
Early Emergence of 5′ Terminally Deleted Coxsackievirus-B3 RNA Forms Is Associated with Acute and Persistent Infections in Mouse Target Tissues

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus Strain, and Reagents
2.2. Virus Production and Titration
2.3. Ethic Statement
2.4. Infection of Mice
2.5. Histology and Immunohistochemistry Assays
2.6. Total RNA Isolation and qRT-PCR
2.7. Positive- and Negative-Strand RNA Ratio
2.8. Rapid Amplification of cDNA Ends-PCR (RACE PCR)
2.9. Cytokines Quantification
2.10. Statistical Analysis
3. Results
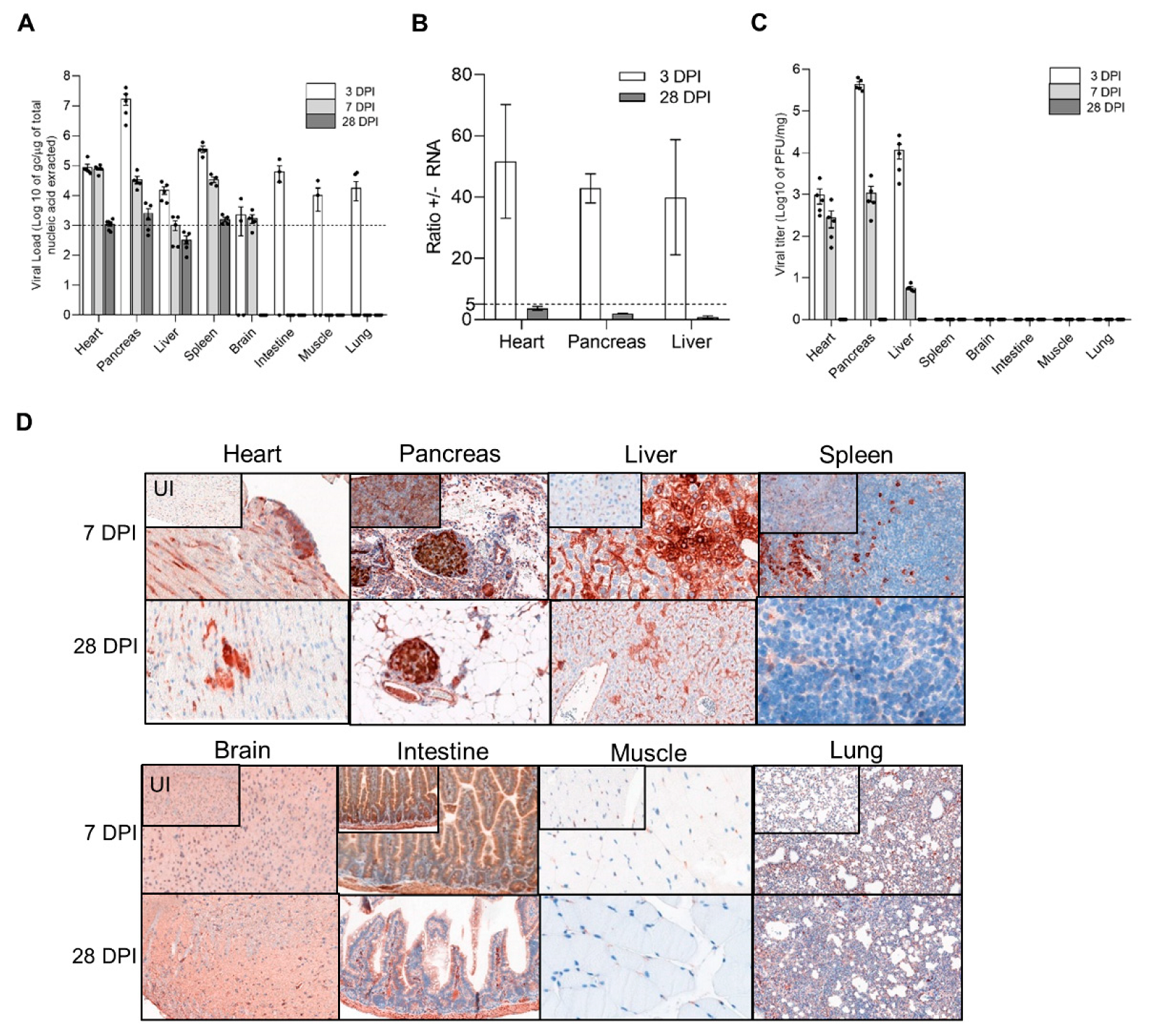
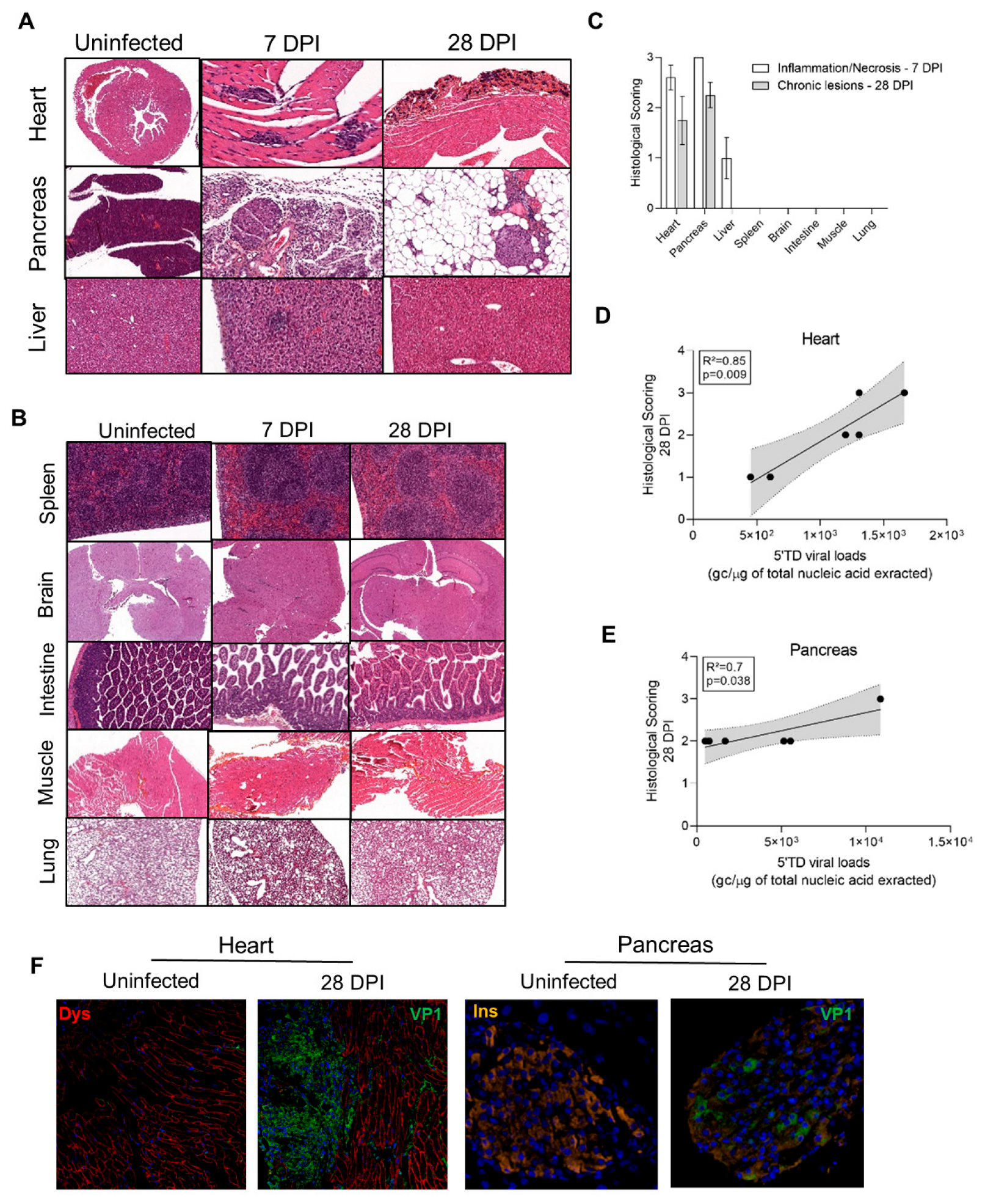
3.1. Identification of DBA/2J Mice Target Tissues with Natural CVB3/28 Acute and Persistent Infections
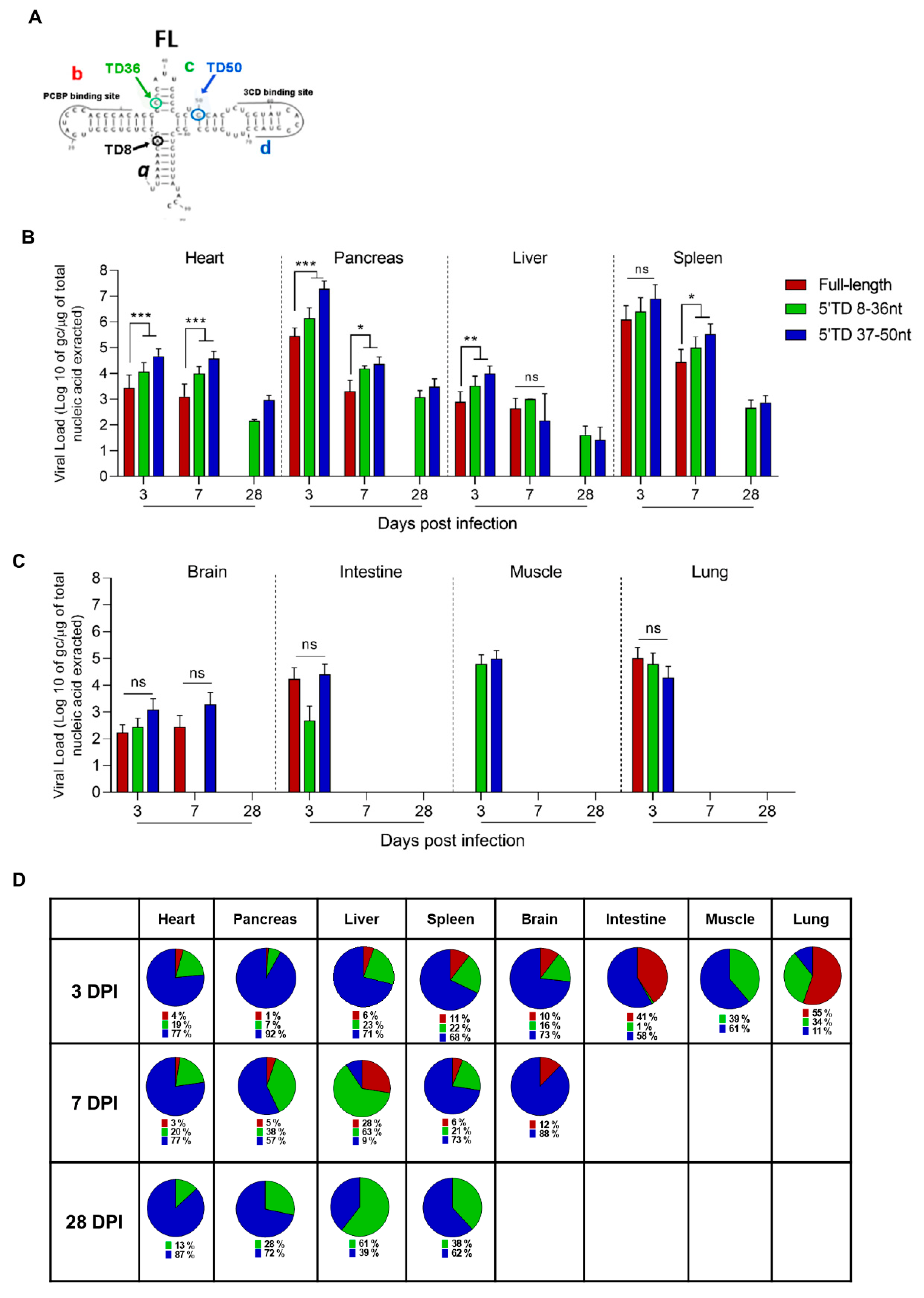
3.2. Dynamics of Emergence of 5′TD Viral Populations in Organs of Mice Infected by CVB3/28 Strain
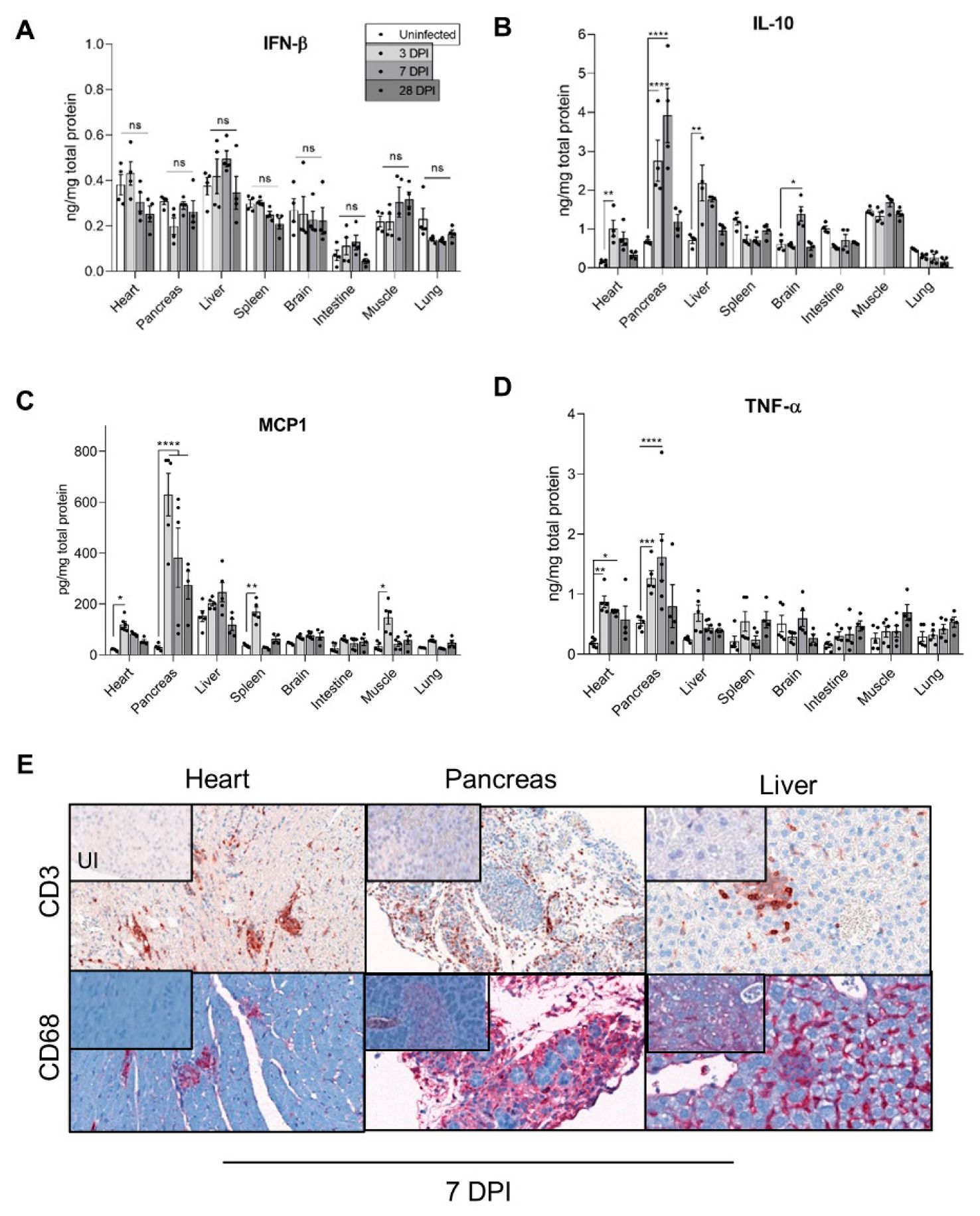
3.3. Inflammatory Cytokines Secretion and Cellular Infiltrates during Acute and Chronic CVB3/28 Infection
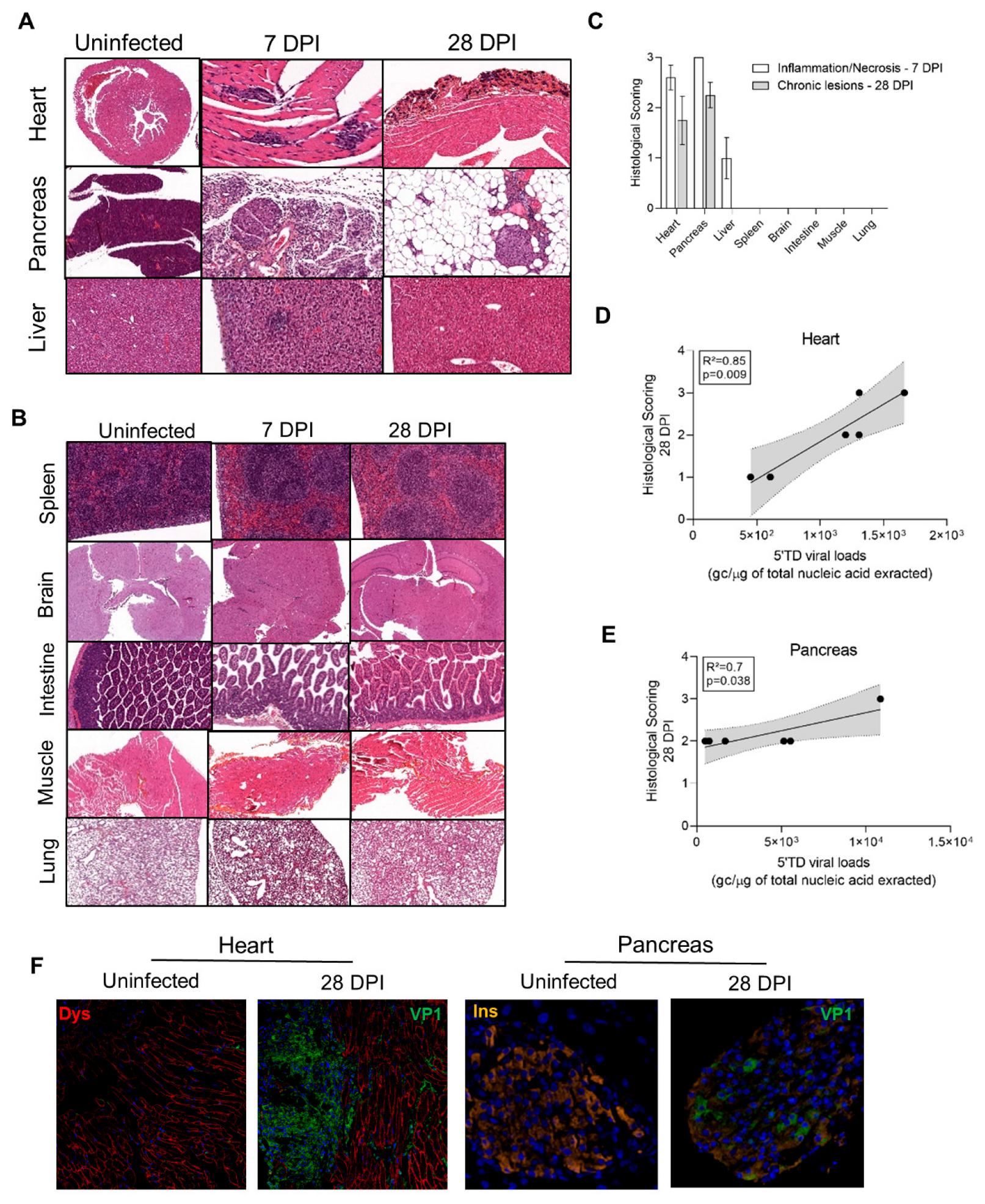
3.4. Impact of 5′TD RNA Forms on Histological Lesions and Cellular Specialized Functions in CVB3/28 Infected Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baggen, J.; Thibaut, H.J.; Strating, J.R.P.M.; van Kuppeveld, F.J.M. The Life Cycle of Non-Polio Enteroviruses and How to Target It. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Genoni, A.; Canducci, F.; Rossi, A.; Broccolo, F.; Chumakov, K.; Bono, G.; Salerno-Uriarte, J.; Salvatoni, A.; Pugliese, A.; Toniolo, A. Revealing Enterovirus Infection in Chronic Human Disorders: An Integrated Diagnostic Approach. Sci. Rep. 2017, 7, 5013. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S.; Smithee, S.; Alhazmi, A.; Chapman, N. Coxsackievirus Can Persist in Murine Pancreas by Deletion of 5′ Terminal Genomic Sequences: Coxsackievirus Persistence in the Pancreas. J. Med. Virol. 2015, 87, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.E.; Fontana, D.R.; Messner, R.P. Coxsackievirus B1–Induced Chronic Inflammatory Myopathy: Differences in Induction of Autoantibodies to Muscle and Nuclear Antigens by Cloned Myopathic and Amyopathic Viruses. J. Lab. Clin. Med. 2003, 142, 196–204. [Google Scholar] [CrossRef]
- Chapman, N.M.; Kim, K.-S.; Drescher, K.M.; Oka, K.; Tracy, S. 5′ Terminal Deletions in the Genome of a Coxsackievirus B2 Strain Occurred Naturally in Human Heart. Virology 2008, 375, 480–491. [Google Scholar] [CrossRef] [Green Version]
- Bouin, A.; Nguyen, Y.; Wehbe, M.; Renois, F.; Fornes, P.; Bani-Sadr, F.; Metz, D.; Andreoletti, L. Major Persistent 5′ Terminally Deleted Coxsackievirus B3 Populations in Human Endomyocardial Tissues. Emerg. Infect. Dis. 2016, 22, 1488–1490. [Google Scholar] [CrossRef]
- Bouin, A.; Gretteau, P.-A.; Wehbe, M.; Renois, F.; N’Guyen, Y.; Leveque, N.; Vu, M.N.; Tracy, S.; Chapman, N.M.; Bruneval, P.; et al. Enterovirus Persistence in Cardiac Cells of Patients Suffering From Idiopathic Dilated Cardiomyopathy Is Linked to 5’ Terminal Genomic RNA-Deleted Viral Populations With Viral-Encoded Proteinase Activities. Circulation 2019, 139, 2326–2338. [Google Scholar] [CrossRef]
- Kim, K.-S.; Tracy, S.; Tapprich, W.; Bailey, J.; Lee, C.-K.; Kim, K.; Barry, W.H.; Chapman, N.M. 5’-Terminal Deletions Occur in Coxsackievirus B3 during Replication in Murine Hearts and Cardiac Myocyte Cultures and Correlate with Encapsidation of Negative-Strand Viral RNA. J. Virol. 2005, 79, 7024–7041. [Google Scholar] [CrossRef] [Green Version]
- Lévêque, N.; Garcia, M.; Bouin, A.; Nguyen, J.H.C.; Tran, G.P.; Andreoletti, L.; Semler, B.L. Functional Consequences of RNA 5′-Terminal Deletions on Coxsackievirus B3 RNA Replication and Ribonucleoprotein Complex Formation. J. Virol. 2017, 91, e00423-17. [Google Scholar] [CrossRef] [Green Version]
- Klingel, K.; Hohenadl, C.; Canu, A.; Albrecht, M.; Seemann, M.; Mall, G.; Kandolf, R. Ongoing Enterovirus-Induced Myocarditis Is Associated with Persistent Heart Muscle Infection: Quantitative Analysis of Virus Replication, Tissue Damage, and Inflammation. Proc. Natl. Acad. Sci. USA 1992, 89, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Glenet, M.; N’Guyen, Y.; Mirand, A.; Henquell, C.; Lebreil, A.-L.; Berri, F.; Bani-Sadr, F.; Lina, B.; Schuffenecker, I.; Andreoletti, L. Major 5′terminally Deleted Enterovirus Populations Modulate Type I IFN Response in Acute Myocarditis Patients and in Human Cultured Cardiomyocytes. Sci. Rep. 2020, 10, 11947. [Google Scholar] [CrossRef]
- Kim, K.-S.; Chapman, N.M.; Tracy, S. Replication of Coxsackievirus B3 in Primary Cell Cultures Generates Novel Viral Genome Deletions. J. Virol. 2008, 82, 2033–2037. [Google Scholar] [CrossRef] [Green Version]
- Glenet, M.; Heng, L.; Callon, D.; Lebreil, A.-L.; Gretteau, P.-A.; Nguyen, Y.; Berri, F.; Andreoletti, L. Structures and Functions of Viral 5′ Non-Coding Genomic RNA Domain-I in Group-B Enterovirus Infections. Viruses 2020, 12, 919. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Olagnier, D.; Chiang, C.; van de Winkel, R.; van Essen, P.; Zoll, J.; Hiscott, J.; van Kuppeveld, F.J.M. Coxsackievirus Cloverleaf RNA Containing a 5′ Triphosphate Triggers an Antiviral Response via RIG-I Activation. PLoS ONE 2014, 9, e95927. [Google Scholar] [CrossRef]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J.M. MDA5 Detects the Double-Stranded RNA Replicative Form in Picornavirus-Infected Cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Cathcart, A.L.; Rozovics, J.M.; Semler, B.L. Cellular MRNA Decay Protein AUF1 Negatively Regulates Enterovirus and Human Rhinovirus Infections. J. Virol. 2013, 87, 10423–10434. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, X.; Zhao, Z.; Liu, X.; Zhang, W. Host Restriction Factor A3G Inhibits the Replication of Enterovirus D68 by Competitively Binding the 5’ Untranslated Region with PCBP1. J. Virol. 2022, 96, e0170821. [Google Scholar] [CrossRef]
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 Inhibits Enterovirus Replication and Promotes Emergence of a Viral Variant with Increased Pathogenicity. Cell 2021, 184, 3410–3425.e17. [Google Scholar] [CrossRef]
- Tu, Z.; Chapman, N.M.; Hufnagel, G.; Tracy, S.; Romero, J.R.; Barry, W.H.; Zhao, L.; Currey, K.; Shapiro, B. The Cardiovirulent Phenotype of Coxsackievirus B3 Is Determined at a Single Site in the Genomic 5’ Nontranslated Region. J. Virol. 1995, 69, 4607–4618. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Aguilera, B.; Banner, J.; Cohle, S.; d’Amati, G.; de Gouveia, R.H.; di Gioia, C.; Fabre, A.; Gallagher, P.J.; Leone, O.; et al. Guidelines for Autopsy Investigation of Sudden Cardiac Death: 2017 Update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017, 471, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Helio, T.; Heymans, S.; Jahns, R.; et al. Current State of Knowledge on Aetiology, Diagnosis, Management, and Therapy of Myocarditis: A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- Andréoletti, L.; Hober, D.; Becquart, P.; Belaich, S.; Copin, M.C.; Lambert, V.; Wattré, P. Experimental CVB3-Induced Chronic Myocarditis in Two Murine Strains: Evidence of Interrelationships between Virus Replication and Myocardial Damage in Persistent Cardiac Infection. J. Med. Virol. 1997, 52, 206–214. [Google Scholar] [CrossRef]
- N’Guyen, Y.; Lesaffre, F.; Metz, D.; Tassan, S.; Saade, Y.; Boulagnon, C.; Fornes, P.; Renois, F.; Andreoletti, L. Enterovirus but Not Parvovirus B19 Is Associated with Idiopathic Dilated Cardiomyopathy and Endomyocardial CD3, CD68, or HLA-DR Expression. J. Med. Virol. 2017, 89, 55–63. [Google Scholar] [CrossRef]
- Lévêque, N.; Renois, F.; Talmud, D.; Nguyen, Y.; Lesaffre, F.; Boulagnon, C.; Bruneval, P.; Fornes, P.; Andréoletti, L. Quantitative Genomic and Antigenomic Enterovirus RNA Detection in Explanted Heart Tissue Samples from Patients with End-Stage Idiopathic Dilated Cardiomyopathy. J. Clin. Microbiol. 2012, 50, 3378–3380. [Google Scholar] [CrossRef] [Green Version]
- Escarmís, C.; Dopazo, J.; Dávila, M.; Palma, E.L.; Domingo, E. Large Deletions in the 5′-Untranslated Region of Foot-and-Mouth Disease Virus of Serotype C. Virus Res. 1995, 35, 155–167. [Google Scholar] [CrossRef]
- Meyer, B.J.; Schmaljohn, C. Accumulation of Terminally Deleted RNAs May Play a Role in Seoul Virus Persistence. J. Virol. 2000, 74, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Höper, D.; et al. Deletion at the 5’-End of Estonian ASFV Strains Associated with an Attenuated Phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef]
- Kamma, H.; Portman, D.S.; Dreyfuss, G. Cell Type-Specific Expression of HnRNP Proteins. Exp. Cell Res. 1995, 221, 187–196. [Google Scholar] [CrossRef]
- Ertel, K.J.; Brunner, J.E.; Semler, B.L. Mechanistic Consequences of HnRNP C Binding to Both RNA Termini of Poliovirus Negative-Strand RNA Intermediates. J. Virol. 2010, 84, 4229–4242. [Google Scholar] [CrossRef] [Green Version]
- Brunner, J.E.; Nguyen, J.H.C.; Roehl, H.H.; Ho, T.V.; Swiderek, K.M.; Semler, B.L. Functional Interaction of Heterogeneous Nuclear Ribonucleoprotein C with Poliovirus RNA Synthesis Initiation Complexes. J. Virol. 2005, 79, 3254–3266. [Google Scholar] [CrossRef] [Green Version]
- Brunner, J.E.; Ertel, K.J.; Rozovics, J.M.; Semler, B.L. Delayed Kinetics of Poliovirus RNA Synthesis in a Human Cell Line with Reduced Levels of HnRNP C Proteins. Virology 2010, 400, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.-P.; Henry, M.; Marchio, A.; Suspène, R.; Aynaud, M.-M.; Guétard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 Editing of Hepatitis B Viral DNA in Cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. APOBEC3 Proteins Mediate the Clearance of Foreign DNA from Human Cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral Quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sesti-Costa, R.; Françozo, M.C.S.; Silva, G.K.; Proenca-Modena, J.L.; Silva, J.S. TLR3 Is Required for Survival Following Coxsackievirus B3 Infection by Driving T Lymphocyte Activation and Polarization: The Role of Dendritic Cells. PLoS ONE 2017, 12, e0185819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.F.; Varela, J.; David, R.M.; Banks, L.; Huang, C.-H.; Li, H.; et al. A Role for Toll-like Receptor 3 Variants in Host Susceptibility to Enteroviral Myocarditis and Dilated Cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.T.; Oldstone, M.B.A. IL-10: Achieving Balance during Persistent Viral Infection. In Interleukin-10 in Health and Disease; Current Topics in Microbiology and Immunology; Fillatreau, S., O’Garra, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 380, pp. 129–144. ISBN 978-3-662-43491-8. [Google Scholar]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Weber, F.; Kochs, G.; Haller, O. Inverse Interference: How Viruses Fight the Interferon System. Viral Immunol. 2004, 17, 498–515. [Google Scholar] [CrossRef]
- Dobrikov, M.I.; Dobrikova, E.Y.; McKay, Z.P.; Kastan, J.P.; Brown, M.C.; Gromeier, M. PKR Binds Enterovirus IRESs, Displaces Host Translation Factors, and Impairs Viral Translation to Enable Innate Antiviral Signaling. mBio 2022, 13, e0085422. [Google Scholar] [CrossRef]
- Cheroni, C.; Donnici, L.; Aghemo, A.; Balistreri, F.; Bianco, A.; Zanoni, V.; Pagani, M.; Soffredini, R.; D’Ambrosio, R.; Rumi, M.G.; et al. Hepatitis C Virus Deletion Mutants Are Found in Individuals Chronically Infected with Genotype 1 Hepatitis C Virus in Association with Age, High Viral Load and Liver Inflammatory Activity. PLoS ONE 2015, 10, e0138546. [Google Scholar] [CrossRef] [Green Version]
- Cahour, A.; Pletnev, A.; Vazielle-Falcoz, M.; Rosen, L.; Lai, C.J. Growth-Restricted Dengue Virus Mutants Containing Deletions in the 5’ Noncoding Region of the RNA Genome. Virology 1995, 207, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Kühl, U.; Lassner, D.; von Schlippenbach, J.; Poller, W.; Schultheiss, H.-P. Interferon-Beta Improves Survival in Enterovirus-Associated Cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1295–1296. [Google Scholar] [CrossRef] [Green Version]
- Badorff, C.; Lee, G.-H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral Protease 2A Cleaves Dystrophin: Evidence of Cytoskeletal Disruption in an Acquired Cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [CrossRef]
- Baden, M.Y.; Fukui, K.; Hosokawa, Y.; Iwahashi, H.; Imagawa, A.; Shimomura, I. Examination of a Viral Infection Mimetic Model in Human IPS Cell-Derived Insulin-Producing Cells and the Anti-Apoptotic Effect of GLP-1 Analogue. PLoS ONE 2015, 10, e0144606. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Bertin, A.; Sane, F.; Gimeno, J.-P.; Fournier, I.; Salzet, M.; Engelmann, I.; Alidjinou, E.K.; Hober, D. Persistence of Coxsackievirus B4 in Pancreatic β Cells Disturbs Insulin Maturation, Pattern of Cellular Proteins, and DNA Methylation. Microorganisms 2021, 9, 1125. [Google Scholar] [CrossRef]
- Lim, B.-K.; Peter, A.K.; Xiong, D.; Narezkina, A.; Yung, A.; Dalton, N.D.; Hwang, K.-K.; Yajima, T.; Chen, J.; Knowlton, K.U. Inhibition of Coxsackievirus-Associated Dystrophin Cleavage Prevents Cardiomyopathy. J. Clin. Investig. 2013, 123, 5146–5151. [Google Scholar] [CrossRef] [Green Version]
- Xiong, D.; Lee, G.-H.; Badorff, C.; Dorner, A.; Lee, S.; Wolf, P.; Knowlton, K.U. Dystrophin Deficiency Markedly Increases Enterovirus-Induced Cardiomyopathy: A Genetic Predisposition to Viral Heart Disease. Nat. Med. 2002, 8, 872–877. [Google Scholar] [CrossRef]
- Alidjinou, E.K.; Sané, F.; Engelmann, I.; Geenen, V.; Hober, D. Enterovirus persistence as a mechanism in the pathogenesis of type 1 diabetes. Discov. Med. 2014, 18, 273–282. [Google Scholar]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J.; et al. Detection of a Low-Grade Enteroviral Infection in the Islets of Langerhans of Living Patients Newly Diagnosed With Type 1 Diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef] [Green Version]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jørgensen, K.; Flodström-Tullberg, M.; Hyöty, H.; Insel, R.A.; Lernmark, A.; Lloyd, R.E.; et al. Rationale for Enteroviral Vaccination and Antiviral Therapies in Human Type 1 Diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callon, D.; Lebreil, A.-L.; Bouland, N.; Fichel, C.; Fornès, P.; Andreoletti, L.; Berri, F. Early Emergence of 5′ Terminally Deleted Coxsackievirus-B3 RNA Forms Is Associated with Acute and Persistent Infections in Mouse Target Tissues. Vaccines 2022, 10, 1203. https://doi.org/10.3390/vaccines10081203
Callon D, Lebreil A-L, Bouland N, Fichel C, Fornès P, Andreoletti L, Berri F. Early Emergence of 5′ Terminally Deleted Coxsackievirus-B3 RNA Forms Is Associated with Acute and Persistent Infections in Mouse Target Tissues. Vaccines. 2022; 10(8):1203. https://doi.org/10.3390/vaccines10081203
Chicago/Turabian StyleCallon, Domitille, Anne-Laure Lebreil, Nicole Bouland, Caroline Fichel, Paul Fornès, Laurent Andreoletti, and Fatma Berri. 2022. "Early Emergence of 5′ Terminally Deleted Coxsackievirus-B3 RNA Forms Is Associated with Acute and Persistent Infections in Mouse Target Tissues" Vaccines 10, no. 8: 1203. https://doi.org/10.3390/vaccines10081203
APA StyleCallon, D., Lebreil, A.-L., Bouland, N., Fichel, C., Fornès, P., Andreoletti, L., & Berri, F. (2022). Early Emergence of 5′ Terminally Deleted Coxsackievirus-B3 RNA Forms Is Associated with Acute and Persistent Infections in Mouse Target Tissues. Vaccines, 10(8), 1203. https://doi.org/10.3390/vaccines10081203