
Adenovirus DNA Polymerase Loses Fidelity on a Stretch of Eleven Homocytidines during Pre-GMP Vaccine Preparation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recombinant DNA and Gel Electrophoresis
2.2. M9 Cells and Rescue of the ChAdOx1.tHIVconsv6 Virus
2.3. Single Virion-Infection (SVI) Purifications
2.4. Identity, Flank-to-Flank and Purity PCRs
2.5. SDS-PAGE and Western Blotting
2.6. Virus Preparation
2.7. DNA Sequencing of the Transgene
2.8. Animals, Vaccinations and Preparation of Splenocytes
2.9. Peptides
2.10. IFN-γ ELISPOT Assay
2.11. Cell-Free Transcription/Translation
3. Results
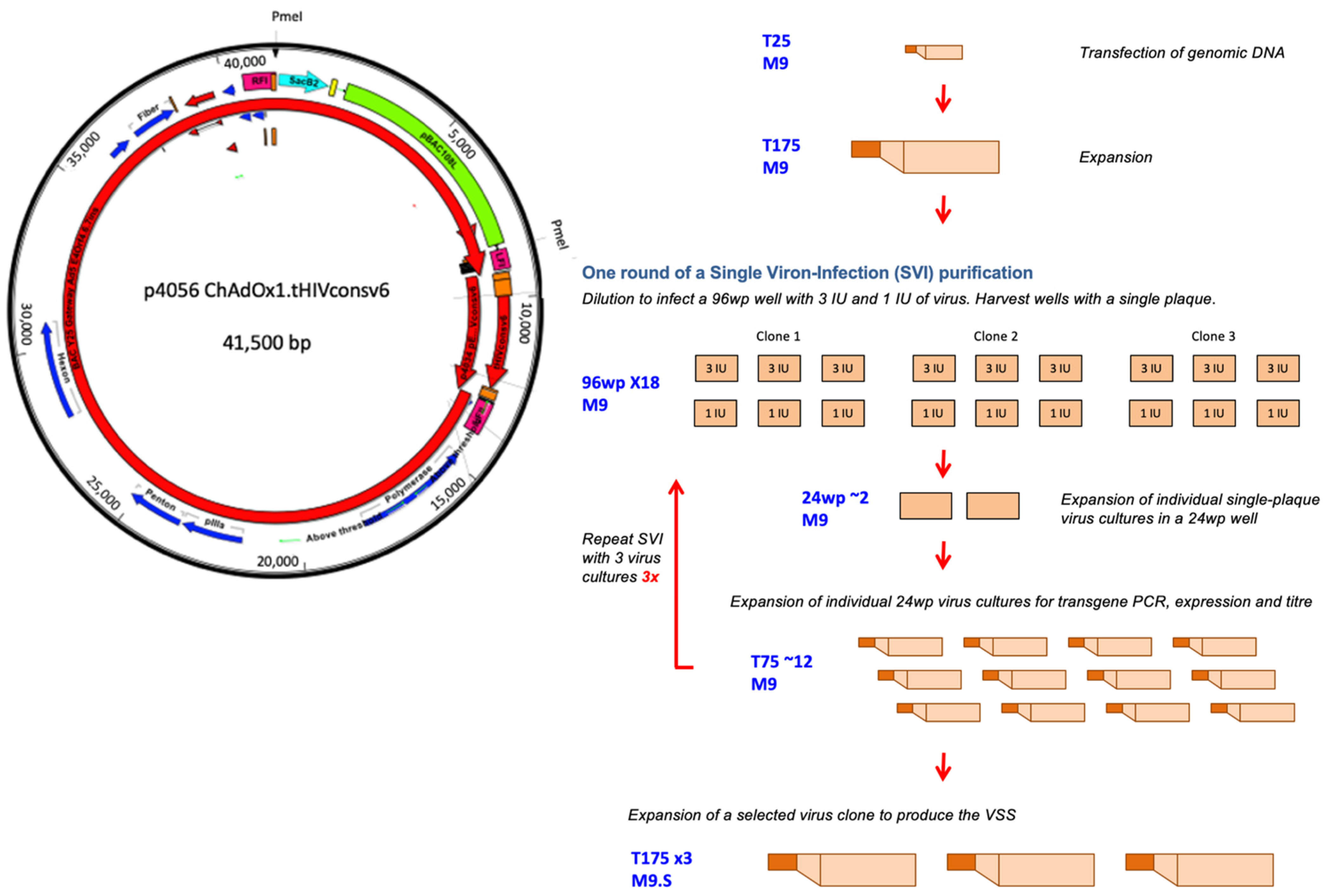
3.1. Construction of the BAC Carrying the ChAdOx1.tHIVconsv6 Vaccine Genome
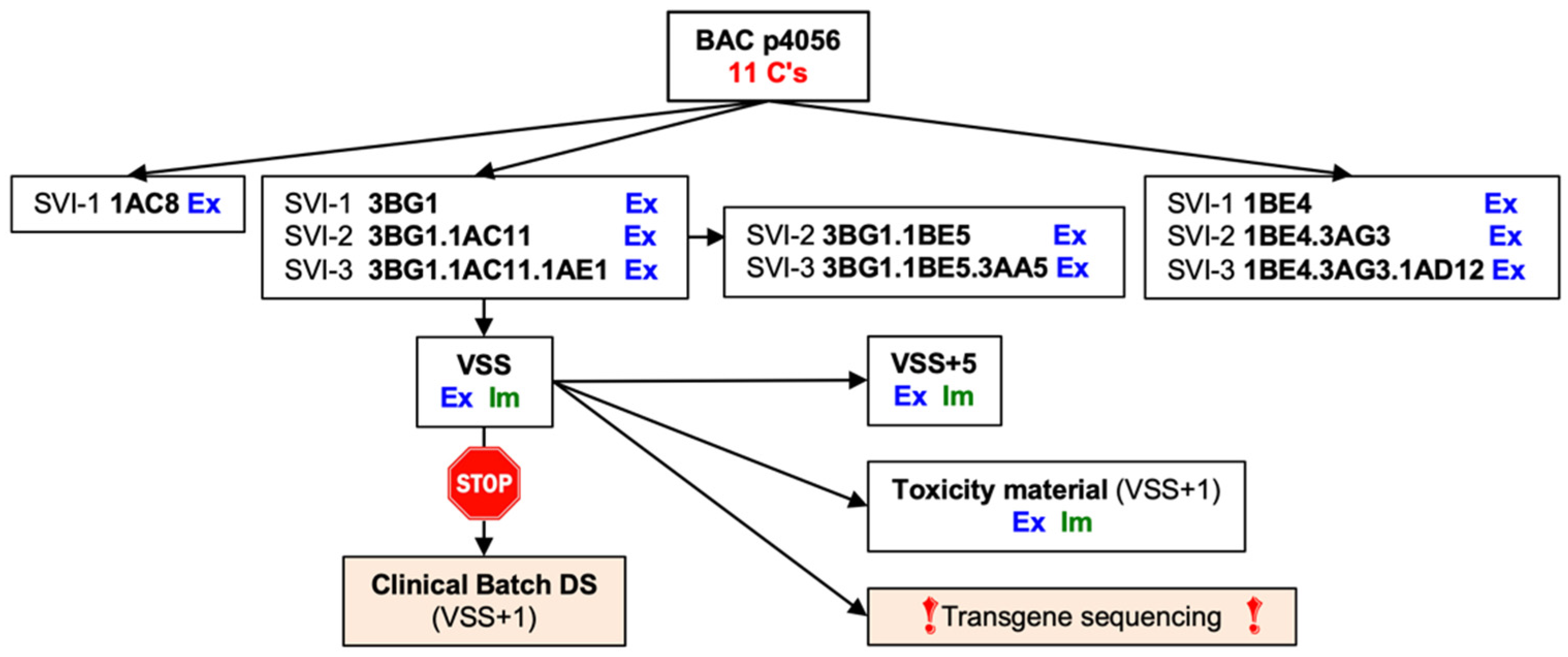
3.2. Rescue, Clonal Purification and Preparation of the Virus Seed Stock of the Vaccine Virus
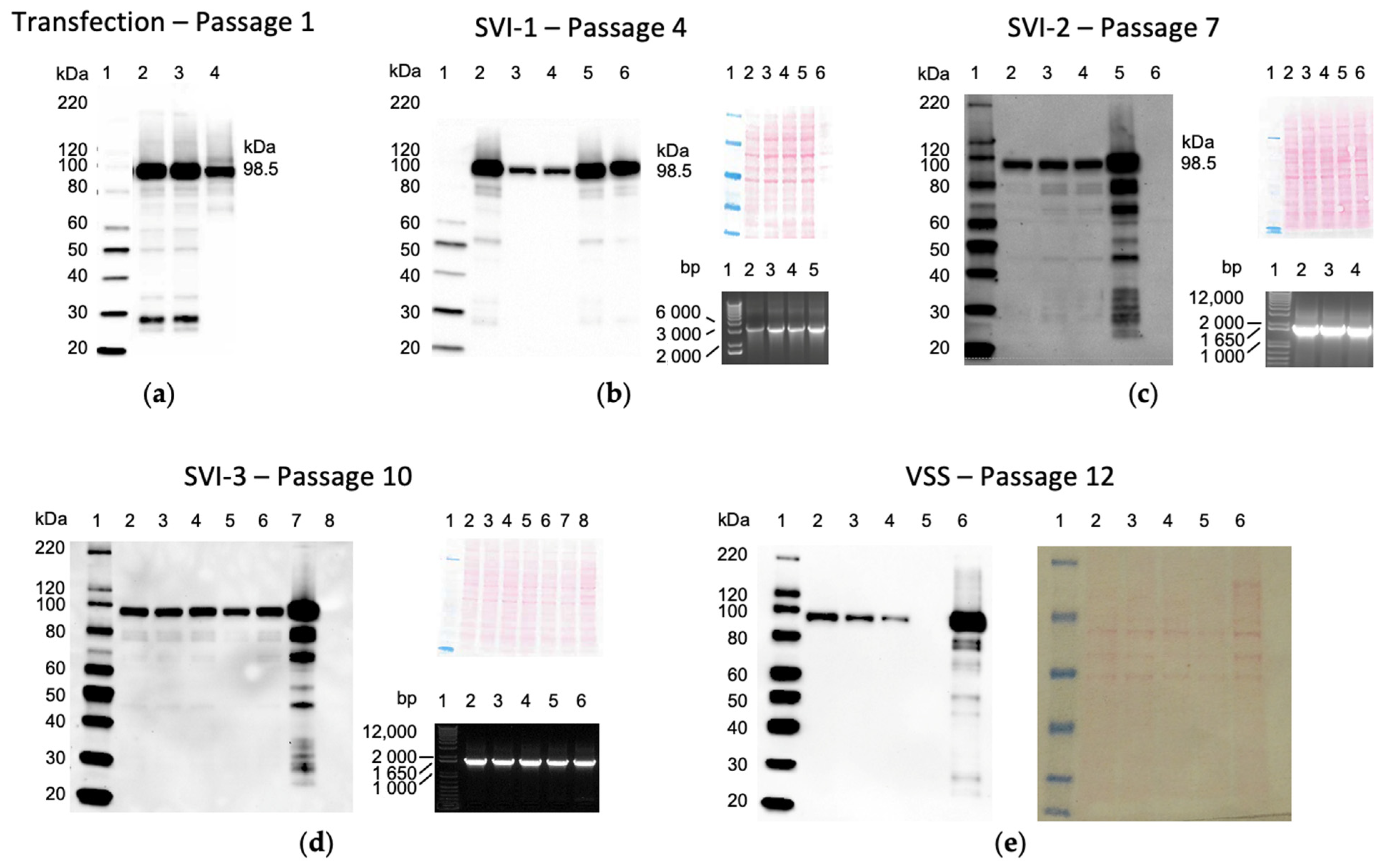
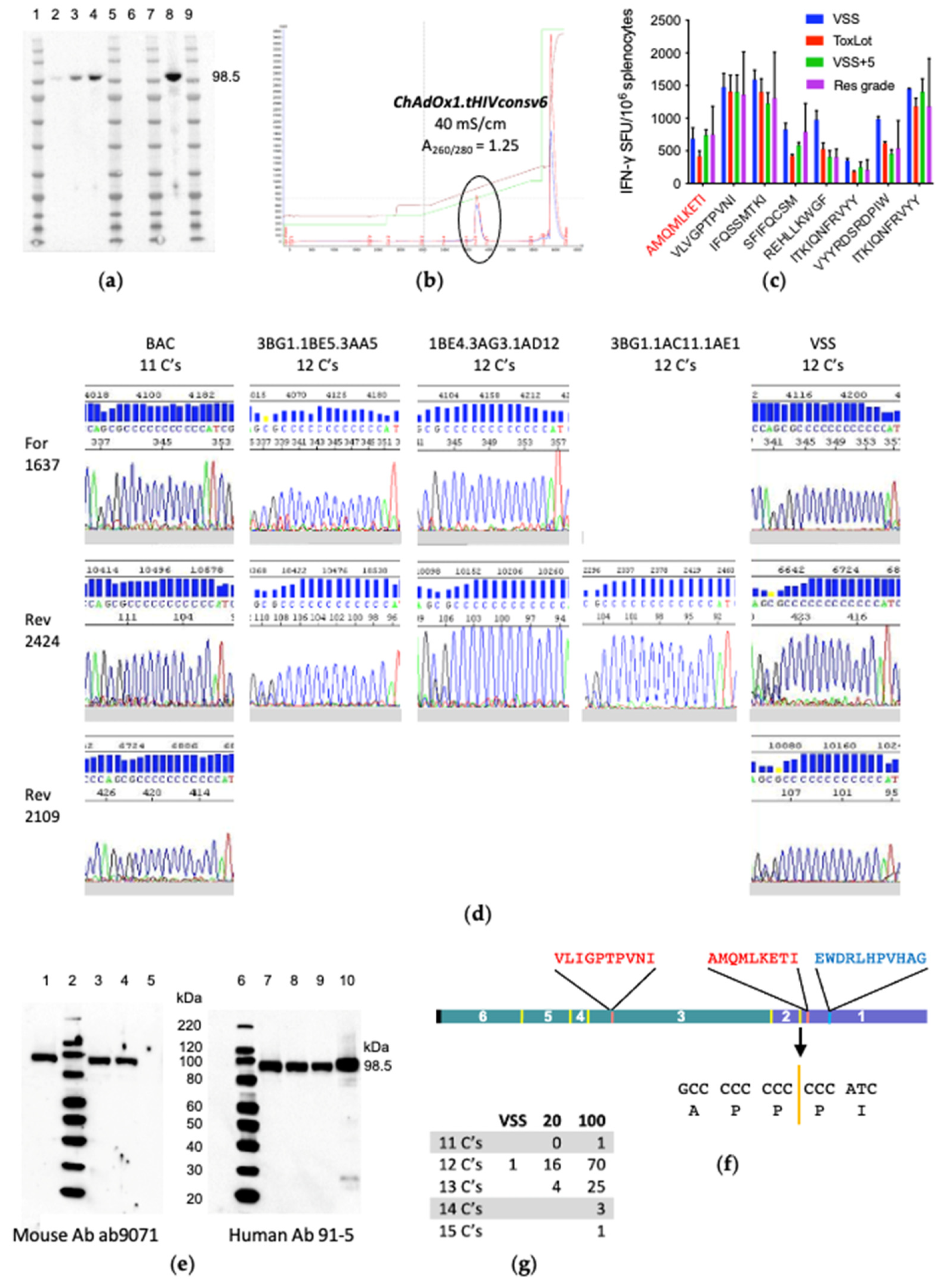
3.3. ChAdOx1.tHIVconsv6 VSS Passed All but the DNA Sequence Quality Tests
3.4. The VSS Is a Mixed Population
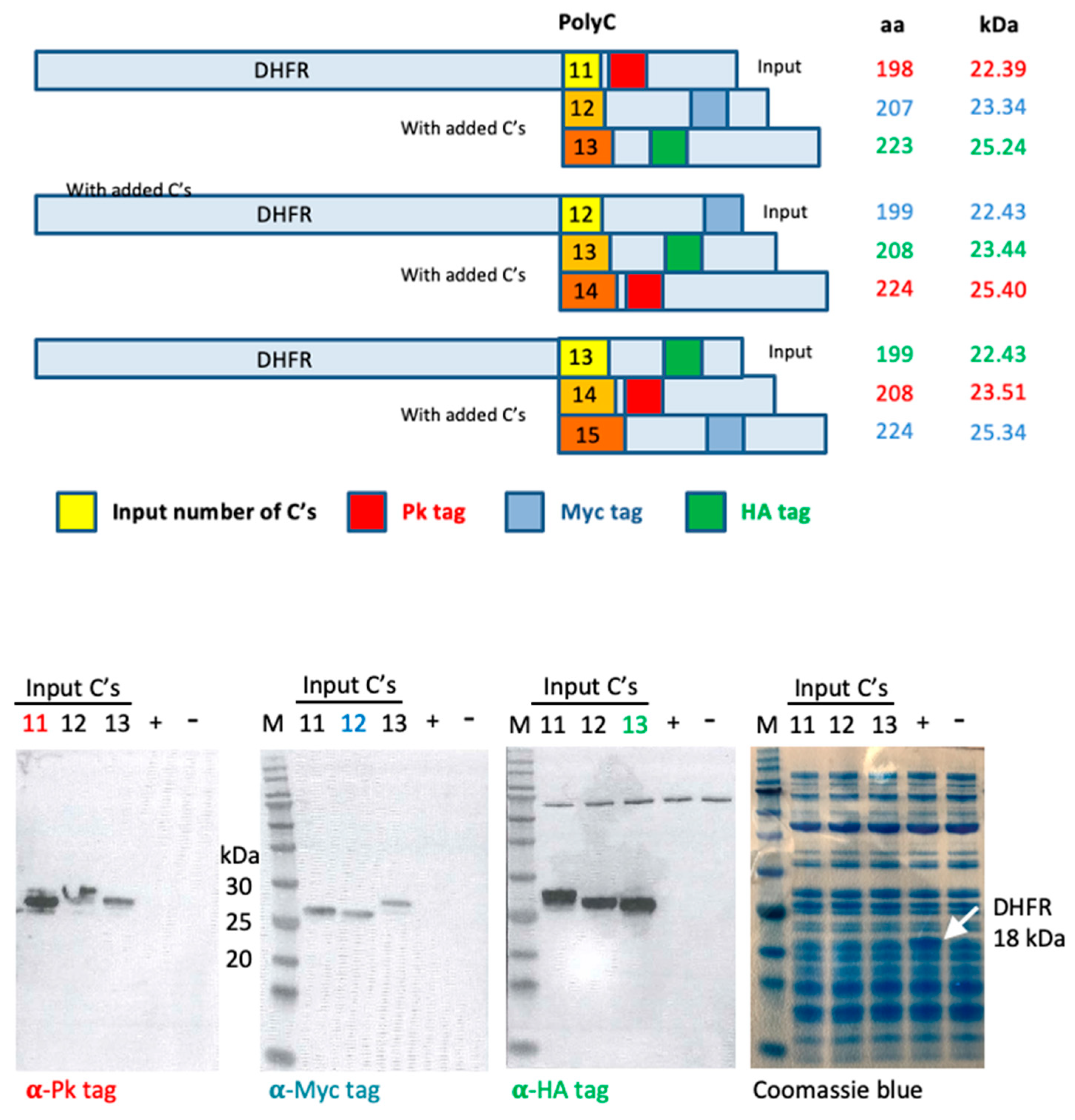
3.5. In Vitro Transcription/Translation System Slips Regularly on Long Polycytidines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plotkin, S.A. Vaccines: The fourth century. Clin. Vaccine Immunol. 2009, 16, 1709–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.A.; Plotkin, S. A Short History of Vaccination, 5th ed.; Elsevier-Saunders: Philadelphia, PA, USA, 2008. [Google Scholar]
- Barouch, D.H. Novel adenovirus vector-based vaccines for HIV-1. Curr. Opin. HIV AIDS 2010, 5, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Barouch, D.H.; Baden, L.R. Nonreplicating vectors in HIV vaccines. Curr. Opin. HIV AIDS 2013, 8, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.S.F.; Kim, M.; Limoli, M.; Obscherning, E.; Wu, P.; Feisee, L.; Nakashima, N.; Lim, J.C.W. Measuring progress of regulatory convergence and cooperation among Asia–Pacific Economic Cooperation (APEC) member economies in the context of the COVID-19 pandemic. Ther. Innov. Regul. Sci. 2021, 55, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, J.; Wang, J.; Gao, Q.; Ma, Z.; Xu, S.; Zhang, L.; Cai, J.; Zhou, W. Reshaping cell line development and CMC strategy for fast responses to pandemic outbreak. Biotechnol. Prog. 2021, 37, e3186. [Google Scholar] [CrossRef]
- Fischer, W.; Perkins, S.J.; Theiler, J.; Bhattacharya, T.; Yusim, K.; Funkhouser, R.; Kuiken, C.L.; Haynes, B.F.; Letvin, N.L.; Walker, B.D.; et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 2006, 13, 100–106. [Google Scholar] [CrossRef]
- Hanke, T. Aiming for protective T-cell responses: A focus on the first generation conserved-region HIVconsv vaccines in preventive and therapeutic clinical trials. Expert Rev. Vaccines 2019, 18, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Korber, B.; Fischer, W. T cell-based strategies for HIV-1 vaccines. Hum. Vaccines Immunother. 2019, 16, 713–722. [Google Scholar] [CrossRef]
- McMichael, A.J.; Haynes, B.F. Lessons learned from HIV-1 vaccine trials: New priorities and directions. Nat. Immunol. 2012, 13, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Ondondo, B.; Murakoshi, H.; Clutton, G.; Abdul-Jawad, S.; Wee, E.G.-T.; Gatanaga, H.; Oka, S.; McMichael, A.J.; Takiguchi, M.; Korber, B.; et al. Novel conserved-region T-cell mosaic vaccine with high global HIV-1 coverage is recognized by protective responses in untreated infection. Mol. Ther. 2016, 24, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, N.; Ahmed, T.; Ondondo, B.; Hayes, P.; Rose, A.; Ebrahimsa, U.; Hayton, E.-J.; Black, A.; Bridgeman, A.; Rosario, M.; et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol. Ther. 2014, 22, 464–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, S.; Seed, B.; Eberle, J.; Schraut, W.; Bultmann, A.; Haas, J. Increased immune response elicited by DNA vaccination with a synthetic gp120 sequence with optimized codon usage. J. Virol. 1998, 72, 1497–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Chavez, F. Most used codons per amino acid and per genome in the code of man compared to other organisms according to the rotating circular genetic code. NeuroQuantology 2011, 9, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab, D.; Graf, M.; Notka, F.; Schödl, T.; Wagner, R. The GeneOptimizer Algorithm: Using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst. Synth. Biol. 2010, 4, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Farfan-Arribas, D.J.; Shen, S.; Chou, T.-H.W.; Hirsch, A.; He, F.; Lu, S. Relative contributions of codon usage, promoter efficiency and leader sequence to the antigen expression and immunogenicity of HIV-1 Env DNA vaccine. Vaccine 2006, 24, 4531–4540. [Google Scholar] [CrossRef]
- Coughlan, L.; Sridhar, S.; Payne, R.; Edmans, M.; Milicic, A.; Venkatraman, N.; Lugonja, B.; Clifton, L.; Qi, C.; Folegatti, P.M.; et al. Heterologous two-dose vaccination with simian adenovirus and poxvirus vectors elicits long-lasting cellular immunity to influenza virus a in healthy adults. EBioMedicine 2018, 29, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, S.C.; Schneider, J.; Hannan, C.M.; Hu, J.T.; Plebanski, M.; Sinden, R.; Hill, A.V. Enhanced CD8 T cell immunogenicity and protective efficacy in a mouse malaria model using a recombinant adenoviral vaccine in heterologous prime–boost immunisation regimes. Vaccine 2001, 20, 1039–1045. [Google Scholar] [CrossRef]
- Moore, A.C.; Hill, A.V.S. Progress in DNA-based heterologous prime-boost immunization strategies for malaria. Immunol. Rev. 2004, 199, 126–143. [Google Scholar] [CrossRef]
- Mothe, B.; Manzardo, C.; Snachez-Bernabeau, A.; Coll, P.; Moron-Lopez, S.; Puertas, M.C.; Rosas, M.; Cobarsi, P.; Escrig, N.; Perez-Alvarez, N.; et al. Therapeutic vaccination refocused T-cell responses to conserved regions of HIV-1 in early reated individuals (BCN 01 study). Lancet eClinMed. 2019, 1, 65–80. [Google Scholar]
- Mutua, G.; Farah, B.; Langat, R.; Indangasi, J.; Ogola, S.; Onsembe, B.; Kopycinski, J.T.; Hayes, P.; Borthwick, N.J.; Ashraf, A.; et al. Broad HIV-1 inhibition in vitro by vaccine-elicited CD8+ T cells in African adults. Mol. Ther. Methods Clin. Dev. 2016, 3, 16061. [Google Scholar] [CrossRef]
- Vuola, J.M.; Keating, S.; Webster, D.P.; Berthoud, T.; Dunachie, S.; Gilbert, S.; Hill, A.V.S. Differential immunogenicity of various heterologous prime-boost vaccine regimens using DNA and viral vectors in healthy volunteers. J. Immunol. 2004, 174, 449–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangari, D.S.; Mittal, S.K. Development of nonhuman adenoviruses as vaccine vectors. Vaccine 2006, 24, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Dicks, M.; Spencer, A.; Edwards, N.; Wadell, G.; Bojang, K.; Gilbert, S.; Hill, A.V.S.; Cottingham, M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: Improved systems for vector derivation and comparative immunogenicity. PLoS ONE 2012, 7, e40385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Hay, R.T. Adenovirus DNA replication. In DNA Replication in Eukaryotic Cells; DePamphilis, M.L., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1966; pp. 699–721. [Google Scholar]
- van der Vliet, P.C. Roles of transcription factors in DNA replication. In DNA Replication in Eukaryotic Cells; DePamphilis, M.L., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1996; pp. 87–119. [Google Scholar]
- van der Vliet, P.C.; Hoeben, R.C. Adenovirus. In DNA Replication in Eukaryotic Cells; DePamphilis, M.L., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006; pp. 645–661. [Google Scholar]
- Hoeben, R.C.; Uil, T.G. Adenovirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a013003. [Google Scholar] [CrossRef] [Green Version]
- van Bergen, B.G.; van der Vliet, P.C. Temperature-sensitive initiation and elongation of adenovirus DNA replication in vitro with nuclear extracts from H5ts36-, H5ts149-, and H5ts125-infected HeLa cells. J. Virol. 1983, 46, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Giberson, A.N.; Davidson, A.R.; Parks, R.J. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res. 2011, 40, 2369–2376. [Google Scholar] [CrossRef] [Green Version]
- Hanke, T.; Graham, F.L.; Rosenthal, K.L.; Johnson, D.C. Identification of an immunodominant cytotoxic T-lymphocyte recognition site in glycoprotein B of herpes simplex virus by using recombinant adenovirus vectors and synthetic peptides. J. Virol. 1991, 65, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Tatsis, N.; Ertl, H.C. Adenoviruses as vaccine vectors. Mol. Ther. 2004, 10, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, R.D.; Coughlan, L.; Berthoud, T.K.; Dicks, M.; Hill, A.V.; Lambe, T.; Gilbert, S.C. Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved influenza a antigens. Mol. Ther. 2014, 22, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; Del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the step study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef] [Green Version]
- Hayton, E.J.; Rose, A.; Ibrahimsa, U.; Del Sorbo, M.; Capone, S.; Crook, A.; Black, A.P.; Dorrell, L.; Hanke, T. Safety and tolerability of conserved region vaccines vectored by plasmid DNA, simian adenovirus and modified vaccinia virus ankara administered to human immunodeficiency virus type 1-uninfected adults in a randomized, single-blind phase I trial. PLoS ONE 2014, 9, e101591. [Google Scholar] [CrossRef] [Green Version]
- Mpendo, J.; Mutua, G.; Nyombayire, J.; Ingabire, R.; Nanvubya, A.; Anzala, O.; Karita, E.; Hayes, P.; Kopycinski, J.; Dally, L.; et al. A phase I double blind, placebo-controlled, randomized study of the safety and immunogenicity of electroporated HIV DNA with or without Interleukin 12 in prime-boost combinations with an Ad35 HIV vaccine in healthy HIV-seronegative African adults. PLoS ONE 2015, 10, e0134287. [Google Scholar] [CrossRef] [Green Version]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Maniatis, T.; Fritsch, E.F.; Sambrook, J. Molecular Cloning. A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1986. [Google Scholar]
- Gorny, M.K.; Gianakakos, V.; Sharpe, S.; Zolla-Pazner, S. Generation of human monoclonal antibodies to human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1989, 86, 1624–1628. [Google Scholar] [CrossRef] [Green Version]
- Wee, E.G.; Moyo, N.; Hannoun, Z.; Giorgi, E.E.; Korber, B.; Hanke, T. Effect of epitope variant co-delivery on the depth of CD8 T cell responses induced by HIV-1 conserved mosaic vaccines. Mol. Ther. Methods Clin. Dev. 2021, 21, 741–753. [Google Scholar] [CrossRef]
- Mata, M.; Travers, P.J.; Liu, Q.; Frankel, F.R.; Paterson, Y. The MHC class I-restricted immune response to HIV-gag in BALB/c mice selects a single epitope that does not have a predictable MHC-binding motif and binds to KD through interactions between a glutamine at P3 and pocket D. J. Immunol. 1998, 161, 2985–2993. [Google Scholar]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef]
- Chamberlin, M.; Berg, P. Deoxyribonucleic acid-directed synthesis of ribonucleic acid by an enzyme from Escherichia coli. Proc. Natl. Acad. Sci. USA 1962, 48, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahirov, T.H.; Temiakov, D.; Anikin, M.; Patlan, V.; McAllister, W.T.; Vassylyev, D.G.; Yokoyama, S. Structure of a T7 RNA polymerase elongation complex at 2.9 A. resolution. Nature 2002, 420, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Turnbough, C.L. Regulation of gene expression by reiterative transcription. Curr. Opin. Microbiol. 2011, 14, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, A.; Steele, E.J.; Lindley, R.A. A proposed reverse transcription mechanism for (CAG)n and similar expandable repeats that cause neurological and other diseases. Heliyon 2020, 6, e03258. [Google Scholar] [CrossRef]
- Falvey, A.K.; Weiss, G.B.; Krueger, L.J.; Kantor, J.A.; Anderson, W.F. Transcription of single base oligonucleotides by ribonucleic acid-directed deoxyribonucleic acid polymerase. Nucleic Acids Res. 1976, 3, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Majors, J. Translational frameshift suppression in mouse mammary tumor virus and other retroviruses. Enzyme 1990, 44, 320–331. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannoun, Z.; Wee, E.G.; Crook, A.; Colloca, S.; Di Marco, S.; Hanke, T. Adenovirus DNA Polymerase Loses Fidelity on a Stretch of Eleven Homocytidines during Pre-GMP Vaccine Preparation. Vaccines 2022, 10, 960. https://doi.org/10.3390/vaccines10060960
Hannoun Z, Wee EG, Crook A, Colloca S, Di Marco S, Hanke T. Adenovirus DNA Polymerase Loses Fidelity on a Stretch of Eleven Homocytidines during Pre-GMP Vaccine Preparation. Vaccines. 2022; 10(6):960. https://doi.org/10.3390/vaccines10060960
Chicago/Turabian StyleHannoun, Zara, Edmund G. Wee, Alison Crook, Stefano Colloca, Stefania Di Marco, and Tomáš Hanke. 2022. "Adenovirus DNA Polymerase Loses Fidelity on a Stretch of Eleven Homocytidines during Pre-GMP Vaccine Preparation" Vaccines 10, no. 6: 960. https://doi.org/10.3390/vaccines10060960