
In Vivo Oncolytic Virotherapy in Murine Models of Hepatocellular Carcinoma: A Systematic Review



Abstract
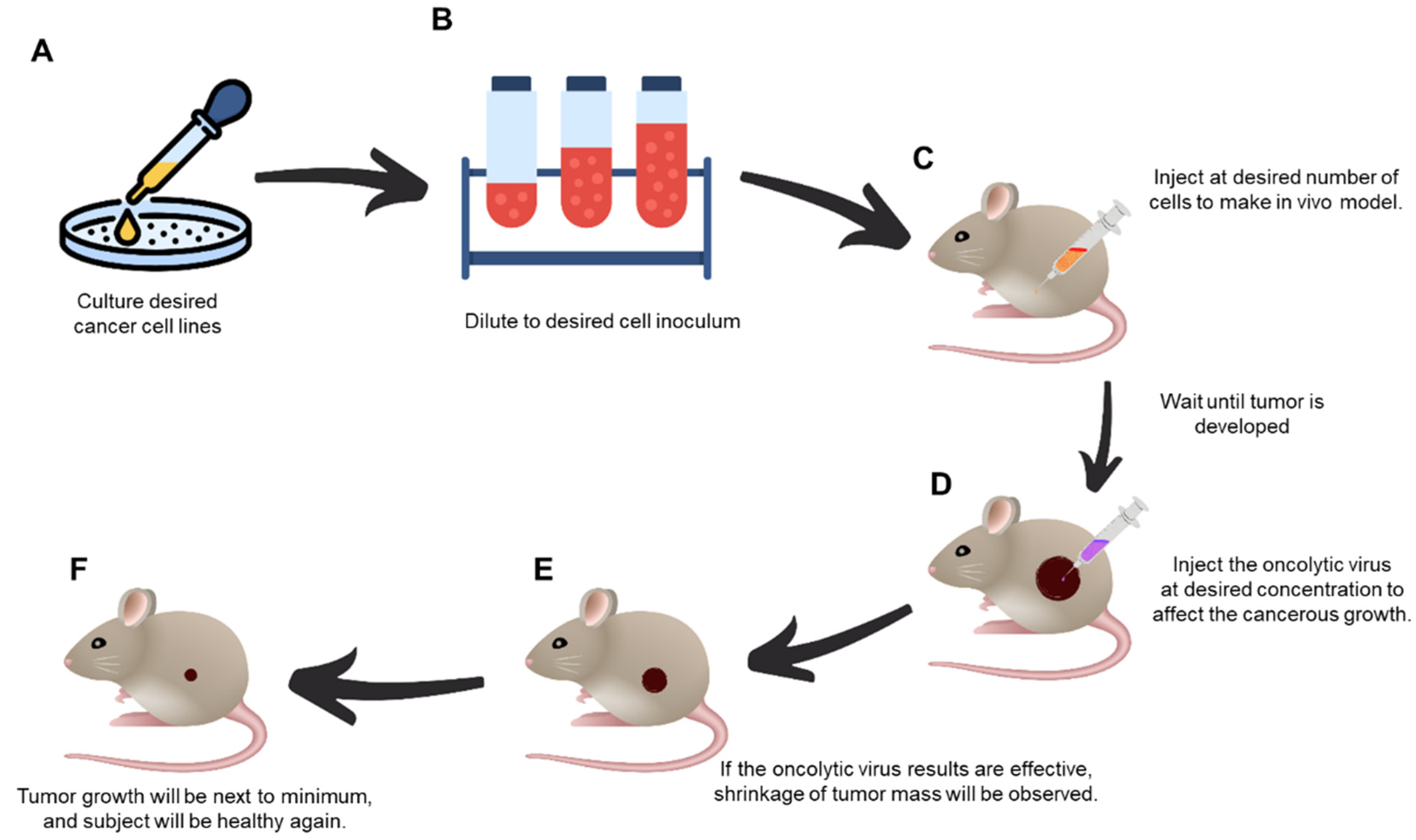
:1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Criteria for Inclusion and Exclusion
2.3. Data Extraction and Assessment of Study Quality
3. Results
3.1. Eligible Studies
3.2. Characteristics of the Studies Included
3.3. In Vivo Oncolytic Virotherapy
3.3.1. Oncolytic Vaccinia Virus (VV)

{kind=link}
{kind=link}
{kind=link}
| OV | Publication Author/Year | Animal, Sex 1, Age 2 | Cell Line | Number of Cancer Cells 3, Route * | Tumor Measurement Frequency (Days) | Tumor Size at the Time of Virotherapy (mm3) | Agent/Virus 4 | Virus Load 5 (PFU), Route * | Outcome (+/−) 6 |
|---|---|---|---|---|---|---|---|---|---|
| Oncolytic vaccinia virus | Wang et al. (2021) [16] | BALB/c nude mice F, 5 | MHCC97-H | 4 × 106 SC | 5 | 80–120 | Luteolin/VV-IL24 | 2 × 107 IP, IT | VV-IL24/Luteolin + (p < 0.001) |
| Zhang et al. (2019) [17] | C57BL6 mice F, 6 | Bel7402 | 2.5 × 106 SC | - | - | VV-IL-37 | 1 × 107 IT | VV-IL-37 + (p < 0.01) | |
| Li et al. (2018) [18] | BALB/c nude mice F, 4–5 | MHCC97-H, | 2.5 × 106 SC | 5 | 120 | OncoVV-TTL | 1 × 107, IT | OncoVV-TTL+ | |
| Gentschev et al. (2011) [19] | Nude-Foxn1 M/F, 6–8 | Huh-7, PLC | 5 × 106 SC | 3.5 | - | GLV-1h68 | 5 × 106 IV | GLV-1h68 PLC+, HuH7 - | |
| Oncolytic adenovirus | Xie et al. (2018) [20] | BALB/c nude mice F, 4 | Huh-7 | 3 × 106 - | - | 80–100 | Ad-sp-VGLL4 | 5 × 108 - | Ad-sp-VGLL4 + (p < 0.01) |
| Zhang et al. (2016) [21] | NOD/SCID mice M, 4 BALB/C nude mice F, 5 | PLC/PRF/5 | 2 × 106 SC | 3 | 100 | GD55 ZD55 | 6 × 108 IT | GD55 + | |
| Chen et al. (2011) [22] | BALB/c nude mice; M/F, 4–6 | Hep3B | 5 × 106 SC | - | 100–150 | Ad5/35EGFP | 1 × 109 IT | SG600-p53- and SG635-p53 + (p < 0.001) | |
| Cao et al. (2011) [23] | athymic nude mice F, 4 | Huh-7 | 5 × 106 SC | 7 | 90–120 | SOCS3, IL-24/OAV | 2 × 109 IT | SOCS3, IL-24/OAV+ (p < 0.001) | |
| Newcastle disease virus | Meng et al. (2020) [24] | C57BL6 mice M, 6 | H22, Hepa 1–6 | 2 × 106, 5 × 106 IP, SC | - | - | Dichloroacetate/NDV | 1 × 107 IT | DCA/NDV + |
| Oncolytic avian reovirus | Cai et al. (2019) [25] | SPF Kunming mice F, 5 | - | - | - | - | ARV S1133 | 3 ×106 O, IM | ARV S1133+, No pathologic damage Apoptosis in cell line HepG2 |
| Oncolytic vesicular stomatitis virus | Altomonte et al. (2008) [26] | Buffalo rats (IC) M, 6 | - | - | - | - | rVSV-gG | 1.3 × 107 IV | rVSV-gG +, No pathologic damage to organs, |
| Altomonte et al. (2009) [27] | Buffalo rats M, 5–7 | - | - | - | - | rVSVUL141, rVSV-F | 1 × 107 IV | rVSV-F-, rVSV-UL141 + (p < 0.001) | |
| Altomonte et al. (2013) [28] | Buffalo rats M, 6 | - | - | - | - | rVSV-LacZ, rVSV (M51R) | 1 × 107 IV | rVSV-LacZ + (p < 0.005) |
3.3.2. Oncolytic Adenovirus (Adv)
3.3.3. Oncolytic Vesicular Stomatitis (VSV)
3.3.4. Newcastle Disease Virus (NDV)
3.3.5. Oncolytic Avian Reovirus (ARV)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Liver, E.A.S. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar]
- Yoo, S.Y.; Badrinath, N.; Woo, H.Y.; Heo, J. Oncolytic Virus-Based Immunotherapies for Hepatocellular Carcinoma. Mediat. Inflamm. 2017, 2017, 5198798. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Jeong, S.N.; Yoo, S.Y. Novel Oncolytic Virus Armed with Cancer Suicide Gene and Normal Vasculogenic Gene for Improved Anti-Tumor Activity. Cancers 2020, 12, 1070. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef]
- Badrinath, N.; Heo, J.; Yoo, S.Y. Viruses as nanomedicine for cancer. Int. J. Nanomed. 2016, 11, 4835–4847. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Badrinath, N.; Lee, H.L.; Heo, J.; Kang, D.H. A Cancer-Favoring, Engineered Vaccinia Virus for Cholangiocarcinoma. Cancers 2019, 11, 1667. [Google Scholar] [CrossRef]
- Truong, C.S.; Yoo, S.Y. Oncolytic Vaccinia Virus in Lung Cancer Vaccines. Vaccines 2022, 10, 240. [Google Scholar] [CrossRef]
- Ailia, M.J.; Jin, Y.K.; Kim, H.K.; Jang, G. Development of in-vitro maturation protocol for rat oocytes; under simple culture vs co-culture with cumulus cell monolayer and its developmental potential via Parthenogenetic/artificial activation. BMC Vet. Res. 2021, 17, 44. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. Embo Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Carter, G.C.; Law, M.; Hollinshead, M.; Smith, G.L. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 2005, 86, 1279–1290. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.F.; Guo, Z.B.; Giehl, E.; Feist, M.; Dai, E.Y.; Liu, W.L.; Storkus, W.J.; He, Y.K.; Liu, Z.Q.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef]
- Wang, C.; Li, Q.; Xiao, B.; Fang, H.; Huang, B.; Huang, F.; Wang, Y. Luteolin enhances the antitumor efficacy of oncolytic vaccinia virus that harbors IL-24 gene in liver cancer cells. J. Clin. Lab. Anal. 2021, 35, e23677. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Zhang, J.W.; Zhang, Y.Y.; Xing, J.Y.; Yu, Z.J. Vaccinia virus expressing IL-37 promotes antitumor immune responses in hepatocellular carcinoma. Cell Biochem. Funct. 2019, 37, 618–624. [Google Scholar] [CrossRef]
- Li, G.; Cheng, J.; Mei, S.; Wu, T.; Ye, T. Tachypleus tridentatus Lectin Enhances Oncolytic Vaccinia Virus Replication to Suppress In Vivo Hepatocellular Carcinoma Growth. Mar. Drugs 2018, 16, 200. [Google Scholar] [CrossRef]
- Gentschev, I.; Müller, M.; Adelfinger, M.; Weibel, S.; Grummt, F.; Zimmermann, M.; Bitzer, M.; Heisig, M.; Zhang, Q.; Yu, Y.A.; et al. Efficient colonization and therapy of human hepatocellular carcinoma (HCC) using the oncolytic vaccinia virus strain GLV-1h68. PLoS ONE 2011, 6, e22069. [Google Scholar] [CrossRef]
- Xie, W.; Hao, J.; Zhang, K.; Fang, X.; Liu, X. Adenovirus armed with VGLL4 selectively kills hepatocellular carcinoma with G2/M phase arrest and apoptosis promotion. Biochem. Biophys. Res. Commun. 2018, 503, 2758–2763. [Google Scholar] [CrossRef]
- Zhang, X.; Meng, S.; Zhang, R.; Ma, B.; Liu, T.; Yang, Y.; Xie, W.; Liu, X.; Huang, F.; Liu, T.; et al. GP73-regulated oncolytic adenoviruses possess potent killing effect on human liver cancer stem-like cells. Oncotarget 2016, 7, 29346–29358. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wu, Y.; Liu, W.; Wang, G.; Wang, X.; Yang, Y.; Chen, W.; Tai, Y.; Lu, M.; Qian, Q.; et al. Enhanced antitumor efficacy of a novel fiber chimeric oncolytic adenovirus expressing p53 on hepatocellular carcinoma. Cancer Lett. 2011, 307, 93–103. [Google Scholar] [CrossRef]
- Cao, X.; Wei, R.; Liu, X.; Zeng, Y.; Huang, H.; Ding, M.; Zhang, K.; Liu, X.Y. Cancer targeting gene-viro-therapy specific for liver cancer by α-fetoprotein-controlled oncolytic adenovirus expression of SOCS3 and IL-24. Acta Biochim. Biophys. Sin. 2011, 43, 813–821. [Google Scholar] [CrossRef]
- Meng, G.; Li, B.; Chen, A.; Zheng, M.; Xu, T.; Zhang, H.; Dong, J.; Wu, J.; Yu, D.; Wei, J. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br. J. Cancer 2020, 122, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Meng, G.; Li, Y.; Wang, W.; Diao, Y.; Zhao, S.; Feng, Q.; Tang, Y. The oncolytic efficacy and safety of avian reovirus and its dynamic distribution in infected mice. Exp. Biol. Med. 2019, 244, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Wu, L.; Chen, L.; Meseck, M.; Ebert, O.; García-Sastre, A.; Fallon, J.; Woo, S.L. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 146–153. [Google Scholar] [CrossRef]
- Altomonte, J.; Wu, L.; Meseck, M.; Chen, L.; Ebert, O.; Garcia-Sastre, A.; Fallon, J.; Mandeli, J.; Woo, S.L. Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther. 2009, 16, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Marozin, S.; De Toni, E.N.; Rizzani, A.; Esposito, I.; Steiger, K.; Feuchtinger, A.; Hellerbrand, C.; Schmid, R.M.; Ebert, O. Antifibrotic properties of transarterial oncolytic VSV therapy for hepatocellular carcinoma in rats with thioacetamide-induced liver fibrosis. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Flatt, J.W. Adenovirus Entry: From Infection to Immunity. Annu. Rev. Virol. 2019, 6, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, W.; Ehrhardt, A. Expanding the Spectrum of Adenoviral Vectors for Cancer Therapy. Cancers 2020, 12, 1139. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhong, L.; Zhao, Y. Oncolytic adenovirus: A tool for reversing the tumor microenvironment and promoting cancer treatment (Review). Oncol. Rep. 2021, 45, 49. [Google Scholar] [CrossRef]
- Lei, Z.G.; Ren, X.H.; Wang, S.S.; Liang, X.H.; Tang, Y.L. Immunocompromised and immunocompetent mouse models for head and neck squamous cell carcinoma. OncoTargets Ther. 2016, 9, 545–555. [Google Scholar] [CrossRef]
- Yang, Y.G.; Sykes, M. Xenotransplantation: Current status and a perspective on the future. Nat. Rev. Immunol. 2007, 7, 519–531. [Google Scholar] [CrossRef]
- McKenna, M.K.; Rosewell-Shaw, A.; Suzuki, M. Modeling the Efficacy of Oncolytic Adenoviruses In Vitro and In Vivo: Current and Future Perspectives. Cancers 2020, 12, 619. [Google Scholar] [CrossRef]
- Naik, S.; Nace, R.; Federspiel, M.J.; Barber, G.N.; Peng, K.W.; Russell, S.J. Curative one-shot systemic virotherapy in murine myeloma. Leukemia 2012, 26, 1870–1878. [Google Scholar] [CrossRef]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef]
- Breitbach, C.J.; De Silva, N.S.; Falls, T.J.; Aladl, U.; Evgin, L.; Paterson, J.; Sun, Y.Y.; Roy, D.G.; Rintoul, J.L.; Daneshmand, M.; et al. Targeting tumor vasculature with an oncolytic virus. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 886–894. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. The Enigma of miRNA Regulation in Cancer. Adv. Cancer Res. 2017, 135, 25–52. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Talukdar, S.; Bhoopathi, P.; Shen, X.N.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. mda-7/IL-24 Mediates Cancer Cell-Specific Death via Regulation of miR-221 and the Beclin-1 Axis. Cancer Res. 2017, 77, 949–959. [Google Scholar] [CrossRef]
- Scarlatti, F.; Maffei, R.; Beau, I.; Codogno, P.; Ghidoni, R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008, 15, 1318–1329. [Google Scholar] [CrossRef]
- Wu, T.; Xiang, Y.L.; Liu, T.T.; Wang, X.; Ren, X.Y.; Ye, T.; Li, G.C. Oncolytic Vaccinia Virus Expressing Aphrocallistes vastus Lectin as a Cancer Therapeutic Agent. Mar. Drugs 2019, 17, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzwedel, A.O.; Han, J.; LaRocca, C.J.; Shanley, R.; Yamamoto, M.; Davydova, J. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget 2018, 9, 18041–18052. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ji, W.; Hu, H.; Ma, J.; Li, X.; Mei, W.; Xu, Y.; Hu, H.; Yan, Y.; Song, Q.; et al. Survivin promoter-regulated oncolytic adenovirus with Hsp70 gene exerts effective antitumor efficacy in gastric cancer immunotherapy. Oncotarget 2014, 5, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Kubo, S.; Fukumoto, M.; Yano, A.; Mawatari-Furukawa, Y.; Okamura, H.; Tomita, N. Whole cell vaccination using immunogenic cell death by an oncolytic adenovirus is effective against a colorectal cancer model. Mol. Ther.-Oncolytics 2016, 3, 16031. [Google Scholar] [CrossRef]
- Xu, Y.N.; Chu, L.; Yuan, S.J.; Yang, Y.Q.; Yang, Y.; Xu, B.; Zhang, K.J.; Liu, X.Y.; Wang, R.W.; Fang, L.; et al. RGD-modified oncolytic adenovirus-harboring shPKM2 exhibits a potent cytotoxic effect in pancreatic cancer via autophagy inhibition and apoptosis promotion. Cell Death Dis. 2017, 8, e2835. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef]
- Kim, M.; Zinn, K.R.; Barnett, B.G.; Sumerel, L.A.; Krasnykh, V.; Curiel, D.T.; Douglas, J.T. The therapeutic efficacy of adenoviral vectors for cancer gene therapy is limited by a low level of primary adenovirus receptors on tumour cells. Eur. J. Cancer 2002, 38, 1917–1926. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shariat, S.F.; Ayala, G.E.; Rauen, K.A.; Lerner, S.P. Loss of coxsackie and adenovirus receptor expression is associated with features of aggressive bladder cancer. Urology 2005, 66, 441–446. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, F.; Sakamoto, H.; Nakajima, Y.; Une, Y.; Hinoda, Y.; Imai, K. Association of reduced cell adhesion regulator messenger RNA expression with tumor progression in human hepatocellular carcinoma. Int. J. Cancer 1997, 74, 251–254. [Google Scholar] [CrossRef]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Zhang, W.J.; Gao, Y.J.; Li, P.X.; Shi, Z.B.; Guo, T.; Li, F.; Han, X.K.; Feng, Y.; Zheng, C.; Wang, Z.Y.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2021, 31, 1137. [Google Scholar] [CrossRef]
- Thaci, B.; Ulasov, I.V.; Ahmed, A.U.; Ferguson, S.D.; Han, Y.; Lesniak, M.S. Anti-angiogenic therapy increases intratumoral adenovirus distribution by inducing collagen degradation. Gene Ther. 2013, 20, 318–327. [Google Scholar] [CrossRef]
- Liu, X.Y. The Excellent Anti-Tumour Strategy (CTGVT, OV-gene) and the Excellent Anti-Tumor Gene (IL-24). Int. J. Biomed. Sci. 2012, 8, 87–93. [Google Scholar]
- Mansour, M.; Palese, P.; Zamarin, D. Oncolytic Specificity of Newcastle Disease Virus Is Mediated by Selectivity for Apoptosis-Resistant Cells. J. Virol. 2011, 85, 6015–6023. [Google Scholar] [CrossRef]
- Fiola, C.; Peeters, B.; Fournier, P.; Arnold, A.; Bucur, M.; Schirrmacher, V. Tumor selective replication of Newcastle disease virus: Association with defects of tumor cells in antiviral defence. Int. J. Cancer 2006, 119, 328–338. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Schirrmacher, V. Clinical trials of antitumor vaccination with an autologous tumor cell vaccine modified by virus infection: Improvement of patient survival based on improved antitumor immune memory. Cancer Immunol. Immunother. 2005, 54, 587–598. [Google Scholar] [CrossRef]
- Schirrmacher, V. Oncolytic Newcastle disease virus as a prospective anti-cancer therapy. A biologic agent with potential to break therapy resistance. Expert Opin. Biol. Ther. 2015, 15, 1757–1771. [Google Scholar] [CrossRef]
- Schwaiger, T.; Knittler, M.R.; Grund, C.; Roemer-Oberdoerfer, A.; Kapp, J.F.; Lerch, M.M.; Mettenleiter, T.C.; Mayerle, J.; Blohm, U. Newcastle disease virus mediates pancreatic tumor rejection via NK cell activation and prevents cancer relapse by prompting adaptive immunity. Int. J. Cancer 2017, 141, 2505–2516. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra232. [Google Scholar] [CrossRef]
- Ricca, J.M.; Oseledchyk, A.; Walther, T.; Liu, C.; Mangarin, L.; Merghoub, T.; Wolchok, J.D.; Zamarin, D. Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Beneteau, M.; Zunino, B.; Jacquin, M.A.; Meynet, O.; Chiche, J.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.E. Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc. Natl. Acad. Sci. USA 2012, 109, 20071–20076. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef]
- Li, C.Y.; Meng, G.; Su, L.; Chen, A.P.; Xia, M.; Xu, C.; Yu, D.C.; Jiang, A.Q.; Wei, J.W. Dichloroacetate blocks aerobic glycolytic adaptation to attenuated measles virus and promotes viral replication leading to enhanced oncolysis in glioblastoma. Oncotarget 2015, 6, 1544–1555. [Google Scholar] [CrossRef]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.P. Disorders of glucose metabolism in patients infected with human immunodeficiency virus. Clin. Infect. Dis. 2000, 31, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ailia, M.J.; Yoo, S.Y. In Vivo Oncolytic Virotherapy in Murine Models of Hepatocellular Carcinoma: A Systematic Review. Vaccines 2022, 10, 1541. https://doi.org/10.3390/vaccines10091541
Ailia MJ, Yoo SY. In Vivo Oncolytic Virotherapy in Murine Models of Hepatocellular Carcinoma: A Systematic Review. Vaccines. 2022; 10(9):1541. https://doi.org/10.3390/vaccines10091541
Chicago/Turabian StyleAilia, Muhammad Joan, and So Young Yoo. 2022. "In Vivo Oncolytic Virotherapy in Murine Models of Hepatocellular Carcinoma: A Systematic Review" Vaccines 10, no. 9: 1541. https://doi.org/10.3390/vaccines10091541