
Immunotherapeutic Strategies for Head and Neck Squamous Cell Carcinoma (HNSCC): Current Perspectives and Future Prospects


Abstract
:1. Introduction
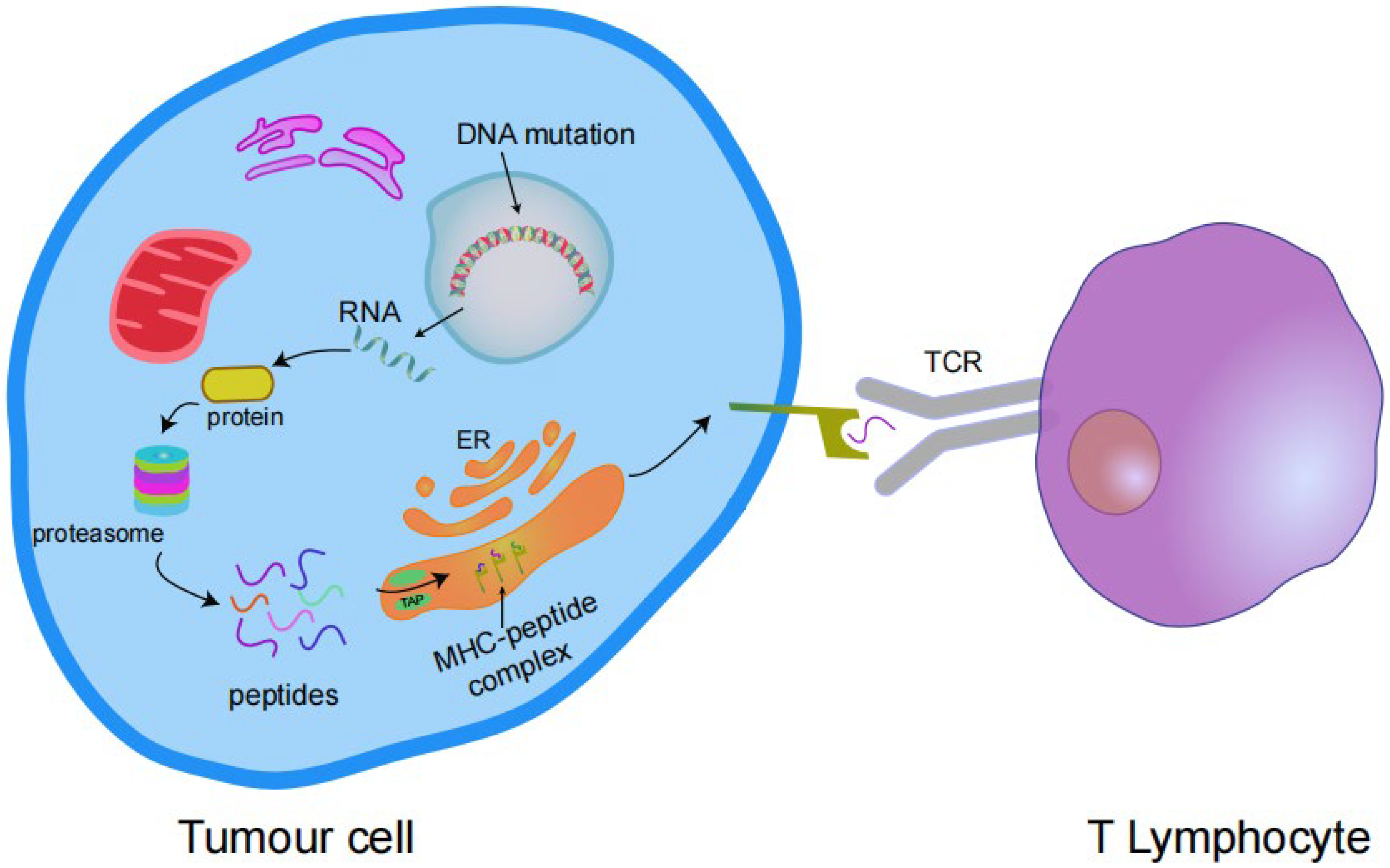
2. Advantages of Targeting Neoantigens
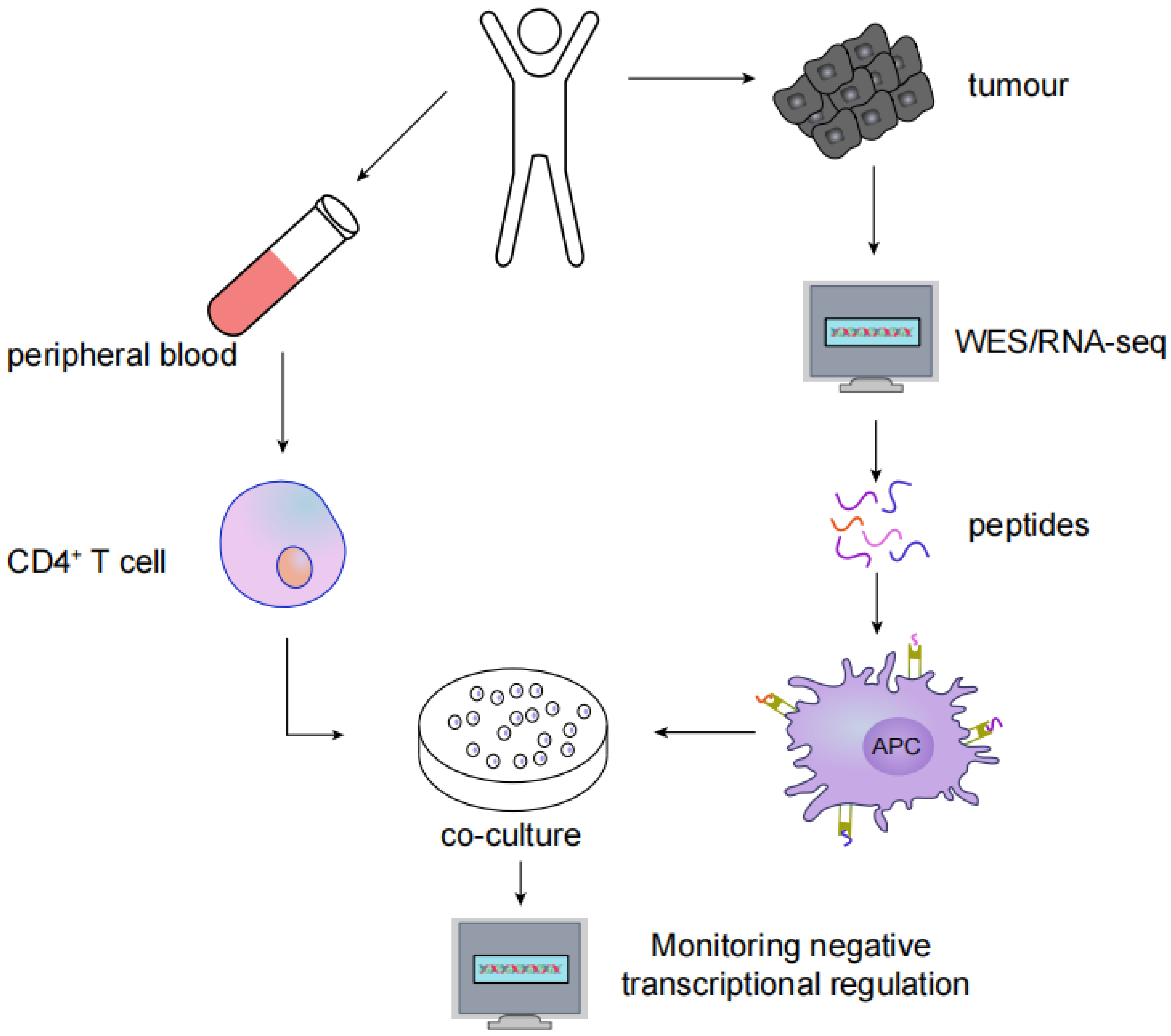
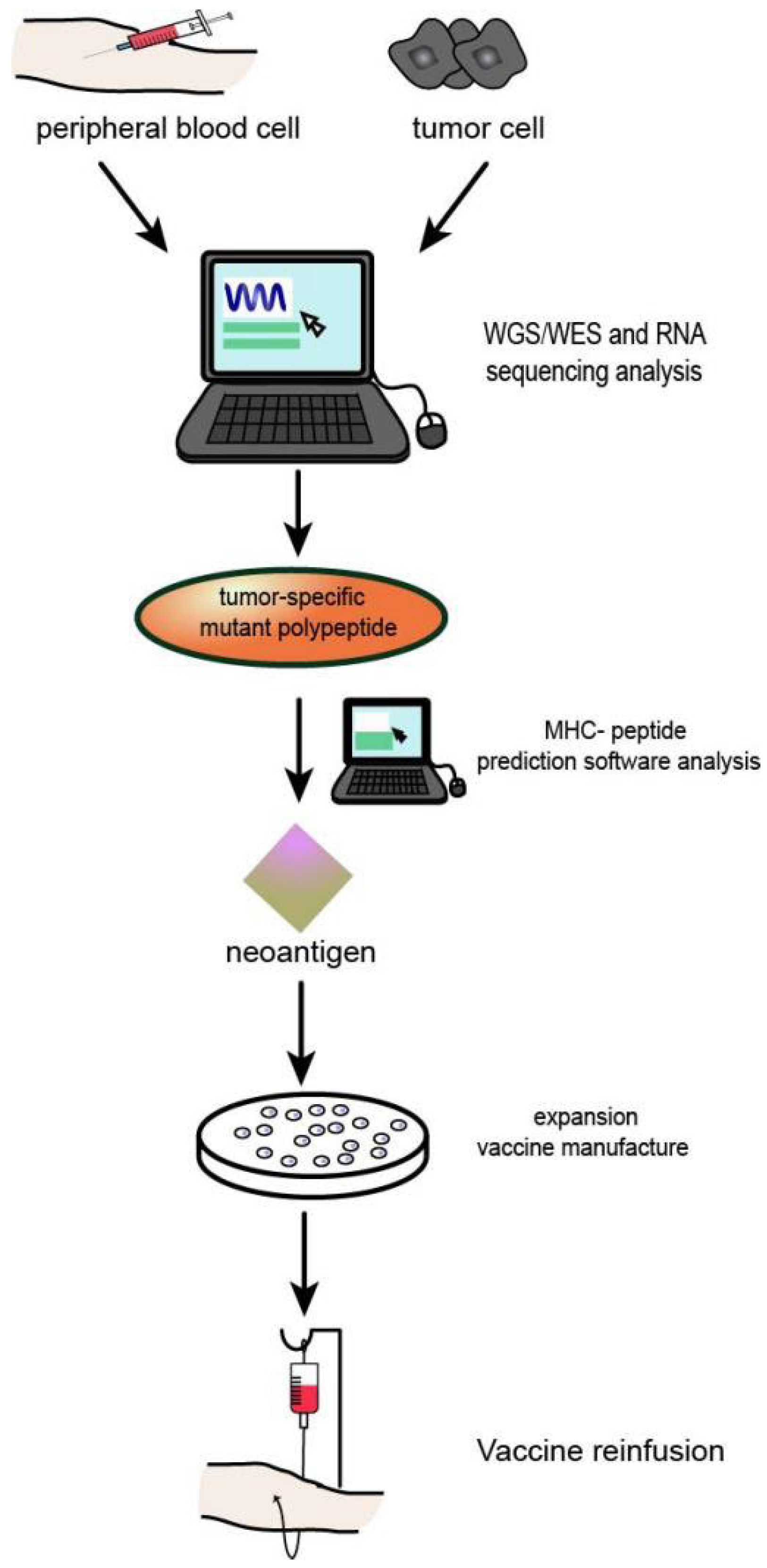
3. Identification and Screening of Neoantigens
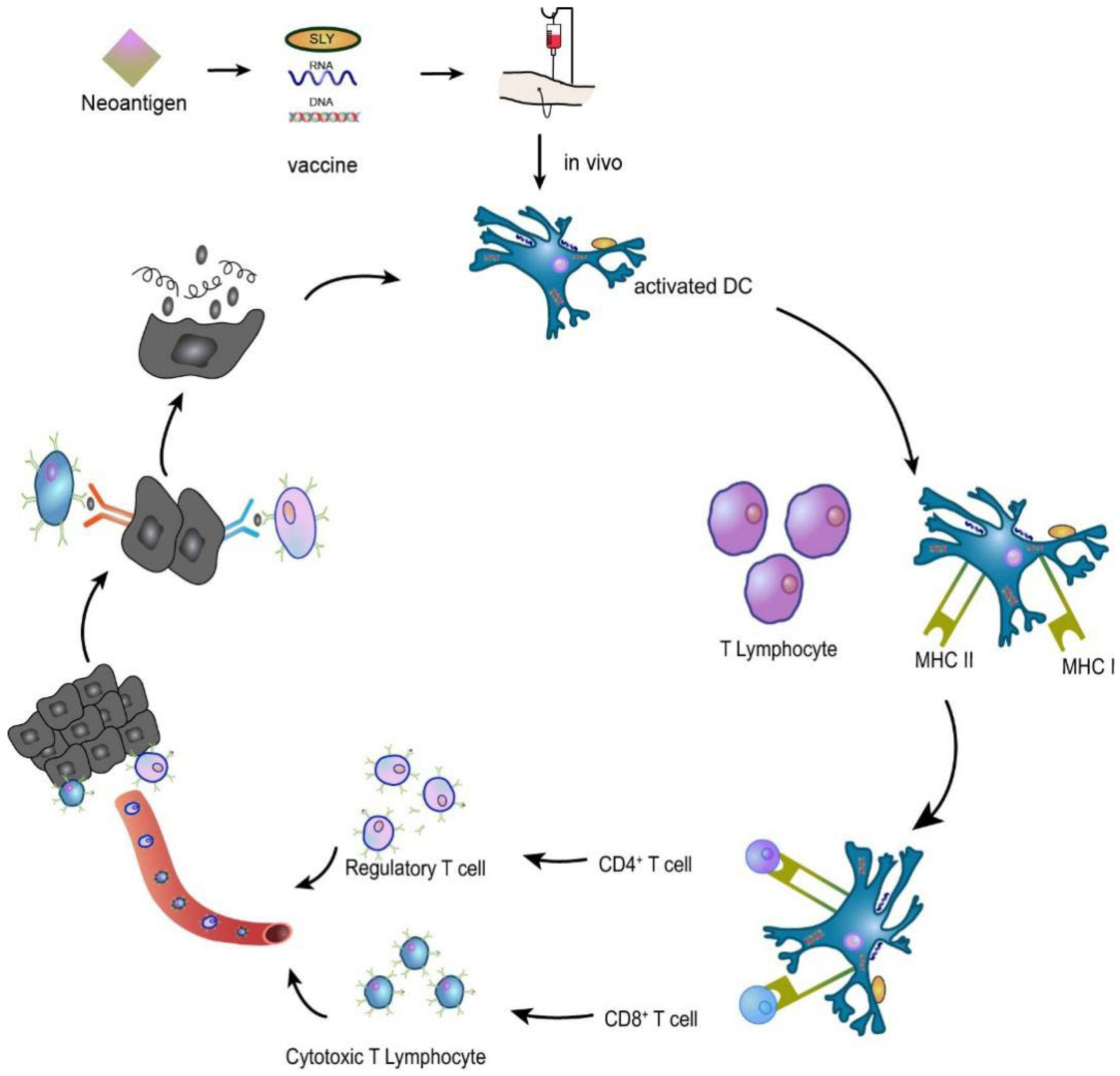
4. HNSCC Neoantigen Therapy
4.1. HNSCC Vaccination Studies
4.2. HNSCC ACT
4.2.1. CAR-T-Cell Therapy
4.2.2. NK-Cell Therapy
4.2.3. TCR-T-Cell Therapy
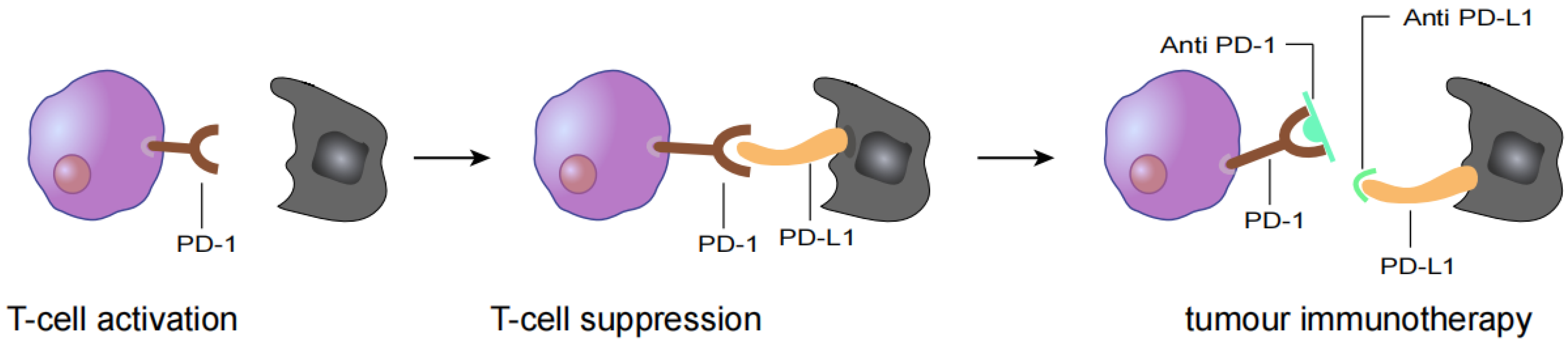
5. HNSCC Immunotherapy
5.1. PD-1 Checkpoint Inhibitor Drug
5.1.1. Nivolumab
5.1.2. Pembrolizumab
5.1.3. PD-1-Targeting Therapy Combined with Neoantigen Therapy
5.2. PD-L1 Checkpoint Inhibitor Drugs
5.2.1. Durvalumab
5.2.2. Atezolizumab
5.2.3. PD-L1 Inhibitors Combined with Neoantigen Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Paver, E.C.; Currie, A.M.; Gupta, R.; Dahlstrom, J.E. Human papilloma virus related squamous cell carcinomas of the head and neck: Diagnosis, clinical implications and detection of HPV. Pathology 2020, 52, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Spuldaro, T.R.; Wagner, V.P.; Nör, F.; Gaio, E.J.; Squarize, C.H.; Carrard, V.C.; Rösing, C.K.; Castilho, R.M. Periodontal disease affects oral cancer progression in a surrogate animal model for tobacco exposure. Int. J. Oncol. 2022, 60, 77. [Google Scholar] [CrossRef]
- Jung, K.; Narwal, M.; Min, S.Y.; Keam, B.; Kang, H. Squamous cell carcinoma of head and neck: What internists should know. Korean J. Intern. Med. 2020, 35, 1031–1044. [Google Scholar] [CrossRef]
- Gelwan, E.; Malm, I.J.; Khararjian, A.; Fakhry, C.; Bishop, J.A.; Westra, W.H. Nonuniform Distribution of High-risk Human Papillomavirus in Squamous Cell Carcinomas of the Oropharynx: Rethinking the Anatomic Boundaries of Oral and Oropharyngeal Carcinoma From an Oncologic HPV Perspective. Am. J. Surg. Pathol. 2017, 41, 1722–1728. [Google Scholar] [CrossRef]
- Brennan, S.; Baird, A.-M.; O’Regan, E.; Sheils, O. The Role of Human Papilloma Virus in Dictating Outcomes in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2021, 8, 677900. [Google Scholar] [CrossRef]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef]
- Argiris, A.; Harrington, K.J.; Tahara, M.; Schulten, J.; Chomette, P.; Ferreira Castro, A.; Licitra, L. Evidence-Based Treatment Options in Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck. Front. Oncol. 2017, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, M.L.; Goh, G.; Chiosea, S.I.; Bauman, J.E.; Freilino, M.L.; Zeng, Y.; Wang, L.; Diergaarde, B.B.; Gooding, W.E.; Lui, V.W.; et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Investig. 2016, 126, 1606. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Kitamura, N.; Sento, S.; Yoshizawa, Y.; Sasabe, E.; Kudo, Y.; Yamamoto, T. Yamamoto. Current Trends and Future Prospects of Molecular Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 22, 240. [Google Scholar] [CrossRef]
- Horton, J.D.; Knochelmann, H.M.; Day, T.A.; Paulos, C.M.; Neskey, D.M. Immune Evasion by Head and Neck Cancer: Foundations for Combination Therapy. Trends Cancer 2019, 5, 208–232. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef]
- Supabphol, S.; Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Neoantigen vaccine platforms in clinical development: Understanding the future of personalized immunotherapy. Expert Opin. Investig. Drugs 2021, 30, 529–541. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Von Witzleben, A.; Wang, C.; Laban, S.; Savelyeva, N.; Ottensmeier, C.H. HNSCC: Tumour Antigens and Their Targeting by Immunotherapy. Cells 2020, 9, 2103. [Google Scholar] [CrossRef]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef]
- Chen, R.; Fulton, K.M.; Twine, S.M.; Li, J. Identification of MHC peptides using mass spectrometry for neoantigen discovery and cancer vaccine development. Mass Spectrom. Rev. 2021, 40, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Heubeck, B.; Wendler, O.; Bumm, K.; Schäfer, R.; Müller-Vogt, U.; Häusler, M.; Meese, E.; Iro, H.; Steinhart, H. Tumor-associated antigenic pattern in squamous cell carcinomas of the head and neck—Analysed by SEREX. Eur. J. Cancer 2013, 49, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Baldin, A.V.; Isayev, O.; Werner, J.; Zamyatnin, A.A., Jr.; Bazhin, A.V. Cancer Vaccines: Antigen Selection Strategy. Vaccines 2021, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.-L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Nik-Zainal. Mutational signatures: Emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 2021, 21, 619–637. [Google Scholar] [CrossRef]
- Walk, E.E.; Yohe, S.L.; Beckman, A.; Schade, A.; Zutter, M.M.; Pfeifer, J.; Berry, A.B. The Cancer Immunotherapy Biomarker Testing Landscape. Arch. Pathol. Lab. Med. 2020, 144, 706–724. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-Generation Sequencing Technologies. Cold Spring. Harb. Perspect. Med. 2019, 9, a036798. [Google Scholar] [CrossRef]
- Ang, M.Y.; Low, T.Y.; Lee, P.Y.; Nazarie, W.F.W.M.; Guryev, V.; Jamal, R. Proteogenomics: From next-generation sequencing (NGS) and mass spectrometry-based proteomics to precision medicine. Clin. Chim. Acta 2019, 498, 38–46. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Lancaster, E.M.; Jablons, D.; Kratz, J.R. Applications of Next-Generation Sequencing in Neoantigen Prediction and Cancer Vaccine Development. Genet. Test. Mol. Biomark. 2020, 24, 59–66. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Szeto, C.; Lobos, C.A.; Nguyen, A.T.; Gras, S. TCR Recognition of Peptide-MHC-I: Rule Makers and Breakers. Int. J. Mol. Sci. 2020, 22, 68. [Google Scholar] [CrossRef]
- He, Q.; Jiang, X.; Zhou, X.; Weng, J. Targeting cancers through TCR-peptide/MHC interactions. J. Hematol. Oncol. 2019, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Luo, H.; Kong, Y.; Lai, W.-F.; Cui, L.; Zhu, X. Cancer neoantigen: Boosting immunotherapy. Biomed. Pharmacother. 2020, 131, 110640. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Taniuchi, I. CD4 Helper and CD8 Cytotoxic T Cell Differentiation. Annu. Rev. Immunol. 2018, 36, 579–601. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Haabeth, O.A.W.; Fauskanger, M.; Manzke, M.; Lundin, K.U.; Corthay, A.; Bogen, B.; Tveita, A.A. CD4(+) T-cell-Mediated Rejection of MHC Class II-Positive Tumor Cells Is Dependent on Antigen Secretion and Indirect Presentation on Host APCs. Cancer Res. 2018, 78, 4573–4585. [Google Scholar] [CrossRef] [Green Version]
- Stifter, K.; Dekhtiarenko, I.; Krieger, J.; Tissot, A.C.; Seufferlein, T.; Wagner, M.; Schirmbeck, R. A tumor-specific neoepitope expressed in homologous/self or heterologous/viral antigens induced comparable effector CD8(+) T-cell responses by DNA vaccination. Vaccine 2020, 38, 3711–3719. [Google Scholar] [CrossRef]
- Axelrod, M.L.; Cook, R.S.; Johnson, D.B.; Balko, J.M. Biological Consequences of MHC-II Expression by Tumor Cells in Cancer. Clin. Cancer Res. 2019, 25, 2392–2402. [Google Scholar] [CrossRef]
- Matsuda-Lennikov, M.; Ohigashi, I.; Takahama, Y. Tissue-specific proteasomes in generation of MHC class I peptides and CD8(+) T cells. Curr. Opin. Immunol. 2022, 77, 102217. [Google Scholar] [CrossRef]
- Veatch, J.R.; Lee, S.M.; Shasha, C.; Singhi, N.; Szeto, J.L.; Moshiri, A.S.; Kim, T.S.; Smythe, K.; Kong, P.; Fitzgibbon, M.; et al. Neoantigen-specific CD4(+) T cells in human melanoma have diverse differentiation states and correlate with CD8(+) T cell, macrophage, and B cell function. Cancer Cell 2022, 40, 393–409. [Google Scholar] [CrossRef]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Gwin, W.R., 3rd; Mitchell, D.A. Vaccine Therapies for Cancer: Then and Now. Target Oncol. 2021, 16, 121–152. [Google Scholar] [CrossRef]
- Igarashi, Y.; Sasada, T. Cancer Vaccines: Toward the Next Breakthrough in Cancer Immunotherapy. J. Immunol. Res. 2020, 2020, 5825401. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Xu, N.; Saito, S.; Zhou, L.; Ozgenc, I.; Webb, J.; Fu, C.; Zolkind, P.; Egloff, A.M.; Uppaluri, R. Uppaluri. Integrating CD4(+) T cell help for therapeutic cancer vaccination in a preclinical head and neck cancer model. Oncoimmunology 2021, 10, 1958589. [Google Scholar] [CrossRef] [PubMed]
- Zolkind, P.; Przybylski, D.; Marjanovic, N.; Nguyen, L.; Lin, T.; Johanns, T.; Alexandrov, A.; Zhou, L.; Allen, C.T.; Miceli, A.P.; et al. Cancer immunogenomic approach to neoantigen discovery in a checkpoint blockade responsive murine model of oral cavity squamous cell carcinoma. Oncotarget 2018, 9, 4109–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafferji, M.S.; Yang, J.C. Adoptive T-Cell Therapy for Solid Malignancies. Surg. Oncol. Clin. N. Am. 2019, 28, 465–479. [Google Scholar] [CrossRef]
- Gong, Y.; Wolterink, R.G.J.K.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell Therapy With TILs: Training and Taming T Cells to Fight Cancer. Front. Immunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Liu, D. CAR-T “the living drugs”, immune checkpoint inhibitors, and precision medicine: A new era of cancer therapy. J. Hematol. Oncol. 2019, 12, 113. [Google Scholar] [CrossRef]
- Ecsedi, M.; McAfee, M.S.; Chapuis, A.G. The Anticancer Potential of T Cell Receptor-Engineered T Cells. Trends Cancer 2021, 7, 48–56. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Markowitz, L.E.; Naleway, A.L.; Klein, N.P.; Lewis, R.M.; Crane, B.; Querec, T.D.; Hsiao, A.; Aukes, L.; Timbol, J.; Weinmann, S.; et al. Human Papillomavirus Vaccine Effectiveness Against HPV Infection: Evaluation of One, Two, and Three Doses. J. Infect. Dis. 2020, 221, 910–918. [Google Scholar] [CrossRef]
- Tumban, E. A Current Update on Human Papillomavirus-Associated Head and Neck Cancers. Viruses 2019, 11, 922. [Google Scholar] [CrossRef] [Green Version]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients With Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Jeffrey Melson Clarke, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; Robert-Tissot, C.; et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Delord, J.-P.; Block, M.S.; Ottensmeier, C.; Colon-Otero, G.; Le Tourneau, C.; Lalanne, A.; Jamet, C.; Lantz, O.; Knutson, K.L.; Lacoste, G.; et al. Phase 1 studies of personalized neoantigen vaccine TG4050 in ovarian carcinoma(OC) and head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2022, 40, 2637. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Caushi, J.X.; Zhang, J.; Ji, Z.; Vaghasia, A.; Zhang, B.; Hsiue, E.H.-C.; Mog, B.J.; Hou, W.; Justesen, S.; Blosser, R.; et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 2021, 596, 126–132. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Pan, C.; Liu, H.; Robins, E.; Song, W.; Liu, D.; Li, Z.; Zheng, L. Next-generation immuno-oncology agents: Current momentum shifts in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 29. [Google Scholar] [CrossRef] [Green Version]
- Moreno, V.; Hernandez, T.; de Miguel, M.; Doger, B.; Calvo, E. Adoptive cell therapy for solid tumors: Chimeric antigen receptor T cells and beyond. Curr. Opin. Pharmacol. 2021, 59, 70–84. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, J.; Liu, M.; Xing, H.; Wang, Z.; Huang, L.; Mellor, A.L.; Wang, W.; Wu, S. Adoptive CD8(+) T cell therapy against cancer: Challenges and opportunities. Cancer Lett. 2019, 462, 23–32. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Zhang, K.; Lam, A.K.; Huang, J.; Qiu, F.; Qiao, B.; Zhang, Y. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med. 2020, 9, 640–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.P.; Jin, L.; Bennett, K.B.; Wang, D.; Fredenburg, K.M.; Tseng, J.E.; Chang, L.-J.; Huang, J.; Chan, E.K. CD70 as a target for chimeric antigen receptor T cells in head and neck squamous cell carcinoma. Oral Oncol. 2018, 78, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Hyrenius-Wittsten, A.; Su, Y.; Park, M.; Garcia, J.M.; Alavi, J.; Perry, N.; Montgomery, G.; Liu, B.; Roybal, K.T. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med. 2021, 13, eabd8836. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef]
- Charap, A.J.; Enokida, T.; Brody, R.; Sfakianos, J.; Miles, B.; Bhardwaj, N.; Horowitz, A. Landscape of natural killer cell activity in head and neck squamous cell carcinoma. J. Immunother. Cancer 2020, 8, e001523. [Google Scholar] [CrossRef]
- Friedman, J.; Padget, M.; Lee, J.; Schlom, J.; Hodge, J.; Allen, C. Direct and antibody-dependent cell-mediated cytotoxicity of head and neck squamous cell carcinoma cells by high-affinity natural killer cells. Oral Oncol. 2019, 90, 38–44. [Google Scholar] [CrossRef]
- Lim, C.M.; Liou, A.; Poon, M.; Koh, L.P.; Tan, L.K.; Loh, K.S.; Petersson, B.F.; Ting, E.; Campana, D.; Goh, B.C.; et al. Phase I study of expanded natural killer cells in combination with cetuximab for recurrent/metastatic nasopharyngeal carcinoma. Cancer Immunol. Immunother. 2022. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef]
- Huang, R.-S.; Lai, M.-C.; Shih, H.-A.; Lin, S. A robust platform for expansion and genome editing of primary human natural killer cells. J. Exp. Med. 2021, 218, e20201529. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; De Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef]
- Jan, C.-I.; Huang, S.-W.; Canoll, P.; Bruce, J.N.; Lin, Y.-C.; Pan, C.-M.; Lu, H.-M.; Chiu, S.-C.; Cho, D.-Y. Targeting human leukocyte antigen G with chimeric antigen receptors of natural killer cells convert immunosuppression to ablate solid tumors. J. Immunother. Cancer 2021, 9, e003050. [Google Scholar] [CrossRef]
- Zhao, Q.; Jiang, Y.; Xiang, S.; Kaboli, P.J.; Shen, J.; Zhao, Y.; Wu, X.; Du, F.; Li, M.; Cho, C.H.; et al. Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front. Immunol. 2021, 12, 658753. [Google Scholar] [CrossRef]
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: A systematic review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef]
- Wei, T.; Leisegang, M.; Xia, M.; Kiyotani, K.; Li, N.; Zeng, C.; Deng, C.; Jiang, J.; Harada, M.; Agrawal, N.; et al. Generation of neoantigen-specific T cells for adoptive cell transfer for treating head and neck squamous cell carcinoma. Oncoimmunology 2021, 10, 1929726. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Lim, L.H.K.; Cheung, F.S.G. The role of the tumour microenvironment in immunotherapy. Endocr.-Relat. Cancer 2017, 24, T283–T295. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Gao, J.; Li, P.; Wang, Y.; Qi, Q.; Liu, X.; Li, J.; Wang, C.; Du, L. Pyroptosis, metabolism, and tumor immune microenvironment. Clin. Transl. Med. 2021, 11, e492. [Google Scholar] [CrossRef] [PubMed]
- GGordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Zamani, M.R.; Aslani, S.; Salmaninejad, A.; Javan, M.R.; Rezaei, N. PD-1/PD-L and autoimmunity: A growing relationship. Cell Immunol. 2016, 310, 27–41. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Solomon, B.; Young, R.J.; Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin. Cancer Biol. 2018, 52, 228–240. [Google Scholar] [CrossRef]
- Yokota, T.; Homma, A.; Kiyota, N.; Tahara, M.; Hanai, N.; Asakage, T.; Matsuura, K.; Ogawa, T.; Saito, Y.; Sano, D.; et al. Immunotherapy for squamous cell carcinoma of the head and neck. JPN J. Clin. Oncol. 2020, 50, 1089–1096. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef] [Green Version]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.J.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018, 81, 45–51. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, L.G.; Ferrarotto, R. Pembrolizumab in the first-line treatment of advanced head and neck cancer. Expert Rev. Anticancer. Ther. 2021, 21, 1321–1331. [Google Scholar] [CrossRef]
- Van Vugt, M.J.H.; Stone, J.A.; De Greef, H.J.M.M.; Snyder, E.S.; Lipka, L.; Turner, D.C.; Chain, A.; Lala, M.; Li, M.; Robey, S.H.; et al. Immunogenicity of pembrolizumab in patients with advanced tumors. J. Immunother. Cancer 2019, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Twardowski, P.; Johnson, M.; Stein, M.; Vaishampayan, U.; Gillison, M.; McNeil, L.; Dowal, L.; DeOliveira, D.; Jain, M.; Price, J.; et al. A phase I trial of GEN-009, a neoantigen vaccine using ATLAS™, an autologous immune assay, to identify immunogenic and inhibitory tumour mutations. Ann. Oncol. 2019, 30, v479. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Ou, S.-H.I.; Balmanoukian, A.; Fury, M.G.; Massarelli, E.; Brahmer, J.R.; Weiss, J.; Schöffski, P.; Antonia, S.J.; Massard, C.; et al. Safety and efficacy of durvalumab in patients with head and neck squamous cell carcinoma: Results from a phase I/II expansion cohort. Eur. J. Cancer 2019, 109, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.; Haddad, R.; Even, C.; Tahara, M.; Dvorkin, M.; Ciuleanu, T.; Clement, P.; Mesia, R.; Kutukova, S.; Zholudeva, L.; et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann. Oncol. 2020, 31, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Camidge, D.; Iafolla, M.; Rottey, S.; Schuler, M.; Hellmann, M.; Balmanoukian, A.; Dirix Gordon, L.M.; Sullivan, R.; Henick, B.; et al. A phase Ib study to evaluate RO7198457, an individualized neoantigen specific immunotherapy (iNeST), in combination with atezolizumab in patients with locally advanced or metastatic solid tumors. Cancer Res. 2020, 80, CT301. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kim, S.S.; Pang, J.; Gold, K.A.; Gutkind, J.S.; Califano, J.A.; Mell, L.K.; Cohen, E.E.; Sharabi, A.B. Immune Modulation of Head and Neck Squamous Cell Carcinoma and the Tumor Microenvironment by Conventional Therapeutics. Clin. Cancer Res. 2019, 25, 4211–4223. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Number | Study Title | Conditions | Interventions | Phase | Number Enrolled | First Posted | Inclusion Criteria |
|---|---|---|---|---|---|---|---|
| NCT03633110 | Safety, Tolerability, Immunogenicity, and Antitumor Activity of GEN-009 Adjuvanted Vaccine | HNSCC, others | Biological: GEN-009 Adjuvanted Vaccine Drug: Nivolumab Drug: Pembrolizumab | I/II | 24 | 16 August 2018 | 1. Patients beginning pembrolizumab with recurrent or metastatic HNSCC who experienced disease progression while on or after receiving a platinum-based therapy, or those beginning first-line pembrolizumab for recurrent or metastatic tumours. 2. Agree to a tumour biopsy 50 days after first GEN-009 vaccination. |
| NCT05269381 | Personalized Neoantigen Peptide-based Vaccine in Combination with Pembrolizumab for the Treatment of Advanced Solid Tumours, The PNeoVCA Study | HNSCC, others | Drug: Cyclophosphamide Biological: Neoantigen Peptide Vaccine Biological: Pembrolizumab Biological: Sargramostim | I | 36 | 8 March 2022 | 1. Histologically confirmed unresectable locally advanced or meta static solid malignancies. 2. Soft tissue lesion amenable for adequate tissue sampling. 3. Patients with actionable genomic abnormality including but not limited to EGFR, ALK, MET, ROS-1, RET, NTRK, KRAS or BRAF must have also received and progressed while on at least one line of prior FDA-approved targeted therapy. |
| NCT04266730 | Trial of a Personalized and Adaptive Neoantigen Dose-adjusted Vaccine Concurrently with Pembrolizumab | HNSCC, others | Biological: PANDA-VAC Drug: Pembrolizumab | I | 6 | 12 February 2020 | 1. As 1st line treatment for tumours expressing PD-L1 [Combined Positive Score (CPS) ≥ 1] as determined by an FDA-approved test. 2. As a non1st line treatment for patients with recurrent or metastatic HNSCC who experienced disease progression while on or after receiving platinum-containing chemotherapy. |
| NCT01998542 | Safety and Tolerability Study of AlloVaxTM in Patients With Metastatic or Recurrent Cancer of the Head and Neck | HNSCC, HNC | Biological: AlloVax Biological: CRCL Biological: AlloStim | II | 12 | 29 November 2013 | 1. Patients must have a tumour safely accessible for biopsy resulting in a minimum of 0.1 g of tumour sample for CRCL processing. 2. Patients must have visible external tumours measurable with at least one lesion deemed to be safely accessible for serial biopsy. |
| NCT03552718 | QUILT-2.025 NANT Neoepitope Yeast Vaccine (YE-NEO-001): Adjuvant Immunotherapy Using a Personalized Neoepitope Yeast-based Vaccine to Induce T-Cell Responses In Subjects W/Previously Treated Cancers | HNSCC | Biological:YE-NEO-001 | I | 16 | 12 June 2018 | 1. Must have received <6 months of SoC therapy. 2. ECOG performance status of 0 to 2. 3. If cancer recurs while on treatment during this study, the patient must be willing to provide a tumour biopsy specimen for exploratory analyses, if considered safe by the Investigator. |
| Advantage | Disadvantage | |
|---|---|---|
| PD-1 inhibitors | 1. Suitable for all types of malignant tumours. 2. Long-term efficacy. 3. Side effects are relatively minor. | 1. The efficiency is not high, at less than 30%, and even lower when combined with genetic mutations. 2. The onset of action is slow, with a median of three months. 3. Expensive |
| PD-L1 inhibitors | 1. PD-1 inhibitors only block the PD-1-PD-L1 pathway, without affecting the PD-1-PD-L2 pathway, avoiding the occurrence of interstitial pneumonia and other side effects. 2. It can block the coinhibitory function of B7.1 and PD-L1, and fully activate T cell function and cytokine production. | 1. Expensive. 2. A higher dose is required for the same efficacy as PD-1 drugs. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, L.; Zhang, A.; Yang, F.; Du, W. Immunotherapeutic Strategies for Head and Neck Squamous Cell Carcinoma (HNSCC): Current Perspectives and Future Prospects. Vaccines 2022, 10, 1272. https://doi.org/10.3390/vaccines10081272
Gao L, Zhang A, Yang F, Du W. Immunotherapeutic Strategies for Head and Neck Squamous Cell Carcinoma (HNSCC): Current Perspectives and Future Prospects. Vaccines. 2022; 10(8):1272. https://doi.org/10.3390/vaccines10081272
Chicago/Turabian StyleGao, Lei, Anqi Zhang, Fuyuan Yang, and Wei Du. 2022. "Immunotherapeutic Strategies for Head and Neck Squamous Cell Carcinoma (HNSCC): Current Perspectives and Future Prospects" Vaccines 10, no. 8: 1272. https://doi.org/10.3390/vaccines10081272