; Kabiraj, A.; Mondal, A.; Bandopadhyay, R. Overview on the role of heavy metals tolerance on developing antibiotic resistance in both Gram-negative and Gram-positive bacteria. Arch. Microbiol. 2021, 203, 2761–2770. Available online: https://link.springer.com/article/10.1007/s00203-021-02275-w (accessed on 18 June 2021). [CrossRef]
- Caniça, M.; Manageiro, V.; Abriouel, H.; Moran-Gilad, J.; Franz, C.M.A.P. Antibiotic resistance in foodborne bacteria. Trends Food Sci. Technol. 2019, 84, 41–44. [Google Scholar] [CrossRef]
- MacLean, R.C.; San Millan, A. The evolution of antibiotic resistance. Science 2019, 365, 1082–1083. [Google Scholar] [CrossRef]
- Koch, B.J.; Hungate, B.A.; Price, L.B. Food-animal production and the spread of antibiotic resistance: The role of ecology. Front. Ecol. Environ. 2017, 15, 309–318. [Google Scholar] [CrossRef]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Singh, A.P.; Kumar, S.; Giri, B.S.; Kim, K.-H. Antibiotic resistance in major rivers in the world: A systematic review on occurrence, emergence, and management strategies. J. Clean. Prod. 2019, 234, 1484–1505. [Google Scholar] [CrossRef]
- Chin, W.; Zhong, G.; Pu, Q.; Yang, C.; Lou, W.; De Sessions, P.F.; Periaswamy, B.; Lee, A.; Liang, Z.C.; Ding, X. A macromolecular approach to eradicate multidrug resistant bacterial infections while mitigating drug resistance onset. Nat. Commun. 2018, 9, 917. [Google Scholar] [CrossRef] [PubMed]
- Covián, C.; Fernández-Fierro, A.; Retamal-Díaz, A.; Díaz, F.E.; Vasquez, A.E.; Lay, M.K.; Riedel, C.A.; González, P.A.; Bueno, S.M.; Kalergis, A.M. BCG-induced cross-protection and development of trained immunity: Implication for vaccine design. Front. Immunol. 2019, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- Halloran, M.E.; Longini, I.M.; Struchiner, C.J.; Longini, I.M. Design and Analysis of Vaccine Studies; Springer: Berlin/Heidelberg, Germany, 2010; Volume 18. [Google Scholar]
- Ansari, A.; Madan, A.; Prakash, D. Vaccine development—A complex science. EPRA Int. J. Multidiscip. Res. 2021, 7, 34–37. [Google Scholar] [CrossRef]
- Gasperini, G.; Alfini, R.; Arato, V.; Mancini, F.; Aruta, M.G.; Kanvatirth, P.; Pickard, D.; Necchi, F.; Saul, A.; Rossi, O. Salmonella Paratyphi A Outer Membrane Vesicles Displaying Vi Polysaccharide as a Multivalent Vaccine against Enteric Fever. Infect. Immun. 2021, 89, e00699-20. [Google Scholar] [CrossRef]
- Chen, H.; Gao, Z.; Bai, S.; Liu, X.; Han, S.; Xiao, Y.; Liu, F.; Yu, Y.; Sun, H.; Yang, X. Immunogenicity and safety of sabin-strain based inactivated poliovirus vaccine replacing salk-strain based inactivated poliovirus vaccine: An innovative application of different strain-IPVs replacement. Vaccine 2021, 39, 2467–2474. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E. The Tuberculosis Vaccine Development Pipeline: Present and Future Priorities and Challenges for Research and Innovation. In Essential Tuberculosis; Springer: Berlin/Heidelberg, Germany, 2021; pp. 395–405. [Google Scholar]
- Gong, W.; Aspatwar, A.; Wang, S.; Parkkila, S.; Wu, X. COVID-19 pandemic: SARS-CoV-2 specific vaccines and challenges, protection via BCG trained immunity, and clinical trials. Expert Rev. Vaccines 2021, 20, 857–880. Available online: https://www.tandfonline.com/doi/full/10.1080/14760584.2021.1938550 (accessed on 18 June 2021). [CrossRef]
- Dar, H.A.; Ismail, S.; Waheed, Y.; Ahmad, S.; Jamil, Z.; Aziz, H.; Hetta, H.F.; Muhammad, K. Designing a multi-epitope vaccine against Mycobacteroides abscessus by pangenome-reverse vaccinology. Sci. Rep. 2021, 11, 11197. [Google Scholar] [CrossRef]
- Thompson, M.G.; Burgess, J.L.; Naleway, A.L.; Tyner, H.L.; Yoon, S.K.; Meece, J.; Olsho, L.E.W.; Caban-Martinez, A.J.; Fowlkes, A.; Lutrick, K. Interim estimates of vaccine effectiveness of BNT162b2 and mRNA-1273 COVID-19 vaccines in preventing SARS-CoV-2 infection among health care personnel, first responders, and other essential and frontline workers—Eight US locations, December 2020–March 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 495. [Google Scholar] [CrossRef]
- Bidmos, F.A.; Siris, S.; Gladstone, C.A.; Langford, P.R. Bacterial Vaccine Antigen Discovery in the Reverse Vaccinology 2.0 Era: Progress and Challenges. Front. Immunol. 2018, 9, 2315. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Huo, C.; Lang, S.; Caution, K.; Nick, S.T.; Dubey, P.; Deora, R.; Huang, X. Chemical Synthesis and Immunological Evaluation of a Pentasaccharide Bearing Multiple Rare Sugars as a Potential Anti-pertussis Vaccine. Angew. Chem. 2020, 132, 6513–6520. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial Enterobacteriaceae pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. What are the most powerful immunogen design vaccine strategies? Reverse vaccinology 2.0 shows great promise. Cold Spring Harb. Perspect. Biol. 2017, 9, a030262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzler, L.M.; Feavers, I.M.; Gray-Owen, S.D.; Jerse, A.E.; Rice, P.A.; Deal, C.D. Summary and recommendations from the National Institute of Allergy and Infectious Diseases (NIAID) workshop “Gonorrhea Vaccines: The Way forward. ” Clin. Vaccine Immunol. 2016, 23, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalsass, M.; Brozzi, A.; Medini, D.; Rappuoli, R. Comparison of open-source Reverse Vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Yang, Q.; Chan, L.W.; Bhatia, S.N.; Ruoslahti, E.; Sailor, M.J. Fusogenic porous silicon nanoparticles as a broad-spectrum immunotherapy against bacterial infections. Nanoscale Horiz. 2021, 6, 330–340. [Google Scholar] [CrossRef]
- Gagneux-Brunon, A.; Lucht, F.; Launay, O.; Berthelot, P.; Botelho-Nevers, E. Vaccines for healthcare-associated infections: Present, future, and expectations. Expert Rev. Vaccines 2018, 17, 421–433. [Google Scholar] [CrossRef]
- Ahmad, S.; Waheed, Y.; Ismail, S.; Abbasi, S.W.; Najmi, M.H. A computational study to disclose potential drugs and vaccine ensemble for COVID-19 conundrum. J. Mol. Liq. 2021, 324, 114734. [Google Scholar] [CrossRef]
- Yero, D.; Conchillo-Solé, O.; Daura, X. Antigen Discovery in Bacterial Panproteomes. In Vaccine Delivery Technology; Springer: Berlin/Heidelberg, Germany, 2021; pp. 43–62. [Google Scholar]
- Abdullah, M.; Kadivella, M.; Sharma, R.; Faisal, S.M.; Azam, S. Designing of multiepitope-based vaccine against Leptospirosis using Immuno-Informatics approaches. bioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.02.22.431920v1 (accessed on 18 June 2021).
- Santos, A.; Ali, A.; Barbosa, E.; Silva, A.; Miyoshi, A.; Barh, D.; Azevedo, V. The reverse vaccinology—A contextual overview. IIOABJ 2011, 2, 8–15. [Google Scholar]
- Yuan, C.; Wei, Y.; Zhang, S.; Cheng, J.; Cheng, X.; Qian, C.; Wang, Y.; Zhang, Y.; Yin, Z.; Chen, H. Comparative Genomic Analysis Reveals Genetic Mechanisms of the Variety of Pathogenicity, Antibiotic Resistance, and Environmental Adaptation of Providencia Genus. Front. Microbiol. 2020, 11, 572642. [Google Scholar] [CrossRef]
- Sagar, S.; Narasimhaswamy, N.; d’Souza, J. Providencia rettgeri: An emerging nosocomial uropathogen in an indwelling urinary catheterised patient. J. Clin. Diagn. Res. JCDR 2017, 11, DD01–DD02. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Sharma, P.; Soni, P. First case report of Providencia rettgeri neonatal sepsis. BMC Res. Notes 2017, 10, 17–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Jeong, S.H.; Lee, H.; Hong, J.S.; Park, M.-J.; Song, W. Emergence of multidrug-resistant Providencia rettgeri isolates co-producing NDM-1 carbapenemase and PER-1 extended-spectrum β-lactamase causing a first outbreak in Korea. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Koreishi, A.F.; Schechter, B.A.; Karp, C.L. Ocular Infections Caused by Providencia rettgeri. Ophthalmology 2006, 113, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Tshisevhe, V.S.; Lekalakala, M.R.; Tshuma, N.; Janse Van Rensburg, S.; Mbelle, N. Outbreak of carbapenem-resistant Providencia rettgeri in a tertiary hospital. S. Afr. Med. J. 2017, 107, 31–33. [Google Scholar] [CrossRef] [Green Version]
- Iwata, S.; Tada, T.; Hishinuma, T.; Tohya, M.; Oshiro, S.; Kuwahara-Arai, K.; Ogawa, M.; Shimojima, M.; Kirikae, T. Emergence of carbapenem-resistant Providencia rettgeri and Providencia stuartii producing IMP-type metallo-β-lactamase in Japan. Antimicrob. Agents Chemother. 2020, 64, e00382-20. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Assef, A.P.D.; Pereira, P.S.; Albano, R.M.; Berião, G.C.; Chagas, T.P.G.; Timm, L.N.; Da Silva, R.C.F.; Falci, D.R.; Asensi, M.D. Isolation of NDM-producing Providencia rettgeri in Brazil. J. Antimicrob. Chemother. 2013, 68, 2956–2957. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [Green Version]
- Pérez de la Lastra, J.M.; Asensio-Calavia, P.; González-Acosta, S.; Baca-González, V.; Morales-delaNuez, A. Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins. Molecules 2021, 26, 1811. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Zeb, S.; Gulfam, S.M.; Bokhari, H. Comparative core/pan genome analysis of Vibrio cholerae isolates from Pakistan. Infect. Genet. Evol. 2020, 82, 104316. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, H.; Dyer, R.; Severin, A.J.; Rajan, H. Comprehensive Analysis of Non Redundant Protein Database. Res. Sq. 2020. Available online: https://www.researchgate.net/publication/343753529_Comprehensive_Analysis_of_Non_Redundant_Protein_Database (accessed on 18 June 2021).
- Wheeler, C.R.; Scarbrough, D. Implementing Bioinformatic Tools to Predict Vaccine Potential from Prioritized Staphylococcus aureus Antigens. 2020. Available online: https://scholarworks.boisestate.edu/icur/2020/Poster_Session/119/ (accessed on 18 June 2021).
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Abbas, G.; Zafar, I.; Ahmad, S.; Azam, S.S. Immunoinformatics design of a novel multi-epitope peptide vaccine to combat multi-drug resistant infections caused by Vibrio vulnificus. Eur. J. Pharm. Sci. 2020, 142, 105160. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chand, Y.; Singh, S. Prioritization of potential vaccine candidates and designing a multiepitope-based subunit vaccine against multidrug-resistant Salmonella Typhi str. CT18: A subtractive proteomics and immunoinformatics approach. Microb. Pathog. 2021, 159, 105150. [Google Scholar] [CrossRef]
- ProtParam—Expasy. ExPASy-ProtParam Tool. 2017. Available online: https://web.expasy.org/protparam/ (accessed on 2 May 2021).
- Sahoo, P.R.; Sahoo, G.; Behera, P.C. Comparative Insilco physiochemical and phylogenetic analysis of insulin like growth factor 1 receptor (IGF-1R) in domestic animals. Indian J. Anim. Res. 2019, 53, 1033–1035. [Google Scholar] [CrossRef]
- Lüthje, S.; Ramanathan, K. In Silico Analysis of Class III Peroxidases: Hypothetical Structure, Ligand Binding Sites, Posttranslational Modifications, and Interaction with Substrates. In Plant Proteomics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 325–339. [Google Scholar]
- Ferdous, N.; Reza, M.N.; Emon, M.T.H.; Islam, M.S.; Mohiuddin, A.K.M.; Hossain, M.U. Molecular characterization and functional annotation of a hypothetical protein (SCO0618) of Streptomyces coelicolor A3 (2). Genom. Inform. 2020, 18, e28. [Google Scholar] [CrossRef]
- Ahmad, S.; Navid, A.; Farid, R.; Abbas, G.; Ahmad, F.; Zaman, N.; Parvaiz, N.; Azam, S.S. Design of a novel multi epitope-based vaccine for pandemic coronavirus disease (COVID-19) by vaccinomics and probable prevention strategy against avenging zoonotics. Eur. J. Pharm. Sci. 2020, 151, 105387. [Google Scholar] [CrossRef]
- Ong, E.; Cooke, M.F.; Huffman, A.; Xiang, Z.; Wong, M.U.; Wang, H.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign2: The second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 2021, 49, W671–W678. [Google Scholar] [CrossRef]
- Adeoti, O.M. Prediction of multi-epitopic domains of a putative oral vaccine against hepatitis C virus. Int. J. Immunol. Microbiol. 2021, 1, 16–22. [Google Scholar]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [Green Version]
- Girija, A.S.S.; Shoba, G.; Priyadharsini, J.V. Accessing the T-Cell and B-Cell immuno-dominant peptides from A. baumannii biofilm associated protein (bap) as vaccine candidates: A computational approach. Int. J. Pept. Res. Ther. 2021, 27, 37–45. [Google Scholar] [CrossRef]
- Guan, P.; Doytchinova, I.A.; Zygouri, C.; Flower, D.R. MHCPred: A server for quantitative prediction of peptide–MHC binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taiwo, A.A.; Falilat, A.J.; Ezemuel, Y.S. Computational design of peptide vaccine against Acinetobacter baumannii infection using comparative genomic approach. Comput. Biol. Bioinform. 2014, 2, 13–18. [Google Scholar] [CrossRef]
- Ahmad, S.; Azam, S.S. A novel approach of virulome based reverse vaccinology for exploring and validating peptide-based vaccine candidates against the most troublesome nosocomial pathogen: Acinetobacter baumannii. J. Mol. Graph. Model. 2018, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saadi, M.; Karkhah, A.; Nouri, H.R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect. Genet. Evol. 2017, 51, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Jerin, T.; Sharmin, N.; Raihan, S. Silico Functional Annotation and Molecular Characterization of an Uncharacterized Protein MBO_502153 of Mycobacterium Tuberculosis Variant Bovis. Int. J. Sci. Res. Dent. Med. Sci. 2021, 3, 78–85. [Google Scholar]
- Sarkar, M.; Saha, S. Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies. PLoS ONE 2020, 15, e0237300. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Saba Ismail, S.A.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 669812. [Google Scholar] [CrossRef]
- Devi, A.; Chaitanya, N.S.N. In silico designing of multi-epitope vaccine construct against human coronavirus infections. J. Biomol. Struct. Dyn. 2021, 39, 6903–6917. [Google Scholar] [CrossRef]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Krishnan, S.; Joshi, A.; Akhtar, N.; Kaushik, V. Immunoinformatics designed T cell multi epitope dengue peptide vaccine derived from non structural proteome. Microb. Pathog. 2021, 150, 104728. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, T.; Rizarullah, R. Predicting Multi-Epitope Peptide Cancer Vaccine from Novel TAA Topo48. J. Sci. Appl. Technol. 2021, 5, 171–178. [Google Scholar] [CrossRef]
- Debnath, S.; Sen, D. Mushrooms are potential foods against cancer: Identified by molecular docking and molecular dynamics simulation. Nat. Prod. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Shahid, F.; Tahir ul Qamar, M.; Abbasi, S.W.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.S.; Almatroudi, A.; Ullah Saeed, H.F. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef]
- Huang, K.; Luo, S.; Cong, Y.; Zhong, S.; Zhang, J.Z.H.; Duan, L. An accurate free energy estimator: Based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity. Nanoscale 2020, 12, 10737–10750. [Google Scholar] [CrossRef]
- Bhardwaj, A. In silico Multi Subunit Vaccine Design Referring Spike Glycoprotein of SARS-COV-2 (COVID-19): The World Pandemic. Indian J. Pharm. Sci. 2021, 83, 21–31. [Google Scholar] [CrossRef]
- Omoniyi, A.A.; Adebisi, S.S.; Musa, S.A.; Nzalak, J.O.; Danborno, B.; Bauchi, Z.M.; Badmus, I.T.; Olatomide, O.D.; Oladimeji, O.J.; Nyengaard, J.R. Immunoinformatics Analysis and In-Silico Design of Multi-Epitopes Vaccine against Lassa Virus. Available online: https://www.researchgate.net/publication/350394848_Immunoinformatics_Analysis_and_In-silico_Design_of_Multi-_Epitopes_Vaccine_Against_Lassa_Virus (accessed on 16 August 2021).
- Yadav, S.; Kapley, A. Antibiotic resistance: Global health crisis and metagenomics. Biotechnol. Rep. 2021, 29, e00604. [Google Scholar] [CrossRef]
- Idomir, M.E. providencia species-involvement in pathology and multidrug resistance in a Romanian county hospital. Bull. Transilv. Univ. Brasov Med. Sci. Ser. VI 2021, 14, 43–50. [Google Scholar] [CrossRef]
- Micoli, F.; Bagnoli, F.; Rappuoli, R.; Serruto, D. The role of vaccines in combatting antimicrobial resistance. Nat. Rev. Microbiol. 2021, 19, 287–302. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E. V Microbial genome analysis: The COG approach. Brief. Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef]
- Ehsan, N.; Ahmad, S.; Navid, A.; Azam, S.S. Identification of potential antibiotic targets in the proteome of multi-drug resistant Proteus mirabilis. Meta Gene 2018, 18, 167–173. [Google Scholar] [CrossRef]
- Uddin, R.; Jamil, F. Prioritization of potential drug targets against P. aeruginosa by core proteomic analysis using computational subtractive genomics and Protein-Protein interaction network. Comput. Biol. Chem. 2018, 74, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Da Fiúza, T.S.; de Souza, G.A. Identification of core immunogenic peptides of Shigella sonnei for a Peptide-Based Vaccine. Bio-Manguinhos 2021, 5, 102. [Google Scholar]
- Li, J.; Qiu, J.; Huang, Z.; Liu, T.; Pan, J.; Zhang, Q.; Liu, Q. Reverse vaccinology approach for the identifications of potential vaccine candidates against Salmonella. Int. J. Med. Microbiol. 2021, 311, 151508. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Ghufran, M.; Khan, A.; Azam, S.S.; Uddin, R.; Waqas, M.; Saleem, S. The methicillin-resistant S. epidermidis strain RP62A genome mining for potential novel drug targets identification. Gene Rep. 2017, 8, 88–93. [Google Scholar] [CrossRef]
- Paramasivam, N.; Linke, D. ClubSub-P: Cluster-based subcellular localization prediction for Gram-negative bacteria and archaea. Front. Microbiol. 2011, 2, 218. [Google Scholar] [CrossRef] [Green Version]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope prediction by novel immunoinformatics approach: A state-of-the-art review. Int. J. Pept. Res. Ther. 2020, 26, 1155–1163. [Google Scholar] [CrossRef]
- Yasser, E.-M.; Dobbs, D.; Honavar, V.G. In silico prediction of linear B-cell epitopes on proteins. In Prediction of Protein Secondary Structure; Springer: Berlin/Heidelberg, Germany, 2017; pp. 255–264. [Google Scholar]
- Kollmann, T.R. Variation between populations in the innate immune response to vaccine adjuvants. Front. Immunol. 2013, 4, 81. [Google Scholar] [CrossRef] [Green Version]
- Albagi, S.; Ahmed, O.H.; Gumaa, M.A.; Abd_elrahman, K.A.; Abu-Haraz, A.H. Immunoinformatics-peptide driven vaccine and in silico modeling for Duvenhage rabies virus glycoprotein G. J Clin Cell Immunol 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Kumar, A. Designing an efficient multi-epitope vaccine against Campylobacter jejuni using immunoinformatics and reverse vaccinology approach. Microb. Pathog. 2020, 147, 104398. [Google Scholar] [CrossRef] [PubMed]
- Bibi, N.; Zaidi, N.-S.S.; Tahir, M.; Babar, M.M. Vaccinomics driven proteome-wide screening of Haemophilus influenzae for the prediction of common putative vaccine candidates. Can. J. Microbiol. 2021, 67, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Sanami, S.; Zandi, M.; Pourhossein, B.; Mobini, G.-R.; Safaei, M.; Abed, A.; Arvejeh, P.M.; Chermahini, F.A.; Alizadeh, M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020, 164, 871–883. [Google Scholar] [CrossRef]
- Jafari, E.; Mahmoodi, S. Design, expression, and purification of a multi-epitope vaccine against Helicobacter pylori based on Melittin as an adjuvant. Microb. Pathog. 2021, 157, 104970. [Google Scholar] [CrossRef]
- Javadi, M.; Oloomi, M.; Bouzari, S. In Silico Design of a Poly-epitope Vaccine for Urinary Tract Infection Based on Conserved Antigens by Modern Vaccinology. Int. J. Pept. Res. Ther. 2021, 27, 909–921. [Google Scholar] [CrossRef]
- Ismail, M.; Sajid, Z.; Ali, A.; Wu, X.; Muhammad, S.A.; Shaikh, R.S. Prediction of Prophylactic Peptide Vaccine Candidates for Human Papillomavirus (HPV): Immunoinformatics and Reverse Vaccinology Approaches. Curr. Proteom. 2021, 18, 178–192. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Varadarajan, R. Insights into protein structure, stability and function from saturation mutagenesis. Curr. Opin. Struct. Biol. 2018, 50, 117–125. [Google Scholar] [CrossRef]
- Sayed, S.B.; Nain, Z.; Khan, M.S.A.; Abdulla, F.; Tasmin, R.; Adhikari, U.K. Exploring lassa virus proteome to design a multi-epitope vaccine through immunoinformatics and immune simulation analyses. Int. J. Pept. Res. Ther. 2020, 26, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir ul Qamar, M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef]
- Jyotisha, S.S.; Qureshi, I.A. Multi-epitope vaccine against SARS-CoV-2 applying immunoinformatics and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2020. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7682209/ (accessed on 18 June 2021).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Nemati, A.S.; Tafrihi, M.; Sheikhi, F.; Tabari, A.R.; Haditabar, A. Designing a New Multi Epitope-Based Vaccine against COVID-19 Disease: An Immunoinformatic Study Based on Reverse Vaccinology Approach. 2021. Available online: https://assets.researchsquare.com/files/rs-206270/v1/17abe418-b7e9-4ff5-84da-eaae0965ca5d.pdf?c=1631875889 (accessed on 18 June 2021).
- Rizvi, S.M.; Raghavan, M. Direct peptide-regulatable interactions between MHC class I molecules and tapasin. Proc. Natl. Acad. Sci. USA 2006, 103, 18220–18225. [Google Scholar] [CrossRef] [Green Version]
- Bonvin, A.M.J.J. Flexible protein–protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofving, T. Small Intestinal Neuroendocrine Tumours—Disease Models, Tumour Development, and Remedy. Ph.D. Thesis, University of Gothenburg, Gothenburg, Sweden, 2019. Available online: https://gupea.ub.gu.se/bitstream/2077/58488/5/gupea_2077_58488_5.pdf (accessed on 18 June 2021).
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, K.K.; Srivastava, A.; Panda, M.K.; Singh, Y.D.; Maharana, R.; Mandal, K.; Singh, B.S.M.; Singh, D.; Das, M.; Murmu, D. Computational intelligence in vaccine design against COVID-19. In Computational Intelligence Methods in COVID-19: Surveillance, Prevention, Prediction and Diagnosis; Springer: Berlin/Heidelberg, Germany, 2021; pp. 311–329. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagumuthu, M.; Rajpoot, S.; Baig, M.S. Structure-based design of novel peptidomimetics targeting the SARS-CoV-2 spike protein. Cell. Mol. Bioeng. 2021, 14, 177–185. [Google Scholar] [CrossRef]
- Rafi, S.; Yasmin, S.; Uddin, R. A molecular dynamic simulation approach: Development of dengue virus vaccine by affinity improvement techniques. J. Biomol. Struct. Dyn. 2022, 40, 61–76. [Google Scholar] [CrossRef]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct. Dyn. 2021, 39, 3793–3801. [Google Scholar] [CrossRef]
- Mahapatra, S.R.; Dey, J.; Kushwaha, G.S.; Puhan, P.; Mohakud, N.K.; Panda, S.K.; Lata, S.; Misra, N.; Suar, M. Immunoinformatic approach employing modeling and simulation to design a novel vaccine construct targeting MDR efflux pumps to confer wide protection against typhoidal Salmonella serovars. J. Biomol. Struct. Dyn. 2021, 1–13. Available online: https://pubmed.ncbi.nlm.nih.gov/34463211/ (accessed on 16 June 2021). [CrossRef]
- Vrancianu, C.O.; Gheorghe, I.; Czobor, I.B.; Chifiriuc, M.C. Antibiotic resistance profiles, molecular mechanisms and innovative treatment strategies of Acinetobacter baumannii. Microorganisms 2020, 8, 935. [Google Scholar] [CrossRef]
- Rosini, R.; Nicchi, S.; Pizza, M.; Rappuoli, R. Vaccines against antimicrobial resistance. Front. Immunol. 2020, 11, 1048. [Google Scholar] [CrossRef] [PubMed]
- Mat Rahim, N.; Lee, H.; Strych, U.; AbuBakar, S. Facing the challenges of multidrug-resistant Acinetobacter baumannii: Progress and prospects in the vaccine development. Hum. Vaccines Immunother. 2021, 17, 3784–3794. [Google Scholar] [CrossRef]
- Mbelle, N.M.; Osei Sekyere, J.; Amoako, D.G.; Maningi, N.E.; Modipane, L.; Essack, S.Y.; Feldman, C. Genomic analysis of a multidrug-resistant clinical Providencia rettgeri (PR002) strain with the novel integron ln1483 and an A/C plasmid replicon. Ann. N. Y. Acad. Sci. 2020, 1462, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ntshobeni, N.B.; Allam, M.; Ismail, A.; Amoako, D.G.; Essack, S.Y.; Chenia, H.Y. Draft genome sequence of Providencia rettgeri APW139_S1, an NDM-18-producing clinical strain originating from hospital effluent in South Africa. Microbiol. Resour. Announc. 2019, 8, e00259-19. [Google Scholar] [CrossRef] [Green Version]
- Principi, N.; Esposito, S. Vaccine-preventable diseases, vaccines and Guillain-Barre’syndrome. Vaccine 2019, 37, 5544–5550. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Samuels, E.L.; Bocchini Jr, J.A. Vaccine development: From laboratory to policy. Pediatr. Ann. 2020, 49, e509–e515. [Google Scholar] [CrossRef] [PubMed]
- Palatnik-de-Sousa, C.B. What Would Jenner and Pasteur Have Done About COVID-19 Coronavirus? The Urges of a Vaccinologist. Front. Immunol. 2020, 11, 2173. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sette, A.; Rappuoli, R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Noé, A.; Cargill, T.N.; Nielsen, C.M.; Russell, A.J.C.; Barnes, E. The application of single-cell RNA sequencing in vaccinology. J. Immunol. Res. 2020, 2020, 8624963. [Google Scholar] [CrossRef]
- Goumari, M.M.; Farhani, I.; Nezafat, N.; Mahmoodi, S. Multi-epitope vaccines (MEVs), as a novel strategy against infectious diseases. Curr. Proteom. 2020, 17, 354–364. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine Target | Physiochemical Properties | |||||
|---|---|---|---|---|---|---|
| Extracellular Proteins | Amino Acid | GRAVY | Aliphatic Index | Instability Index | PI | MW |
| >core/4909/2/Org2_Gene2897 (fimbrial protein) | 176 | 0.152 | 94.37 | 15.92 | 5.03 | 18.07 |
| >core/1455/14/Org14_Gene1001(flagellar hook protein FlgE) | 422 | −0.405 | 68.41 | 20.52 | 4.53 | 44.53 |
| Outer Membrane Proteins | ||||||
| >core/3432/1/Org1_Gene4222 (flagellar basal body L-ring protein FlgH | 260 | −0.229 | 85.54 | 37.68 | 9.21 | 27.93 |
| Periplasmic Proteins | ||||||
| >core/6354/1/Org1_Gene719 (flagellar hook-basal body complex protein FliE) | 114 | −0.179 | 91.4 | 40.73 | 5.29 | 12.72 |
| >core/4058/2/Org2_Gene3304 (flagellar basal body P-ring formation protein FlgA) | 234 | −0.259 | 89.57 | 34.84 | 9.68 | 26.13 |
| >core/3402/4/Org4_Gene1551(Gram-negative pili assembly chaperone domain proteins | 273 | −0.478 | 79.3 | 35.34 | 9.45 | 30.48 |
| Selected Epitopes | DRB*0101 IC50 Predicted Score | Antigenicity | Solubility | ToxinPred | Allergenicity |
|---|---|---|---|---|---|
| KALPSAGST | 20.94 | 0.6431 | Soluble | Non-toxic | Non-allergen |
| NFKDGPITR | 88.92 | 0.9418 | |||
| FDVDNPDDS | 4.2 | 0.5464 | |||
| SVYFVKTAD | 94.19 | 0.6127 | |||
| YAKDANDTA | 22.13 | 0.9316 | |||
| GDDPVVTPI | 35.97 | 0.8934 | |||
| PGDDPVVTP | 52.48 | 0.8771 | |||
| ESSTISQQQ | 22.8 | 1.0044 | |||
| YFRKIHGKQ | 21.33 | 1.3281 | |||
| TSSMVRRPW | 20.45 | 0.695 |
| Vaccine Complex | Interactive Residues |
|---|---|
| MHC-I | GLU128, ARG111, ARG65, LYS68, HIS168, GLU4, LYS3, GLN155, HIS67, GLY8, TYR80, YR116, ALA117, ARG144, TYR1, ALA69, VAL67, LYS68, GLU19, VAL76, GLN72, PRO5TYR73, VAL6, TYR159, TRP147, LYS146, ALA150, TYRE84. |
| MHC-II | SER77, ARG66, TYR32, ASN33, ILU82, ASN84, THR83, THR133, PRO86, PRO87, PHE145, LEU138, ASP57, PRO61, PHE109, ARG72, SER110, PHE116, TYR107, GLE113, CYS115, ARG220, TYR104, TYR101, THR100, CYS138, PRO137, TYR139, TYR136, SER42, ASP41, VAL44, ARG25, GLU141, HIS16, ASP116, HIS143, GLN36, ARG40, LEU144, LYS111, PRO86, ASN62, TYR160 |
| TLR-4 | GLN599, ARG589, GLU60, GLY617, GLU485, PRO88, LEU87, ILE80, LYS91, LEU78, VAL135, ASP70, LYS84, GLU87, ARG88, THR92, ALA131, LYS130, SER134, PRO140, GLY143, PHE145, ASP147, ILE150, ALA97, ASP189, ALA187, ALA190, THR193, GLY181, GLY171, ASN65, VAL33, GLU31, LEU37, ILE80, LEU78, LYS91, GLU143, LYS89, ASN86, PHE151, GLU48, ALA462, PRO88, TYR540, ASN526, GLN37, THR11, GLN597, ASP596, VAL604, GLN37, VAL8, GLU 03, ASP29, PHE10, ASP580, GLN547 |
| Energy Parameter | TLR-4–Vaccine Complex | MHC-I–Vaccine Complex | MHC-II–Vaccine Complex |
|---|---|---|---|
| MM-GBSA | |||
| VDWAALS | −78.13 | −72.88 | −70.18 |
| EEL | −69.74 | −51.12 | −60.57 |
| EGB | 65.10 | 57.08 | 60.11 |
| ESURF | −8.13 | −8.15 | −7.18 |
| Delta G gas | −147.87 | −124 | −130.75 |
| Delta G solv | 56.97 | 48.93 | 52.93 |
| Delta Total | −90.9 | −75.07 | −77.82 |
| MM-PBSA | |||
| VDWAALS | −78.13 | −72.88 | −80.24 |
| EEL | −69.74 | −51.12 | −62.58 |
| EPB | 69.10 | 48.97 | 56.67 |
| ENPOLAR | −6.40 | −7.10 | −9.18 |
| Delta G gas | −147.87 | −124 | −130.75 |
| Delta G solv | 62.7 | 41.87 | 47.49 |
| Delta Total | −85.17 | −82.13 | −83.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gul, S.; Ahmad, S.; Ullah, A.; Ismail, S.; Khurram, M.; Tahir ul Qamar, M.; Hakami, A.R.; Alkhathami, A.G.; Alrumaihi, F.; Allemailem, K.S. Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach. Vaccines 2022, 10, 189. https://doi.org/10.3390/vaccines10020189
Gul S, Ahmad S, Ullah A, Ismail S, Khurram M, Tahir ul Qamar M, Hakami AR, Alkhathami AG, Alrumaihi F, Allemailem KS. Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach. Vaccines. 2022; 10(2):189. https://doi.org/10.3390/vaccines10020189
Chicago/Turabian StyleGul, Saba, Sajjad Ahmad, Asad Ullah, Saba Ismail, Muhammad Khurram, Muhammad Tahir ul Qamar, Abdulrahim R. Hakami, Ali G. Alkhathami, Faris Alrumaihi, and Khaled S. Allemailem. 2022. "Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach" Vaccines 10, no. 2: 189. https://doi.org/10.3390/vaccines10020189