
Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies


Abstract
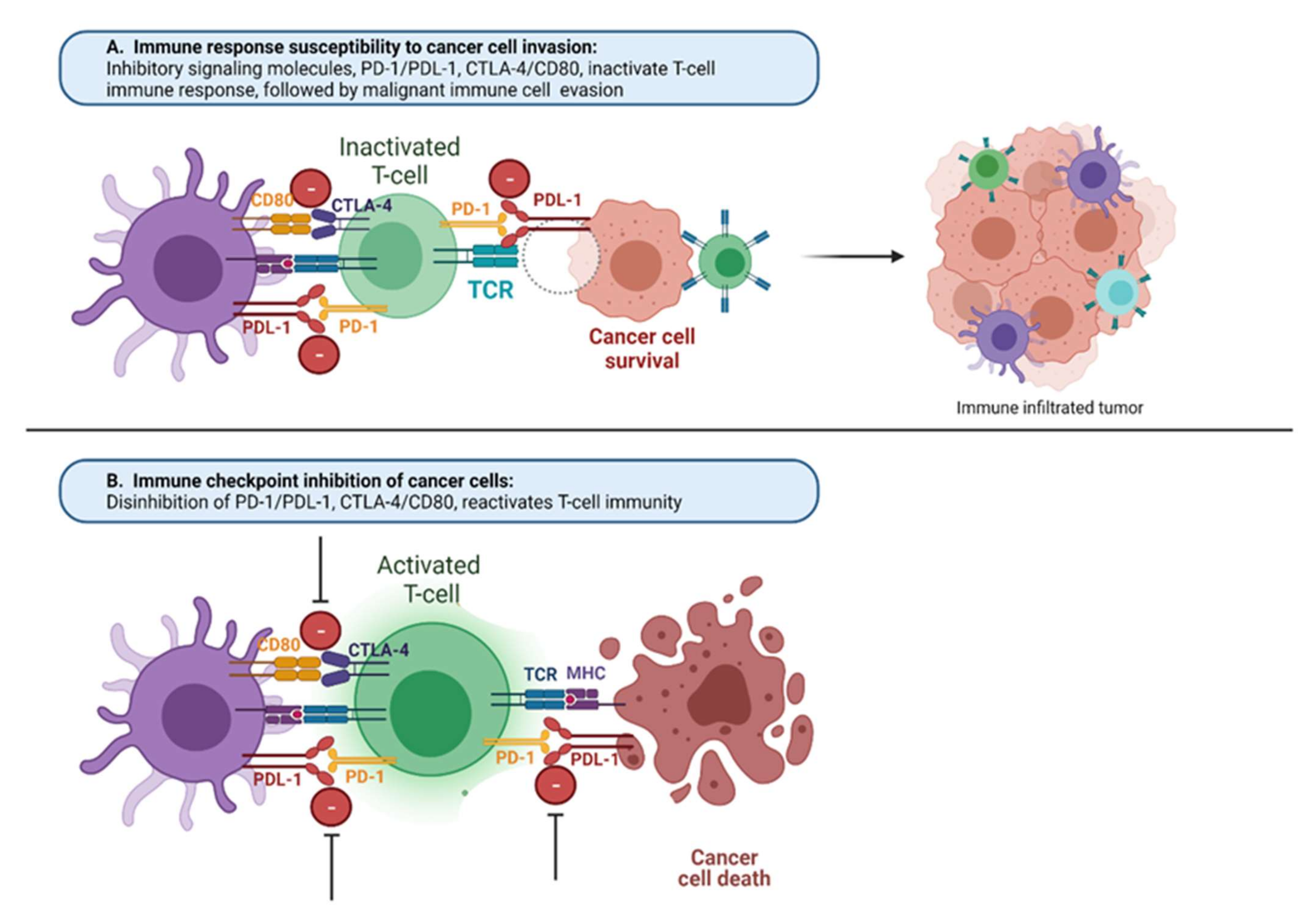
:1. Introduction
2. Adaptive Immune Response Modulation
3. Additional Stimulatory Molecules
4. CTLA4 Pathway
5. PD-1 Pathway
6. Additional Inhibitory Checkpoint Molecules
7. Innate and Antibody-Directed Tumor-Immune Modulation
8. Mechanisms Driving IRAEs
- The release from inhibition of preexisting self-recognizing effector T cells [140];
- A shift toward a more active and less selective innate immune response;
- Direct damage resulting from specific molecule expression on target cells.
9. Cardiotoxicity of ICIs
10. Myocarditis
11. Pericarditis
12. Arrhythmias
13. Atherosclerosis
14. Hypertension
15. Thromboembolism
16. Emerging Treatment of Cardiac IRAEs
17. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Salem, J.-E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef] [Green Version]
- Salem, J.-E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.-P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef]
- Hauck, A.J.; Kearney, D.L.; Edwards, W.D. Evaluation of postmortem endomyocardial biopsy specimens from 38 patients with lymphocytic myocarditis: Implications for role of sampling error. Mayo Clin. Proc. 1989, 64, 1235–1245. [Google Scholar] [CrossRef]
- Agrawal, N.; Khunger, A.; Vachhani, P.; Colvin, T.A.; Hattoum, A.; Spangenthal, E.; Curtis, A.B.; Dy, G.K.; Ernstoff, M.S.; Puzanov, I. Cardiac Toxicity Associated with Immune Checkpoint Inhibitors: Case Series and Review of the Literature. Case Rep. Oncol. 2019, 12, 260–276. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated with Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef]
- Escudier, M.; Cautela, J.; Malissen, N.; Ancedy, Y.; Orabona, M.; Pinto, J.; Monestier, S.; Grob, J.-J.; Scemama, U.; Jacquier, A.; et al. Clinical Features, Management, and Outcomes of Immune Checkpoint Inhibitor-Related Cardiotoxicity. Circulation 2017, 136, 2085–2087. [Google Scholar] [CrossRef] [PubMed]
- Drobni, Z.D.; Zafar, A.; Zubiri, L.; Zlotoff, D.A.; Alvi, R.M.; Lee, C.; Hartmann, S.; Gilman, H.K.; Villani, A.; Nohria, A.; et al. Decreased Absolute Lymphocyte Count and Increased Neutrophil/Lymphocyte Ratio with Immune Checkpoint Inhibitor-Associated Myocarditis. J. Am. Heart Assoc. 2020, 9, e018306. [Google Scholar] [CrossRef] [PubMed]
- Guha, A.; Al-Kindi, S.; Jain, P.; Tashtish, N.; ElAmm, C.; Oliveira, G.H. Association between myocarditis and other immune-related adverse events secondary to immune checkpoint inhibitor use. Int. J. Cancer 2020, 147, 1753–1754. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Yang, E.H.; Molina, J.R.; Bailey, K.R.; Blumenthal, R.S.; Kopecky, S.L. Cardiovascular Health and Outcomes in Cancer Patients Receiving Immune Checkpoint Inhibitors. Am. J. Cardiol. 2020, 125, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Kreileder, M.; Barrett, I.; Bendtsen, C.; Brennan, D.; Kolch, W. Signaling Dynamics Regulating Crosstalks between T-Cell Activation and Immune Checkpoints. Trends Cell Biol. 2021, 31, 224–235. [Google Scholar] [CrossRef]
- González-Navajas, J.M.; Fan, D.D.; Yang, S.; Yang, F.M.; Lozano-Ruiz, B.; Shen, L.; Lee, J. The Impact of Tregs on the Anticancer Immunity and the Efficacy of Immune Checkpoint Inhibitor Therapies. Front. Immunol. 2021, 12, 625783. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- de Simone, M.; Arrigoni, A.; Rossetti, G.; Gruarin, P.; Ranzani, V.; Politano, C.; Bonnal, R.J.P.; Provasi, E.; Sarnicola, M.L.; Panzeri, I.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016, 45, 1135–1147. [Google Scholar] [CrossRef] [Green Version]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y.; et al. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef]
- Jeong, S.; Park, S.-H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 2020, 20, e3. [Google Scholar] [CrossRef]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vence, L.; Bucktrout, S.L.; Fernandez Curbelo, I.; Blando, J.; Smith, B.M.; Mahne, A.E.; Lin, J.C.; Park, T.; Pascua, E.; Sai, T.; et al. Characterization and Comparison of GITR Expression in Solid Tumors. Clin. Cancer Res. 2019, 25, 6501–6510. [Google Scholar] [CrossRef] [Green Version]
- Tran, B.; Carvajal, R.D.; Marabelle, A.; Patel, S.P.; LoRusso, P.M.; Rasmussen, E.; Juan, G.; Upreti, V.V.; Beers, C.; Ngarmchamnanrith, G.; et al. Dose escalation results from a first-in-human, phase 1 study of glucocorticoid-induced TNF receptor-related protein agonist AMG 228 in patients with advanced solid tumors. J. Immunother. Cancer 2018, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, D.A.; Budhu, S.; Liu, C.; Bryson, C.; Malandro, N.; Cohen, A.; Zhong, H.; Yang, X.; Houghton, A.N.; Merghoub, T.; et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol. Res. 2013, 1, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Polesso, F.; Sarker, M.; Weinberg, A.D.; Murray, S.E.; Moran, A.E. OX40 Agonist Tumor Immunotherapy Does Not Impact Regulatory T Cell Suppressive Function. J. Immunol. 2019, 203, 2011–2019. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Etxeberría, I.; Otano, I.; Melero, I. Twists and turns to translating 4-1BB cancer immunotherapy. Sci. Transl. Med. 2019, 11, eaax4738. [Google Scholar] [CrossRef]
- He, Y.; Vlaming, M.; van Meerten, T.; Bremer, E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers 2022, 14, 299. [Google Scholar] [CrossRef]
- Amatore, F.; Gorvel, L.; Olive, D. Role of Inducible Co-Stimulator (ICOS) in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 141–150. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Carlino, M.; Joerger, M.; di Nicola, M.; Meniawy, T.; Rottey, S.; Moreno, V.; Gazzah, A.; Delord, J.-P.; Paz-Ares, L.; et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination with Nivolumab for Patients with Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2020, 6, 100–107. [Google Scholar]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.E.W.; Pishvaian, M.J.; Shepard, D.R.; Wang, D.; Weiss, J.; Johnson, M.L.; Chung, C.H.; Chen, Y.; Huang, B.; Davis, C.B.; et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J. Immunother. Cancer 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Gu-Trantien, C.; Willard-Gallo, K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open 2020, 5, e000544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraehenbuehl, L.; Weng, C.-H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef]
- Speiser, D.E.; Utzschneider, D.T.; Oberle, S.G.; Münz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Gallimore, A.; Hengartner, H.; Zinkernagel, R. Hierarchies of antigen-specific cytotoxic T-cell responses. Immunol. Rev. 1998, 164, 29–36. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining “T cell exhaustion”. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Marengère, L.E.; Waterhouse, P.; Duncan, G.S.; Mittrücker, H.W.; Feng, G.S.; Mak, T.W. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar] [CrossRef]
- Chuang, E.; Fisher, T.S.; Morgan, R.W.; Robbins, M.D.; Duerr, J.M.; vander Heiden, M.G.; Gardner, J.P.; Hambor, J.E.; Neveu, M.J.; Thompson, C.B. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 2000, 13, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Lingel, H.; Wissing, J.; Arra, A.; Schanze, D.; Lienenklaus, S.; Klawonn, F.; Pierau, M.; Zenker, M.; Jänsch, L.; Brunner-Weinzierl, M.C. CTLA-4-mediated posttranslational modifications direct cytotoxic T-lymphocyte differentiation. Cell Death Differ. 2017, 24, 1739–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, D.; Betge, S.; Windisch, A.; Pistulli, R.; Rohm, I.; Fritzenwanger, M.; Jung, C.; Schubert, K.; Theis, B.; Petersen, I.; et al. Recruitment of circulating dendritic cell precursors into the infarcted myocardium and pro-inflammatory response in acute myocardial infarction. Clin. Sci. 2012, 123, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Chen, H.; Liakou, C.I.; Kamat, A.; Pettaway, C.; Ward, J.F.; Tang, D.N.; Sun, J.; Jungbluth, A.A.; Troncoso, P.; Logothetis, C.; et al. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [Green Version]
- Carthon, B.C.; Wolchok, J.D.; Yuan, J.; Kamat, A.; Ng Tang, D.S.; Sun, J.; Ku, G.; Troncoso, P.; Logothetis, C.J.; Allison, J.; et al. Preoperative CTLA-4 blockade: Tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin. Cancer Res. 2010, 16, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef]
- Jost, S.; Altfeld, M. Control of human viral infections by natural killer cells. Annu. Rev. Immunol. 2013, 31, 163–194. [Google Scholar] [CrossRef]
- Judge, S.J.; Dunai, C.; Aguilar, E.G.; Vick, S.C.; Sturgill, I.R.; Khuat, L.T.; Stoffel, K.M.; Van Dyke, J.; Longo, D.L.; Darrow, M.A.; et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig. 2020, 130, 3051–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, P.; Guethlein, L.A. Genetics of Natural Killer Cells in Human Health, Disease, and Survival. Annu. Rev. Immunol. 2018, 36, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [Green Version]
- Youngnak, P.; Kozono, Y.; Kozono, H.; Iwai, H.; Otsuki, N.; Jin, H.; Omura, K.; Yagita, H.; Pardoll, D.M.; Chen, L.; et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem. Biophys. Res. Commun. 2003, 307, 672–677. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry R v Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Molawi , K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Xu, W.; Guo, Q.; Jiang, Z.; Wang, P.; Yue, Y.; Xiong, S. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 2009, 105, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Thibult, M.-L.; Mamessier, E.; Gertner-Dardenne, J.; Pastor, S.; Just-Landi, S.; Xerri, L.; Chetaille, B.; Olive, D. PD-1 is a novel regulator of human B-cell activation. Int. Immunol. 2013, 25, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-M.; Li, C.-W.; Lai, Y.-J.; Hung, M.-C. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-C.; Chen, W.-D.; Alvarez, J.B.; Jia, K.; Shi, L.; Wang, Q.; Zou, N.; He, K.; Zhu, H. Cancer immune checkpoint blockade therapy and its associated autoimmune cardiotoxicity. Acta Pharmacol. Sin. 2018, 39, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Scala, E.; Carbonari, M.; del Porto, P.; Cibati, M.; Tedesco, T.; Mazzone, A.M.; Paganelli, R.; Fiorilli, M. Lymphocyte activation gene-3 (LAG-3) expression and IFN-gamma production are variably coregulated in different human T lymphocyte subpopulations. J. Immunol. 1998, 161, 489–493. [Google Scholar]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Takamura, S.; Tsuji-Kawahara, S.; Yagita, H.; Akiba, H.; Sakamoto, M.; Chikaishi, T.; Kato, M.; Miyazawa, M. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J. Immunol. 2010, 184, 4696–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. Available online: https://pubmed.ncbi.nlm.nih.gov/25363763/ (accessed on 1 January 2022). [CrossRef] [PubMed] [Green Version]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef]
- Sabatos, C.A.; Chakravarti, S.; Cha, E.; Schubart, A.; Sánchez-Fueyo, A.; Zheng, X.X.; Coyle, A.J.; Strom, T.B.; Freeman, G.J.; Kuchroo, V.K. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003, 4, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-T.; Chen, C.-H.; Ku, K.-L.; Hsiao, M.; Chiang, C.-P.; Hsu, T.-L.; Chen, M.H.; Wong, C.H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Tekle, C.; Nygren, M.K.; Chen, Y.-W.; Dybsjord, I.; Nesland, J.M.; Maelandsmo, G.M.; Fodstad, O. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int. J. Cancer 2012, 130, 2282–2290. [Google Scholar] [CrossRef]
- Chen, C.; Shen, Y.; Qu, Q.-X.; Chen, X.-Q.; Zhang, X.-G.; Huang, J.-A. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp. Cell Res. 2013, 319, 96–102. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Toor, S.M.; Sasidharan Nair, V.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2020, 65, 1–12. [Google Scholar] [CrossRef]
- Gavrieli, M.; Watanabe, N.; Loftin, S.K.; Murphy, T.L.; Murphy, K.M. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem. Biophys. Res. Commun. 2003, 312, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.W.; Gill, D.M.; Pal, S.K.; Agarwal, N. The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy 2017, 9, 681–692. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. Available online: https://pubmed.ncbi.nlm.nih.gov/31873309/ (accessed on 1 January 2022). [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Chin, R.; Chen, C.-H.; Budhani, P.; Ai, M.; Reilley, M.J.; Sebastian, M.M.; Hong, D.S.; et al. Activation of 4-1BB on Liver Myeloid Cells Triggers Hepatitis via an Interleukin-27-Dependent Pathway. Clin. Cancer Res. 2018, 24, 1138–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfahani, K.; Elkrief, A.; Calabrese, C.; Lapointe, R.; Hudson, M.; Routy, B.; Miller, W.H., Jr.; Calabrese, L. Moving towards personalized treatments of immune-related adverse events. Nat. Rev. Clin. Oncol. 2020, 17, 504–515. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 437, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, X.J.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity 2019, 51, 1043–1058.e4. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855.e5. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chikuma, S.; Hori, S.; Fagarasan, S.; Honjo, T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl. Acad. Sci. USA 2016, 113, 8490–8495. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Granito, A.; Muratori, L.; Lalanne, C.; Quarneti, C.; Ferri, S.; Guidi, M.; Lenzi, M.; Muratori, P. Hepatocellular carcinoma in viral and autoimmune liver diseases: Role of CD4+ CD25+ Foxp3+ regulatory T cells in the immune microenvironment. World J. Gastroenterol. 2021, 27, 2994–3009. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- da Gama Duarte, J.; Parakh, S.; Andrews, M.C.; Woods, K.; Pasam, A.; Tutuka, C.; Ostrouska, S.; Blackburn, J.M.; Behren, A.; Cebon, J. Autoantibodies May Predict Immune-Related Toxicity: Results from a Phase I Study of Intralesional Bacillus Calmette-Guérin followed by Ipilimumab in Patients with Advanced Metastatic Melanoma. Front. Immunol. 2018, 9, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Bar, N.; Ferreira, M.; Newman, A.M.; Zhang, L.; Bailur, J.K.; Bacchiocchi, A.; Kluger, H.; Wei, W.; Halaban, R.; et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J. Clin. Investig. 2018, 128, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Gowen, M.F.; Giles, K.M.; Simpson, D.; Tchack, J.; Zhou, H.; Moran, U.; Dawood, Z.; Pavlick, A.C.; Hu, S.; Wilson, M.A.; et al. Baseline antibody profiles predict toxicity in melanoma patients treated with immune checkpoint inhibitors. J. Transl. Med. 2018, 16, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, S.A.; Gao, J.; Miura, Y.; Blando, J.; Tidwell, R.S.S.; Zhao, H.; Subudhi, S.K.; Tawbi, H.; Keung, E.; Wargo, J.; et al. Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc. Natl. Acad. Sci. USA 2019, 116, 22246–22251. [Google Scholar] [CrossRef]
- Iwama, S.; de Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci. Transl. Med. 2014, 6, 230ra45. [Google Scholar] [CrossRef]
- Esfahani, K.; Meti, N.; Miller, W.H.; Hudson, M. Adverse events associated with immune checkpoint inhibitor treatment for cancer. CMAJ Can. Med. Assoc. J. 2019, 191, E40–E46. [Google Scholar] [CrossRef] [Green Version]
- Kurimoto, C.; Inaba, H.; Ariyasu, H.; Iwakura, H.; Ueda, Y.; Uraki, S.; Takeshima, K.; Furukawa, Y.; Morita, S.; Yamamoto, Y.; et al. Predictive and sensitive biomarkers for thyroid dysfunctions during treatment with immune-checkpoint inhibitors. Cancer Sci. 2020, 111, 1468–1477. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, P.; Wood, K.; Wassmann, K.; Christenfeld, A.M.; Bhardwaj, N.; Oh, W.K. A whole-blood RNA transcript-based gene signature is associated with the development of CTLA-4 blockade-related diarrhea in patients with advanced melanoma treated with the checkpoint inhibitor tremelimumab. J. Immunother. Cancer 2018, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Hu, H.; Zakharov, P.N.; Peterson, O.J.; Unanue, E.R. Cytocidal macrophages in symbiosis with CD4 and CD8 T cells cause acute diabetes following checkpoint blockade of PD-1 in NOD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 31319–31330. [Google Scholar] [CrossRef]
- Gudd, C.L.C.; Au, L.; Triantafyllou, E.; Shum, B.; Liu, T.; Nathwani, R.; Kumar, N.; Mukherjee, S.; Dhar, A.; Woollard, K.J.; et al. Activation and transcriptional profile of monocytes and CD8+ T cells are altered in checkpoint inhibitor-related hepatitis. J. Hepatol. 2021, 75, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Mihic-Probst, D.; Reinehr, M.; Dettwiler, S.; Kolm, I.; Britschgi, C.; Kudura, K.; Maggio, E.M.; Lenggenhager, D.; Rushing, E.J. The role of macrophages type 2 and T-regs in immune checkpoint inhibitor related adverse events. Immunobiology 2020, 225, 152009. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Naidoo, J.; Zhong, Q.; Xiong, Y.; Mammen, J.; de Flores, M.V.; Cappelli, L.; Balaji, A.; Palmer, A.; Forde, P.M.; et al. The alveolar immune cell landscape is dysregulated in checkpoint inhibitor pneumonitis. J. Clin. Investig. 2019, 129, 4305–4315. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; H Yang, J.Y.; Ghazanfar, S.; Kefford, R.F.; et al. Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1-Based Immunotherapy. Clin. Cancer Res. 2019, 25, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Gerber, D.E. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin. Cancer Biol. 2020, 64, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Albarel, F.; Castinetti, F.; Brue, T. Management of endocrine disease: Immune check point inhibitors-induced hypophysitis. Eur. J. Endocrinol. 2019, 181, R107–R118. [Google Scholar] [CrossRef] [Green Version]
- Nishino, M.; Sholl, L.M.; Hodi, F.S.; Hatabu, H.; Ramaiya, N.H. Anti-PD-1-Related Pneumonitis during Cancer Immunotherapy. N. Engl. J. Med. 2015, 373, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Rapisuwon, S.; Izar, B.; Batenchuk, C.; Avila, A.; Mei, S.; Sorger, P.; Parks, J.M.; Cooper, S.J.; Wagner, D.; Zeck, J.C.; et al. Exceptional response and multisystem autoimmune-like toxicities associated with the same T cell clone in a patient with uveal melanoma treated with immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Berner, F.; Bomze, D.; Diem, S.; Ali, O.H.; Fässler, M.; Ring, S.; Niederer, R.; Ackermann, C.J.; Baumgaertner, P.; Pikor, N.; et al. Association of Checkpoint Inhibitor-Induced Toxic Effects With Shared Cancer and Tissue Antigens in Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, A.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Calle, N.; Rodriguez-Otero, P.; Villar, S.; Mejías, L.; Melero, I.; Prosper, F.; Marinello, P.; Paiva, B.; Idoate, M.; San-Miguel, J. Anti-PD1 associated fulminant myocarditis after a single pembrolizumab dose: The role of occult pre-existing autoimmunity. Haematologica 2018, 103, e318–e321. [Google Scholar] [CrossRef]
- Maher, V.E.; Fernandes, L.L.; Weinstock, C.; Tang, S.; Agarwal, S.; Brave, M.; Ning, Y.M.; Singh, H.; Suzman, D.; Xu, J.; et al. Analysis of the Association Between Adverse Events and Outcome in Patients Receiving a Programmed Death Protein 1 or Programmed Death Ligand 1 Antibody. J. Clin. Oncol. 2019, 37, 2730–2737. [Google Scholar] [CrossRef]
- Indini, A.; di Guardo, L.; Cimminiello, C.; Prisciandaro, M.; Randon, G.; de Braud, F.; del Vecchio, M. Immune-related adverse events correlate with improved survival in patients undergoing anti-PD1 immunotherapy for metastatic melanoma. J. Cancer Res. Clin. Oncol. 2019, 145, 511–521. [Google Scholar] [CrossRef]
- Sato, K.; Akamatsu, H.; Murakami, E.; Sasaki, S.; Kanai, K.; Hayata, A.; Tokudome, N.; Akamatsu, K.; Koh, Y.; Ueda, H.; et al. Correlation between immune-related adverse events and efficacy in non-small cell lung cancer treated with nivolumab. Lung Cancer 2018, 115, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Quandt, Z.; Bluestone, J.A. The Balancing Act between Cancer Immunity and Autoimmunity in Response to Immunotherapy. Cancer Immunol. Res. 2018, 6, 1445–1452. [Google Scholar] [CrossRef] [Green Version]
- Xing, P.; Zhang, F.; Wang, G.; Xu, Y.; Li, C.; Wang, S.; Guo, Y.; Cai, S.; Wang, Y.; Li, J. Incidence rates of immune-related adverse events and their correlation with response in advanced solid tumours treated with NIVO or NIVO+IPI: A systematic review and meta-analysis. J. Immunother. Cancer 2019, 7, 341. [Google Scholar] [CrossRef]
- Ricciuti, B.; Genova, C.; de Giglio, A.; Bassanelli, M.; Dal Bello, M.G.; Metro, G.; Brambilla, M.; Baglivo, S.; Grossi, F.; Chiari, R. Impact of immune-related adverse events on survival in patients with advanced non-small cell lung cancer treated with nivolumab: Long-term outcomes from a multi-institutional analysis. J. Cancer Res. Clin. Oncol. 2019, 145, 479–485. [Google Scholar] [CrossRef]
- Horvat, T.Z.; Adel, N.G.; Dang, T.-O.; Momtaz, P.; Postow, M.A.; Callahan, M.K.; Carvajal, R.D.; Dickson, M.A.; D’Angelo, S.P.; Woo, K.M.; et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J. Clin. Oncol. 2015, 33, 3193–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.H.; Wei, L.; Bertino, E.M.; Edd, T.; Villalona-Calero, M.A.; He, K.; Shields, P.G.; Carbone, D.P.; Otterson, G.A. Incidence, Risk Factors, and Effect on Survival of Immune-related Adverse Events in Patients with Non-Small-cell Lung Cancer. Clin. Lung Cancer 2018, 19, e893–e900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Infante, N.; Ramírez-Flores, Y.A.; Castillo, E.C.; Lozano, O.; García-Rivas, G.; Torre-Amione, G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: A meta-analysis. Eur. J. Heart Fail. 2021, 23, 1739–1747. [Google Scholar] [CrossRef]
- Dolladille, C.; Akroun, J.; Morice, P.-M.; Dompmartin, A.; Ezine, E.; Sassier, M.; Da-Silva, A.; Plane, A.-F.; Legallois, D.; L’Orphelin, J.-M.; et al. Cardiovascular immunotoxicities associated with immune checkpoint inhibitors: A safety meta-analysis. Eur. Heart J. 2021, 42, 4964–4977. [Google Scholar] [CrossRef] [PubMed]
- Agostinetto, E.; Eiger, D.; Lambertini, M.; Ceppi, M.; Bruzzone, M.; Pondé, N.; Plummer, C.; Awada, A.H.; Santoro, A.; Piccart-Gebhart, M.; et al. Cardiotoxicity of immune checkpoint inhibitors: A systematic review and meta-analysis of randomised clinical trials. Eur. J. Cancer 2021, 148, 76–91. [Google Scholar] [CrossRef]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 2012, 7, e36814. [Google Scholar]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.-S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 2009, 206, 497–505. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [PubMed]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1233-42. [Google Scholar] [PubMed]
- Tarrio, M.L.; Grabie, N.; Bu, D.; Sharpe, A.H.; Lichtman, A.H. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J. Immunol. 2012, 188, 4876–4884. [Google Scholar] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar]
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K.; et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Heart J. 2021, 14, 316–329. [Google Scholar]
- Jain, V.; Bahia, J.; Mohebtash, M.; Barac, A. Cardiovascular Complications Associated with Novel Cancer Immunotherapies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 36. [Google Scholar]
- Zimmer, L.; Goldinger, S.M.; Hofmann, L.; Loquai, C.; Ugurel, S.; Thomas, I.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.S.; Göppner, D.; et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur. J. Cancer 2016, 60, 210–225. [Google Scholar]
- Zamami, Y.; Niimura, T.; Okada, N.; Koyama, T.; Fukushima, K.; Izawa-Ishizawa, Y.; Ishizawa, K. Factors Associated With Immune Checkpoint Inhibitor-Related Myocarditis. JAMA Oncol. 2019, 5, 1635–1637. [Google Scholar]
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J. Transl. Med. 2018, 16, 94. [Google Scholar]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378. [Google Scholar] [PubMed]
- Wang, C.; Dehghani, B.; Li, Y.; Kaler, L.J.; Vandenbark, A.A.; Offner, H. Oestrogen modulates experimental autoimmune encephalomyelitis and interleukin-17 production via programmed death 1. Immunology 2009, 126, 329–335. [Google Scholar] [PubMed]
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.-A.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625. [Google Scholar] [PubMed]
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 2019, 58 (Suppl. S7), vii59–vii67. [Google Scholar] [PubMed] [Green Version]
- Ji, C.; Roy, M.D.; Golas, J.; Vitsky, A.; Ram, S.; Kumpf, S.W.; Martin, M.; Barletta, F.; Meier, W.A.; Hooper, A.T.; et al. Myocarditis in Cynomolgus Monkeys Following Treatment with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2019, 25, 4735–4748. [Google Scholar]
- Ganatra, S.; Neilan, T.G. Immune Checkpoint Inhibitor-Associated Myocarditis. Oncologist 2018, 23, 879–886. [Google Scholar]
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Martin, M.; Barletta, F.; Meier, W.A.; Hooper, A.T.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar]
- Keir, M.E.; Freeman, G.J.; Sharpe, A.H. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J. Immunol. 2007, 179, 5064–5070. [Google Scholar]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar]
- Parmacek, M.S.; Solaro, R.J. Biology of the troponin complex in cardiac myocytes. Prog. Cardiovasc. Dis. 2004, 47, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.A.; Menke, J.; Rabacal, W.A.; Schoen, F.J.; Sharpe, A.H.; Kelley, V.R. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J. Immunol. 2008, 181, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okazaki, I.-M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Love, V.A.; Grabie, N.; Duramad, P.; Stavrakis, G.; Sharpe, A.; Lichtman, A. CTLA-4 ablation and interleukin-12 driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ. Res. 2007, 101, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Okazaki, I.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; Baba, M.H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Quagliariello, V.; Passariello, M.; Rea, D.; Barbieri, A.; Iovine, M.; Bonelli, A.; Caronna, A.; Botti, G.; Lorenzo, C.D.; Maurea, N. Evidences of CTLA-4 and PD-1 Blocking Agents-Induced Cardiotoxicity in Cellular and Preclinical Models. J. Pers. Med. 2020, 10, 179. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Galdiero, M.R.; Varricchi, G. Cardiac Toxicity in Patients Treated with Immune Checkpoint Inhibitors: It Is Now Time for Cardio-Immuno-Oncology. J. Am. Coll. Cardiol. 2018, 71, 1765–1767. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, e000247. [Google Scholar] [CrossRef] [Green Version]
- Xing, Q.; Zhang, Z.-W.; Lin, Q.-H.; Shen, L.-H.; Wang, P.-M.; Zhang, S.; Fan, M.; Zhu, B. Myositis-myasthenia gravis overlap syndrome complicated with myasthenia crisis and myocarditis associated with anti-programmed cell death-1 (sintilimab) therapy for lung adenocarcinoma. Ann. Transl. Med. 2020, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Yanase, T.; Moritoki, Y.; Kondo, H.; Ueyama, D.; Akita, H.; Yasui, T. Myocarditis and myasthenia gravis by combined nivolumab and ipilimumab immunotherapy for renal cell carcinoma: A case report of successful management. Urol. Case Rep. 2021, 34, 101508. [Google Scholar] [CrossRef] [PubMed]
- Fazel, M.; Jedlowski, P.M. Severe Myositis, Myocarditis, and Myasthenia Gravis with Elevated Anti-Striated Muscle Antibody following Single Dose of Ipilimumab-Nivolumab Therapy in a Patient with Metastatic Melanoma. Case Rep. Immunol. 2019, 2019, 2539493. [Google Scholar]
- Kimura, T.; Fukushima, S.; Miyashita, A.; Aoi, J.; Jinnin, M.; Kosaka, T.; Ando, Y.; Matsukawa, M.; Inoue, H.; Kiyotani, K.; et al. Myasthenic crisis and polymyositis induced by one dose of nivolumab. Cancer Sci. 2016, 107, 1055–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, E.; Varese, P.; Agosti, S.; Celli, L.; Ghiglione, E.; Pappalardo, I.; Zaccone, G.; Paglia, A.; Morelli, N. Concomitant myasthenia gravis, myositis, myocarditis and polyneuropathy, induced by immune-checkpoint inhibitors: A life-threatening continuum of neuromuscular and cardiac toxicity. eNeurologicalSci 2019, 14, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-T.; Chen, Y.-P.; Lin, W.-C.; Su, W.-C.; Sun, Y.-T. Immune Checkpoint Inhibitor-Induced Myasthenia Gravis. Front. Neurol. 2020, 11, 634. [Google Scholar] [CrossRef] [PubMed]
- Palaskas, N.; Morgan, J.; Daigle, T.; Banchs, J.; Durand, J.-B.; Hong, D.; Naing, A.; Le, H.; Hassan, S.A.; Karimzad, K.; et al. Targeted Cancer Therapies With Pericardial Effusions Requiring Pericardiocentesis Focusing on Immune Checkpoint Inhibitors. Am. J. Cardiol. 2019, 123, 1351–1357. [Google Scholar] [CrossRef]
- Kolla, B.C.; Patel, M.R. Recurrent pleural effusions and cardiac tamponade as possible manifestations of pseudoprogression associated with nivolumab therapy—A report of two cases. J. Immunother. Cancer 2016, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Canale, M.L.; Camerini, A.; Casolo, G.; Lilli, A.; Bisceglia, I.; Parrini, I.; Lestuzzi, C.; Meglio, J.D.; Puccetti, C.; Camerini, L.; et al. Incidence of Pericardial Effusion in Patients with Advanced Non-Small Cell Lung Cancer Receiving Immunotherapy. Adv. Ther. 2020, 37, 3178–3184. [Google Scholar] [CrossRef]
- Altan, M.; Toki, M.I.; Gettinger, S.N.; Carvajal-Hausdorf, D.E.; Zugazagoitia, J.; Sinard, J.H.; Herbst, R.S.; Rimm, D.L. Immune Checkpoint Inhibitor-Associated Pericarditis. J. Thorac. Oncol. 2019, 14, 1102–1108. [Google Scholar] [CrossRef]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef] [PubMed]
- Mir, H.; Alhussein, M.; Alrashidi, S.; Alzayer, H.; Alshatti, A.; Valettas, N.; Mukherjee, S.D.; Nair, V.; Leong, D.P. Cardiac Complications Associated With Checkpoint Inhibition: A Systematic Review of the Literature in an Important Emerging Area. Can. J. Cardiol. 2018, 34, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, M.; Nielsen, D.; Svane, I.M.; Iversen, K.; Rasmussen, P.V.; Madelaire, C.; Fosbøl, E.; Køber, L.; Gustafsson, F.; Andersson, C.; et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: A nationwide Danish study. Eur. Heart J. 2021, 42, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zheng, Q.; Lee, J.; Ma, Z.; Zhu, Q.; Wang, Z. PD-1/PD-L1 expression on CD(4+) T cells and myeloid DCs correlates with the immune pathogenesis of atrial fibrillation. J. Cell. Mol. Med. 2015, 19, 1223–1233. [Google Scholar] [CrossRef]
- Vuong, J.T.; Stein-Merlob, A.F.; Nayeri, A.; Sallam, T.; Neilan, T.G.; Yang, E.H. Immune Checkpoint Therapies and Atherosclerosis: Mechanisms and Clinical Implications: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 577–593. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Zhang, Q.; Li, H.-J.; Michot, J.M.; Liu, H.-B.; Zhan, P.; Lv, T.-F.; Song, Y. Evaluation of rare but severe immune related adverse effects in PD-1 and PD-L1 inhibitors in non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2017, 6 (Suppl. S1), S8–S20. [Google Scholar] [CrossRef] [Green Version]
- Cautela, J.; Rouby, F.; Salem, J.-E.; Alexandre, J.; Scemama, U.; Dolladille, C.; Cohen, A.; Paganelli, F.; Ederhy, S.; Thuny, F. Acute Coronary Syndrome With Immune Checkpoint Inhibitors: A Proof-of-Concept Case and Pharmacovigilance Analysis of a Life-Threatening Adverse Event. Can. J. Cardiol. 2020, 36, 476–481. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef]
- Stemme, S.; Faber, B.; Holm, J.; Wiklund, O.; Witztum, J.L.; Hansson, G.K. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1995, 92, 3893–3897. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L.; Zheng, Q. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 2009, 46, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poels, K.; van Leent, M.M.T.; Boutros, C.; Tissot, H.; Roy, S.; Meerwaldt, A.E.; Toner, Y.C.A.; Reiche, M.E.; Kusters, P.J.H.; Malinova, T.; et al. Immune Checkpoint Inhibitor Therapy Aggravates T Cell-Driven Plaque Inflammation in Atherosclerosis. JACC CardioOncol. 2020, 2, 599–610. [Google Scholar] [CrossRef]
- Ewing, M.M.; Karper, J.C.; Abdul, S.; de Jong, R.C.M.; Peters, H.A.B.; de Vries, M.R.; Redeker, A.; Kuiper, J.; Toes, R.E.M.; Arens, R.; et al. T-cell co-stimulation by CD28-CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int. J. Cardiol. 2013, 168, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Poels, K.; van Leent, M.M.T.; Reiche, M.E.; Kusters, P.J.H.; Huveneers, S.; de Winther, M.P.J.; Mulder, W.J.M.; Lutgens, E.; Seijkens, T.T.P. Antibody-Mediated Inhibition of CTLA4 Aggravates Atherosclerotic Plaque Inflammation and Progression in Hyperlipidemic Mice. Cells 2020, 9, 1987. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of Cytotoxic T-Lymphocyte-Associated Antigen-4 Prevents Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1141–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foks, A.C.; Kuiper, J. Immune checkpoint proteins: Exploring their therapeutic potential to regulate atherosclerosis. Br. J. Pharmacol. 2017, 174, 3940–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykl, R.; Fischer, O.; Vykoupil, K.; Taborsky, M. A unique reason for coronary spasm causing temporary ST elevation myocardial infarction (inferior STEMI)—systemic inflammatory response syndrome after use of pembrolizumab. Arch. Med. Sci. Atheroscler. Dis. 2017, 2, e100–e102. [Google Scholar] [CrossRef]
- Otsu, K.; Tajiri, K.; Sakai, S.; Ieda, M. Vasospastic angina following immune checkpoint blockade. Eur. Heart J. 2020, 41, 1702. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hu, X.; Li, Q.; Dai, W.; Cheng, X.; Huang, W.; Yu, W.; Chen, M.; Guo, Y.; Yuan, G. Effectiveness and safety of toripalimab, camrelizumab, and sintilimab in a real-world cohort of hepatitis B virus associated hepatocellular carcinoma patients. Ann. Transl. Med. 2020, 8, 1187. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Aguilera, J.V.; Chintakuntlawar, A.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Icht, O.; Darzi, N.; Shimony, S.; Jacobi, O.; Reinhorn, D.; Landman, Y.; Mutai, R.; Averbuch, I.; Shochat, T.; Spectre, G.; et al. Venous thromboembolism incidence and risk assessment in lung cancer patients treated with immune checkpoint inhibitors. J. Thromb. Haemost. JTH 2021, 19, 1250–1258. [Google Scholar] [CrossRef]
- Solinas, C.; Saba, L.; Sganzerla, P.; Petrelli, F. Venous and arterial thromboembolic events with immune checkpoint inhibitors: A systematic review. Thromb. Res. 2020, 196, 444–453. [Google Scholar] [CrossRef]
- Goel, A.; Khorana, A.; Kartika, T.; Gowda, S.; Tao, D.L.; Thawani, R.; Shatzel, J.J. Assessing the risk of thromboembolism in cancer patients receiving immunotherapy. Eur. J. Haematol. 2021, 108, 271–277. [Google Scholar] [CrossRef]
- Roopkumar, J.; Swaidani, S.; Kim, A.S.; Thapa, B.; Gervaso, L.; Hobbs, B.P.; Wei, W.; Alban, T.J.; Funchain, P.; Kundu, S.; et al. Increased Incidence of Venous Thromboembolism with Cancer Immunotherapy. Med 2021, 2, 423–434.e3. [Google Scholar] [CrossRef]
- Moik, F.; Chan, W.-S.E.; Wiedemann, S.; Hoeller, C.; Tuchmann, F.; Aretin, M.-B.; Fuereder, T.; Zöchbauer-Müller, S.; Preusser, M.; Pabinger, I.; et al. Incidence, risk factors, and outcomes of venous and arterial thromboembolism in immune checkpoint inhibitor therapy. Blood 2021, 137, 1669–1678. [Google Scholar] [CrossRef]
- Guven, D.C.; Aksun, M.S.; Sahin, T.K.; Aktepe, O.H.; Yildirim, H.C.; Taban, H.; Ceylan, F.; Kertmen, N.; Arik, Z.; Dizdar, O.; et al. Poorer baseline performance status is associated with increased thromboembolism risk in metastatic cancer patients treated with immunotherapy. Support. Care Cancer 2021, 29, 5417–5423. [Google Scholar] [CrossRef]
- Gutierrez-Sainz, L.; Martinez-Marin, V.; Viñal, D.; Martinez-Perez, D.; Pedregosa, J.; Garcia-Cuesta, J.A.; Villamayor, J.; Zamora, P.; Pinto, A.; Redondo, A.; et al. Incidence of venous thromboembolic events in cancer patients receiving immunotherapy: A single-institution experience. Clin. Transl. Oncol. 2021, 23, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, X.; Sun, D.; Zhao, J.; Xu, Z.; Zhao, P.; Ma, Z.; Zhang, Y. Thromboembolic events associated with immune checkpoint inhibitors: A real-world study of data from the food and drug administration adverse event reporting system (FAERS) database. Int. Immunopharmacol. 2021, 98, 107818. [Google Scholar] [CrossRef]
- Deschênes-Simard, X.; Richard, C.; Galland, L.; Blais, F.; Desilets, A.; Malo, J.; Cvetkovic, L.; Belkaid, W.; Elkrief, A.; Gagné, A.; et al. Venous thrombotic events in patients treated with immune checkpoint inhibitors for non-small cell lung cancer: A retrospective multicentric cohort study. Thromb. Res. 2021, 205, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Imamura, K.; Sakata, S.; Ikeda, T.; Horio, Y.; Iyama, S.; Akaike, K.; Hamada, S.; Jodai, T.; Nakashima, K.; et al. Disorder of Coagulation-Fibrinolysis System: An Emerging Toxicity of Anti-PD-1/PD-L1 Monoclonal Antibodies. J. Clin. Med. 2019, 8, 762. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.; Moudgil, R.; Lopez-Mattei, J.C.; Karimzad, K.; Mouhayar, E.N.; Somaiah, N.; Conley, A.P.; Patel, S.; Giza, D.E.; Iliescu, C. Progressive and Reversible Conduction Disease With Checkpoint Inhibitors. Can. J. Cardiol. 2017, 33, 1335.e13–1335.e15. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-R.; Florido, R.; Lipson, E.J.; Naidoo, J.; Ardehali, R.; Tocchetti, C.G.; Lyon, A.R.; Padera, R.F.; Johnson, D.B.; Moslehi, J. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc. Res. 2019, 115, 854–868. [Google Scholar] [CrossRef] [Green Version]
- Tay, R.Y.; Blackley, E.; McLean, C.; Moore, M.; Bergin, P.; Gill, S.; Haydon, A. Successful use of equine anti-thymocyte globulin (ATGAM) for fulminant myocarditis secondary to nivolumab therapy. Br. J. Cancer 2017, 117, 921–924. [Google Scholar] [CrossRef]
- Jain, V.; Mohebtash, M.; Rodrigo, M.E.; Ruiz, G.; Atkins, M.B.; Barac, A. Autoimmune Myocarditis Caused by Immune Checkpoint Inhibitors Treated with Antithymocyte Globulin. J. Immunother. 2018, 41, 332–335. [Google Scholar] [CrossRef]
- Wang, D.Y.; Okoye, G.D.; Neilan, T.G.; Johnson, D.B.; Moslehi, J.J. Cardiovascular Toxicities Associated with Cancer Immunotherapies. Curr. Cardiol. Rep. 2017, 19, 21. [Google Scholar] [CrossRef]
- Frigeri, M.; Meyer, P.; Banfi, C.; Giraud, R.; Hachulla, A.-L.; Spoerl, D.; Friedlaender, A.; Pugliesi-Rinaldi, A.; Dietrich, P.-Y. Immune Checkpoint Inhibitor-Associated Myocarditis: A New Challenge for Cardiologists. Can. J. Cardiol. 2018, 34, 92.e1–92.e3. [Google Scholar] [CrossRef] [Green Version]
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648, 2648a–2648d. [Google Scholar] [CrossRef] [PubMed]
- Balanescu, D.V.; Donisan, T.; Palaskas, N.; Lopez-Mattei, J.; Kim, P.Y.; Buja, L.M.; McNamara, D.M.; Kobashigawa, J.A.; Durand, J.-B.; Iliescu, C.A. Immunomodulatory treatment of immune checkpoint inhibitor-induced myocarditis: Pathway toward precision-based therapy. Cardiovasc. Pathol. 2020, 47, 107211. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Morimoto, R.; Okumura, T.; Yamashita, Y.; Haga, T.; Kuwayama, T.; Yokoi, T.; Hiraiwa, H.; Kondo, T.; Sugiura, Y.; et al. Late-Onset Fulminant Myocarditis With Immune Checkpoint Inhibitor Nivolumab. Can. J. Cardiol. 2018, 34, e1–e812. [Google Scholar] [CrossRef] [PubMed]
- Compton, F.; He, L.; Sarode, R.; Wodajo, A.; Usmani, A.; Burner, J.; Berlacher, M.; Simone, N.D. Immune checkpoint inhibitor toxicity: A new indication for therapeutic plasma exchange? J. Clin. Apher. 2021, 36, 645–648. [Google Scholar] [CrossRef]
- Schiopu, S.R.I.; Käsmann, L.; Schönermarck, U.; Fischereder, M.; Grabmaier, U.; Manapov, F.; Rauch, J.; Orban, M. Pembrolizumab-induced myocarditis in a patient with malignant mesothelioma: Plasma exchange as a successful emerging therapy-case report. Transl. Lung Cancer Res. 2021, 10, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Yogasundaram, H.; Alhumaid, W.; Chen, J.W.; Church, M.; Alhulaimi, N.; Kimber, S.; Paterson, D.I.; Senaratne, J.M. Plasma Exchange for Immune Checkpoint Inhibitor-Induced Myocarditis. CJC Open 2021, 3, 379–382. [Google Scholar] [CrossRef]
- Salem, J.-E.; Allenbach, Y.; Vozy, A.; Brechot, N.; Johnson, D.B.; Moslehi, J.J.; Kerneis, M. Abatacept for Severe Immune Checkpoint Inhibitor-Associated Myocarditis. N. Engl. J. Med. 2019, 380, 2377–2379. [Google Scholar] [CrossRef]
- Esfahani, K.; Buhlaiga, N.; Thébault, P.; Lapointe, R.; Johnson, N.A.; Miller, W.H. Alemtuzumab for Immune-Related Myocarditis Due to PD-1 Therapy. N. Engl. J. Med. 2019, 380, 2375–2376. [Google Scholar] [CrossRef]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, E447–E458. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Drug |
|---|---|
| Anti-CTLA4 | Ipilimumab |
| Tremelimumab | |
| Anti-PD-1 | Nivolumab |
| Pembrolizumab | |
| Cemiplimab | |
| Anti-PD-L1 | Atezolizumab |
| Avelumab | |
| Durvalumab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronen, D.; Bsoul, A.; Lotem, M.; Abedat, S.; Yarkoni, M.; Amir, O.; Asleh, R. Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Vaccines 2022, 10, 540. https://doi.org/10.3390/vaccines10040540
Ronen D, Bsoul A, Lotem M, Abedat S, Yarkoni M, Amir O, Asleh R. Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Vaccines. 2022; 10(4):540. https://doi.org/10.3390/vaccines10040540
Chicago/Turabian StyleRonen, Daniel, Aseel Bsoul, Michal Lotem, Suzan Abedat, Merav Yarkoni, Offer Amir, and Rabea Asleh. 2022. "Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies" Vaccines 10, no. 4: 540. https://doi.org/10.3390/vaccines10040540
APA StyleRonen, D., Bsoul, A., Lotem, M., Abedat, S., Yarkoni, M., Amir, O., & Asleh, R. (2022). Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Vaccines, 10(4), 540. https://doi.org/10.3390/vaccines10040540