
Effect of Double Mutation (L452R and E484Q) on the Binding Affinity of Monoclonal Antibodies (mAbs) against the RBD—A Target for Vaccine Development
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Wild-Type and Mutated Ag-Ab Complex
2.2. Trajectory Analysis after Molecular Dynamics (MD) Simulation
2.3. MMGBSA Analysis
3. Results and Discussion
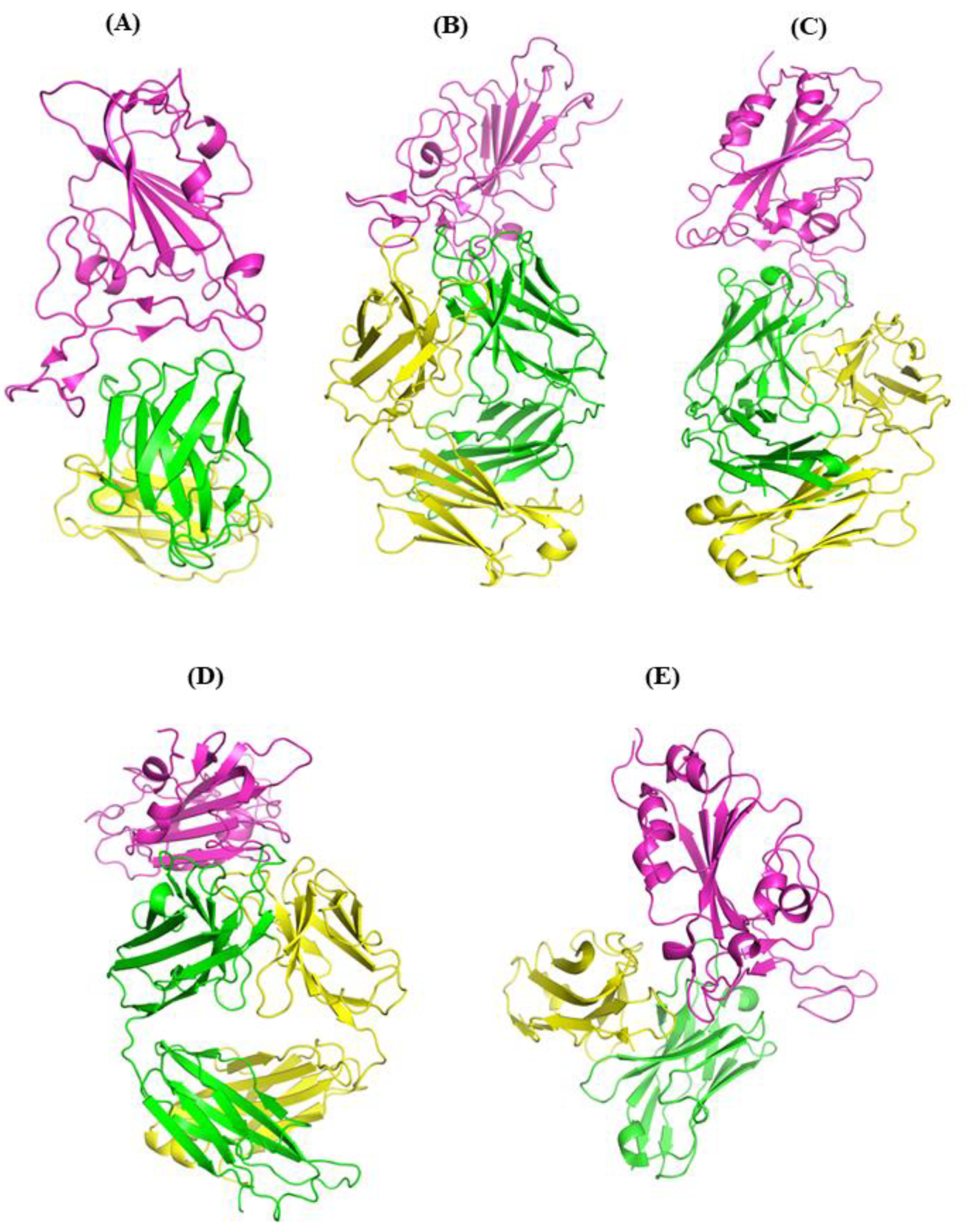
3.1. Comparisons of Biomolecular Interaction between the mAb and RBD Complexes
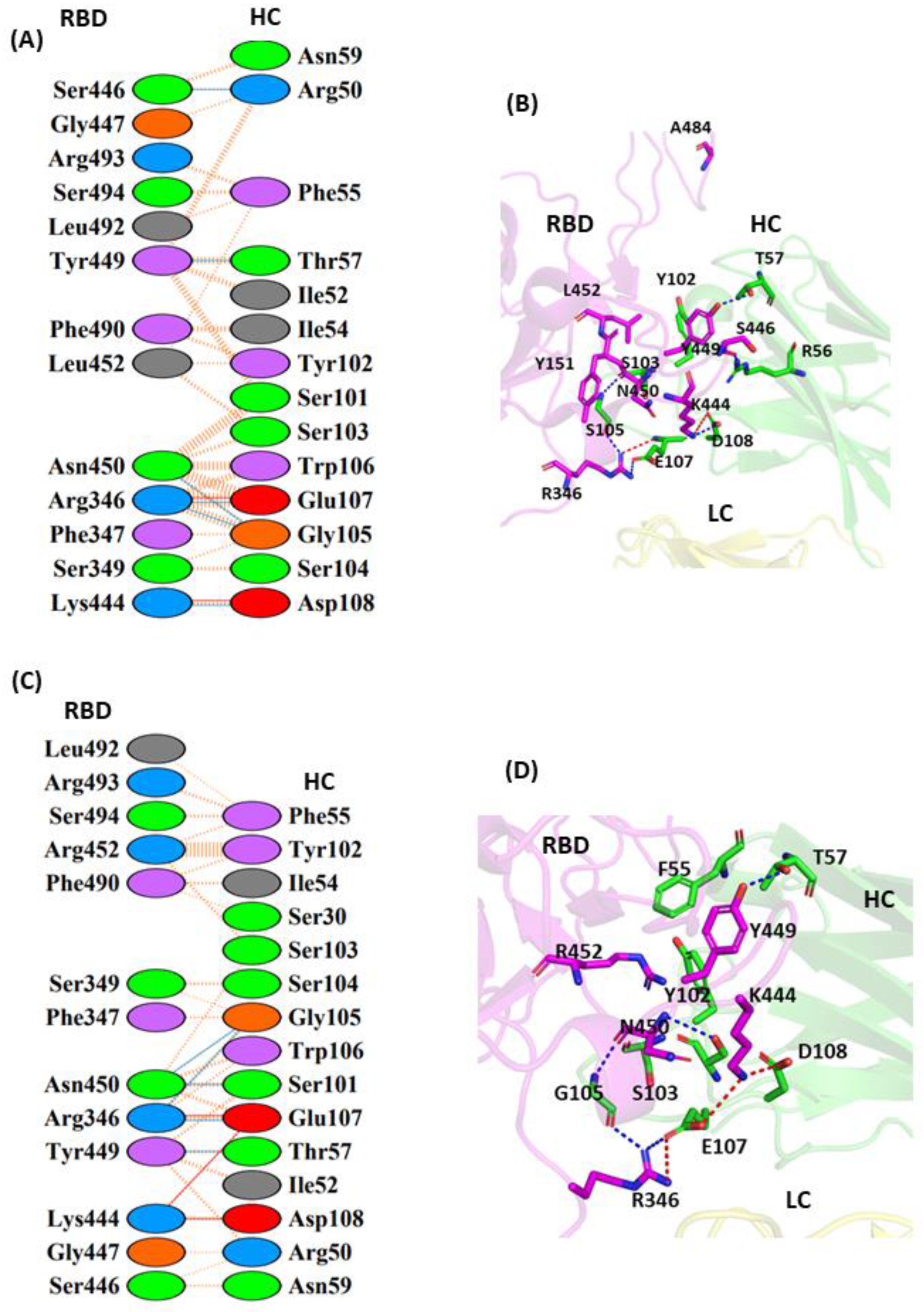
3.1.1. Interaction of REGN10933 with WT and MT-RBD
3.1.2. Interaction of BD-368-2 with WT and MT-RBD
3.1.3. Interaction of S2M11 with WT and MT-RBD
3.1.4. Interaction of EY6A with WT and MT-RBD
3.1.5. Interaction of JMB2002 with WT and MT Omicron-RBD
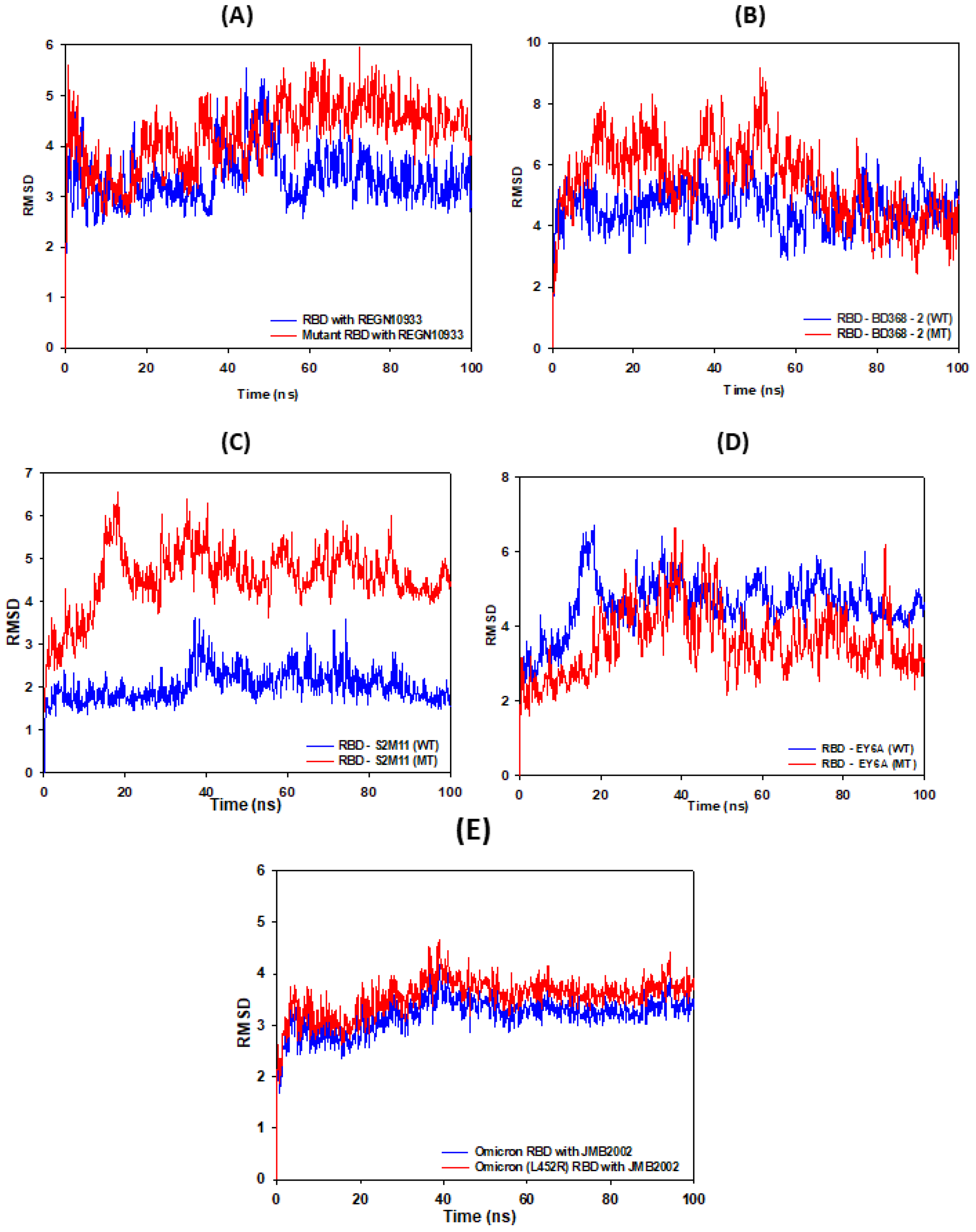
3.2. RMSD Calculations
3.3. RMSF Calculations
3.4. Gibbs Free-Energy Calculations for WT and MT-RBD Complex with mAbs
3.5. Per-Residue Energy Contribution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edara, V.V.; Hudson, W.H.; Xie, X.; Ahmed, R.; Suthar, M.S. Neutralizing Antibodies against SARS-CoV-2 Variants after Infection and Vaccination. JAMA-J. Am. Med. Assoc. 2021, 325, 1896. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhang, L. ACE2 Partially Dictates the Host Range and Tropism of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2020, 18, 4040–4047. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural Basis for SARS-CoV-2 Delta Variant Recognition of ACE2 Receptor and Broadly Neutralizing Antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19); StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and Biochemical Rationale for Enhanced Spike Protein Fitness in Delta and Kappa SARS-CoV-2 Variants. Nat. Commun. 2022, 13, 742. [Google Scholar] [CrossRef]
- Scott, L.; Hsiao, N.Y.; Moyo, S.; Singh, L.; Tegally, H.; Dor, G.; Maes, P.; Pybus, O.G.; Kraemer, M.U.G.; Semenova, E.; et al. Track Omicron’s Spread with Molecular Data. Science 2021, 374, 1454–1455. [Google Scholar] [CrossRef]
- Ingraham, N.E.; Ingbar, D.H. The Omicron Variant of SARS-CoV-2: Understanding the Known and Living with Unknowns. Clin. Transl. Med. 2021, 11, e685. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron Variant of SARS-CoV-2: Genomics, Transmissibility, and Responses to Current COVID-19 Vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- GISAID-Initiative. Available online: https://www.gisaid.org/ (accessed on 2 July 2021).
- Zhang, Y.; Zhang, T.; Fang, Y.; Liu, J.; Ye, Q.; Ding, L. SARS-CoV-2 Spike L452R Mutation Increases Omicron Variant Fusogenicity and Infectivity as Well as Host Glycolysis. Signal Transduct. Target. Ther. 2022, 7, 76. [Google Scholar] [CrossRef]
- Zhang, W.; Davis, B.D.; Chen, S.S.; Sincuir Martinez, J.M.; Plummer, J.T.; Vail, E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA-J. Am. Med. Assoc. 2021, 325, 1324. [Google Scholar] [CrossRef]
- Tang, J.W.; Toovey, O.T.R.; Harvey, K.N.; Hui, D.D.S. Introduction of the South African SARS-CoV-2 Variant 501Y.V2 into the UK. J. Infect. 2021, 82, e8–e10. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Baral, P.; Gerstman, B.S.; Chapagain, P.P. Structural and Dynamical Differences in the Spike Protein RBD in the SARS-CoV-2 Variants B.1.1.7 and B.1.351. J. Phys. Chem. B 2021, 125, 7101–7107. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 Spike L452R Variant Evades Cellular Immunity and Increases Infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Sun, Y.H.; Zhou, J.M.; Ye, Q. The Global Epidemic of SARS-CoV-2 Variants and Their Mutational Immune Escape. J. Med. Virol. 2022, 94, 847–857. [Google Scholar] [CrossRef]
- Rodriguez, J.H. Attractive and Repulsive Residue Fragments at the Interface of SARS-CoV-2 and HACE2. Sci. Rep. 2021, 11, 12567. [Google Scholar] [CrossRef]
- Ku, Z.; Ye, X.; To’a Salazar, G.; Zhang, N.; An, Z. Antibody Therapies for the Treatment of COVID-19. Antib. Ther. 2020, 3, 101–108. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Kumar, S.; Chandele, A.; Sharma, A. Current Status of Therapeutic Monoclonal Antibodies against SARS-CoV-2. PLoS Pathog. 2021, 17, e1009885. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike Trimer with ACE2 and an Anti-Omicron Antibody: Mechanisms for the High Infectivity, Immune Evasion and Antibody Drug Discovery. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gu, C.; Cao, X.; Wang, Z.; Hu, X.; Yao, Y.; Zhou, Y.; Liu, P.; Liu, X.; Gao, G.; Hu, X.; et al. A Human Antibody of Potent Efficacy against SARS-CoV-2 in Rhesus Macaques Showed Strong Blocking Activity to B.1.351. MAbs 2021, 13, 1930636. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Tedla, N.; Bull, R.A. Broadly-Neutralizing Antibodies Against Emerging SARS-CoV-2 Variants. Front. Immunol. 2021, 12, 752003. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Baig, M.H.; Mondal, T.; Alorabi, M.; Sharma, T.; Dong, J.J.; Cho, J.Y. Impact of the Double Mutants on Spike Protein of SARS-CoV-2 B.1.617 Lineage to the Human ACE2 Receptor Binding: A Structural Insight. Viruses 2021, 13, 2295. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in Humanized Mice and Convalescent Humans Yield a SARS-CoV-2 Antibody Cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell 2020, 183, 1013–1023.e13. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent Human Antibodies Protect against SARS-CoV-2 Challenge via Multiple Mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Q.; Ge, J.; Ren, W.; Zhang, R.; Lan, J.; Ju, B.; Su, B.; Yu, F.; Chen, P.; et al. SARS-CoV-2 Variants Resist Antibody Neutralization and Broaden Host ACE2 Usage. bioRxiv 2021. [Google Scholar] [CrossRef]
- Protein Preparation Wizard|Schrödinger. Available online: https://www.schrodinger.com/products/protein-preparation-wizard (accessed on 14 December 2021).
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very Fast Empirical Prediction and Rationalization of Protein PK a Values. Proteins Struct. Funct. Genet. 2005, 61, 704–772. [Google Scholar] [CrossRef]
- Shaw, D. Desmond Molecular Dynamics System; V 3.0. Schrödinger: New York, NY, USA, 2011. [Google Scholar]
- Price, D.J.; Brooks, C.L. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural Summaries of PDB Entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins Struct. Funct. Bioinforma. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Roy, A.; Rawat, R.S.; Alok, A.; Tetala, K.K.R.; Biswas, N.R.; Kaur, P.; Kumar, S. Identification and Structural Studies of Natural Inhibitors against SARS-CoV-2 Viral RNA Methyltransferase (NSP16). J. Biomol. Struct. Dyn. 2021, 40, 13965–13975. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A. ChemInform Abstract: Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. ChemInform 2010, 33, 889–897. [Google Scholar] [CrossRef]
- Deshpande, A.; Harris, B.D.; Martinez-Sobrido, L.; Kobie, J.J.; Walter, M.R. Epitope Classification and RBD Binding Properties of Neutralizing Antibodies Against SARS-CoV-2 Variants of Concern. Front. Immunol. 2021, 12, 691715. [Google Scholar] [CrossRef]
- Cao, Y.; Su, B.; Guo, X.; Sun, W.; Deng, Y.; Bao, L.; Zhu, Q.; Zhang, X.; Zheng, Y.; Geng, C.; et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84.e16. [Google Scholar] [CrossRef] [PubMed]
- Muecksch, F.; Weisblum, Y.; Barnes, C.O.; Schmidt, F.; Schaefer-Babajew, D.; Wang, Z.; Julio, J.C.; Flyak, A.I.; DeLaitsch, A.T.; Huey-Tubman, K.E.; et al. Affinity Maturation of SARS-CoV-2 Neutralizing Antibodies Confers Potency, Breadth, and Resilience to Viral Escape Mutations. Immunity 2021, 54, 1853–1868.e7. [Google Scholar] [CrossRef] [PubMed]
- Kombe Kombe, A.J.; Zahid, A.; Mohammed, A.; Shi, R.; Jin, T. Potent Molecular Feature-Based Neutralizing Monoclonal Antibodies as Promising Therapeutics Against SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 670815. [Google Scholar] [CrossRef]
- Zhou, D.; Duyvesteyn, H.M.E.; Chen, C.P.; Huang, C.G.; Chen, T.H.; Shih, S.R.; Lin, Y.C.; Cheng, C.Y.; Cheng, S.H.; Huang, Y.C.; et al. Structural Basis for the Neutralization of SARS-CoV-2 by an Antibody from a Convalescent Patient. Nat. Struct. Mol. Biol. 2020, 27, 950–958. [Google Scholar] [CrossRef]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front. Immunol. 2020, 11, 576622. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Mohsin, I. The SARS-CoV-2 Spike Glycoprotein as a Drug and Vaccine Target: Structural Insights into Its Complexes with ACE2 and Antibodies. Cells 2020, 9, 2343. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
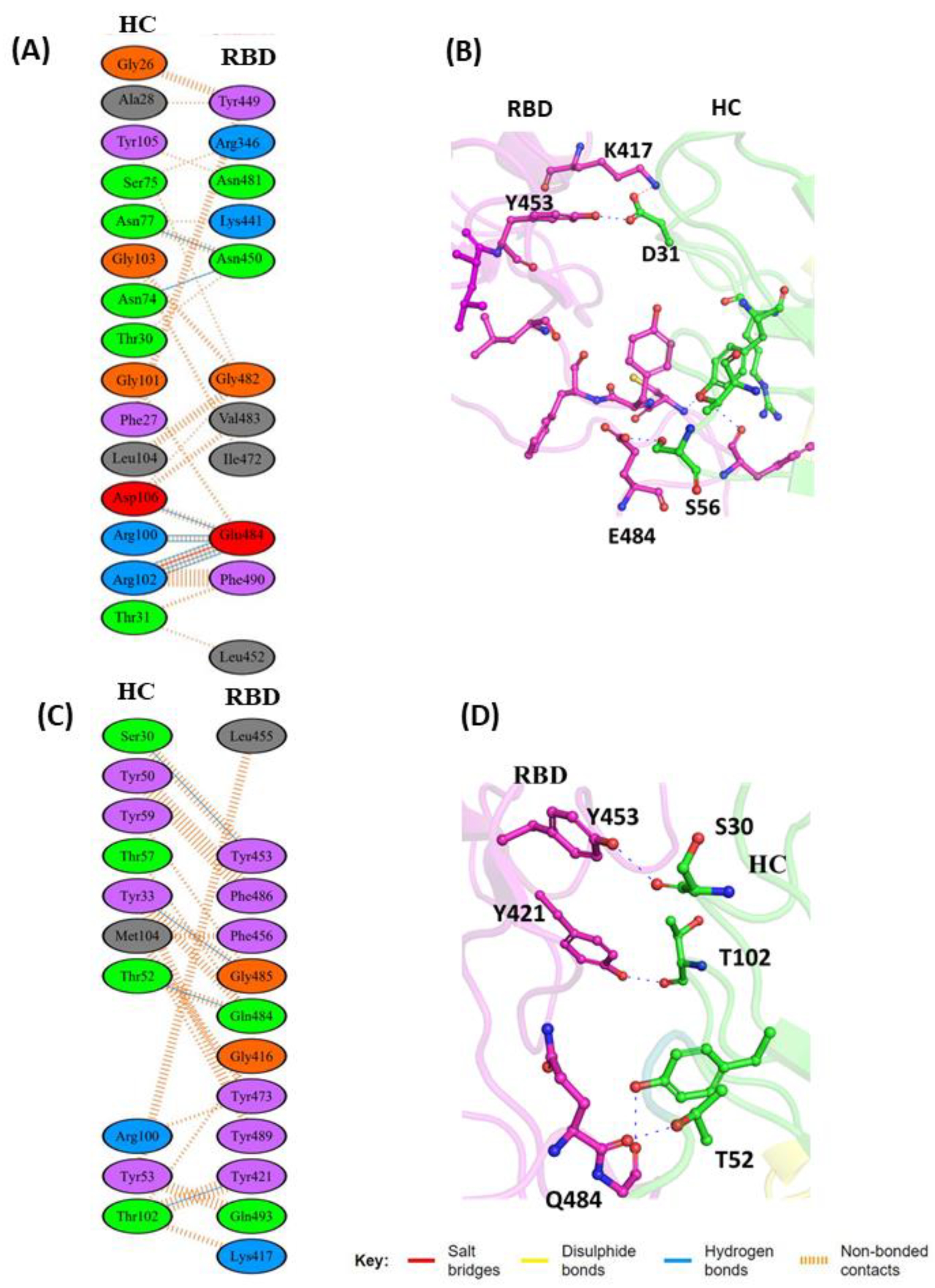

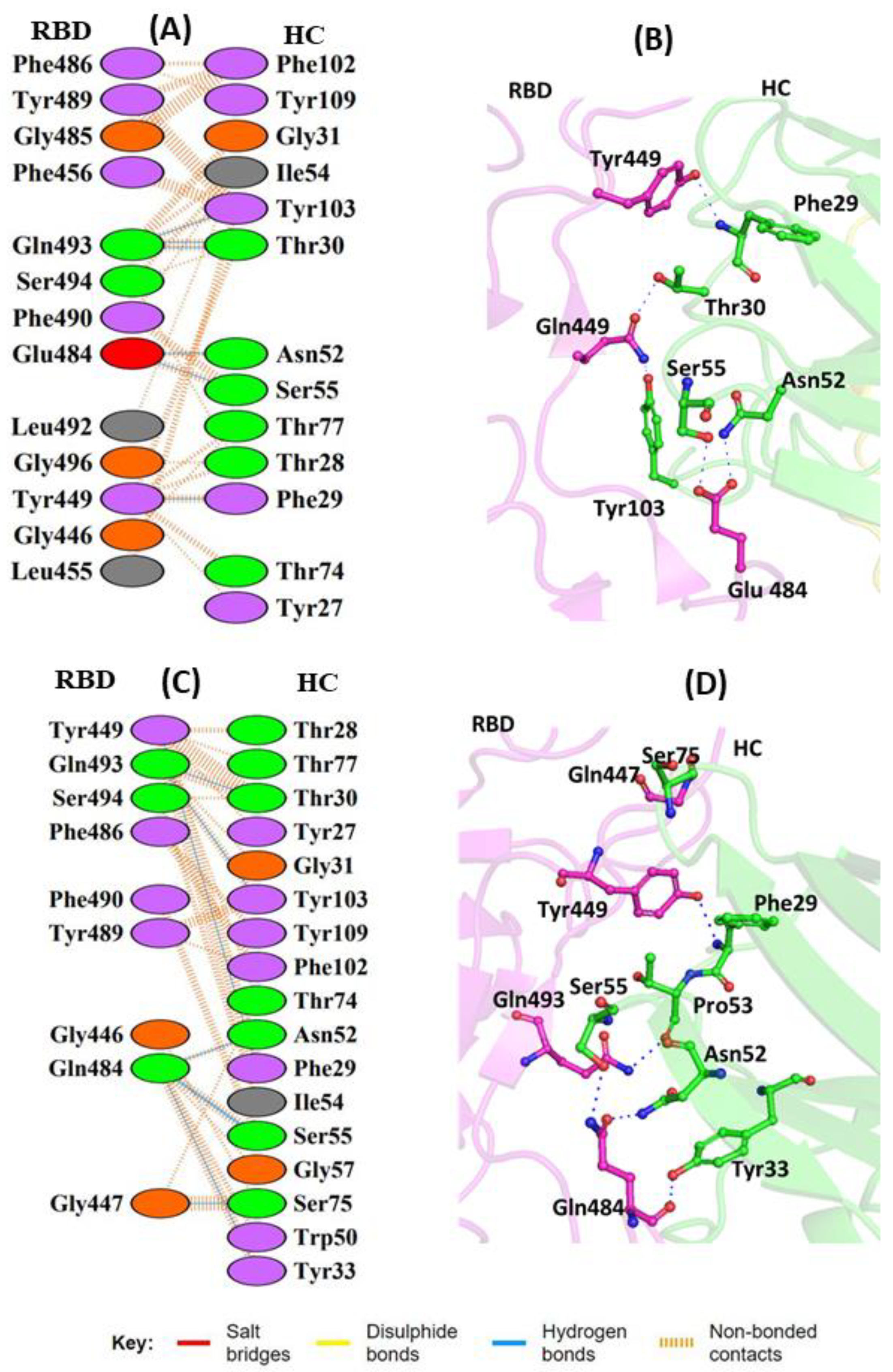


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RBD Complex with mAb | PDB ID | Type of Class |
|---|---|---|
| RBD–REGN10933 | 6XDG | Class I |
| RBD–BD368-2 | 7CHF | Class II |
| RBD–S2M11 | 7K43 | Class III |
| RBD–EY6A | 6ZCZ | Class IV |
| RBD–JMB2002 | 7WRV | Class V |
| RMSF Values of Residues of RBD | RBD REGN10933 | RBD-BD-368-2 | RBD EY6A | RBD S2M11 | RBD JMB2002 |
|---|---|---|---|---|---|
| L452 | 0.997 | 1.05 | 2.24 | 0.842 | 1.2 |
| L452R | 1.29 | 2.58 | 2.84 | 0.943 | 1.8 |
| E484 | 2.0 | 0.981 | 6.421 | 1.85 | - |
| E484Q | 2.5 | 2.14 | 6.9 | 2.26 | - |
| RBD Complex with Antibody | MMGBSA_ΔGBind | Coulomb | Covalent | Bind H Bond | Bind_Lipo | Bind_vdW | Solv_GB |
|---|---|---|---|---|---|---|---|
| RBD–REGN 10933 (WT) | −87.58 ± 3.2 | −40.66 ± 2.5 | −7.0 ± 0.05 | −8.31 ± 0.65 | −26.41 ± 0.16 | −79.2 ± 8.2 | 74.24 ± 9.4 |
| RBD–REGN 10933 (MT) | −76.29 ± 1.7 | −29.28 ± 1.2 | −13.31 ± 0.12 | −9.21 ± 2.25 | −28.18 ± 0.28 | −108.34 ± 10.1 | 94.00 ± 7.0 |
| RBD-BD- 368-2 (WT) | −65.43 ± 1.2 | −89.94 ± 3.4 | −2.8 ± 0.12 | −5.3 ± 0.05 | −21.84 ± 0.21 | −98.01 ± 6.7 | 156.8 ± 16 |
| RBD–BD-368-2 (MT) | −17.96 ± 1.2 | −35.20 ± 2.8 | −0.88 ± 0.03 | −1.45 ± 0.02 | −3.155 ± 0.03 | −10.52 ± 0.24 | 41.04 ± 4.1 |
| RBD–S2M11 (WT) | −78.68 ± 1.5 | −39.58 ± 5.2 | −6.51 ± 0.65 | −1.85 ± 0.05 | −33.27 ± 0.04 | −107.95 ± 11.2 | 109.6 ± 14 |
| RBD–S2M11 (MT) | −60.47 ± 0.09 | −49.03 ± 16.0 | 2.27 ± 0.39 | −1.80 ± 0.09 | −35.21 ± 0.09 | −90.3 ± 3.3 | 92.71 ± 17.2 |
| RBD–EY6A (WT) | −88.23 ± 0.87 | −106.05 ± 9.0 | −5.85 ± 0.34 | −4.43 ± 0.5 | −12.40 ± 0.012 | −59.8 ± 4.23 | 105.61 ± 11.6 |
| RBD–EY6A (MT) | −96.53 ± 6.2 | −74.42 ± 3.5 | −17.95 ± 0.17 | −5.96 ± 0.23 | −18.63 ± 0.8 | −78.41 ± 6.7 | 99.84 ± 9.8 |
| Omicron-RBD–JMB2002 | −82.68 ± 4.2 | −96.53 ± 6.2 | −5.53 ± 0.2 | −4.43 ± 6.2 | −12.53 ± 1.2 | −78.53 ± 6.2 | 107.53 ± 6.2 |
| Omicron (L452R)-RBD–JMB2002 | −76.28 ± 3.2 | −106.53 ± 6.2 | −6.53 ± 0.2 | −4.43 ± 6.2 | −10.53 ± 1.2 | −68.53 ± 6.2 | 112.53 ± 6.2 |
| Residue | dG Bind | Coulomb | Solvation | vdW | H Bond | Lipo | |
|---|---|---|---|---|---|---|---|
| RBD- REGN-10933 | L452 | −0.03 | 0.06 | −0.1 | −0.04 | 0 | 0 |
| L452R | −1.36 | −10.11 | 9.08 | −0.13 | 0.03 | 0 | |
| E484 | 0.51 | 7.68 | −7.49 | −0.03 | −0.42 | 0 | |
| E484Q | −4.93 | −4.95 | 4.33 | −3.68 | −0.41 | −1.33 | |
| RBD-BD-368-2 | L452 | −1.17 | 0.87 | −0.77 | −1.54 | 0 | −0.69 |
| L452R | −0.9 | −5.89 | 5.46 | −0.52 | −0.09 | −0.08 | |
| E484 | −5.34 | −10.81 | 9.55 | −3.73 | −1.75 | −0.76 | |
| E484Q | −0.86 | −1.77 | 1.92 | −1.1 | −0.24 | −0.09 | |
| RBD-S2M11 | L452 | −2.57 | −16.99 | 15.02 | −1.68 | −0.83 | −0.24 |
| L452R | −0.86 | −10.77 | 10.27 | −0.4 | −0.08 | −0.04 | |
| E484 | −2.83 | −2.92 | 3.88 | −3.57 | −0.21 | −0.56 | |
| E484Q | −2.63 | −2.77 | −1.1 | −2.17 | −0.33 | −0.15 | |
| RBD-EY6A | L452 | −0.01 | −0.14 | 0.23 | −0.01 | 0 | 0 |
| L452R | 0.04 | −6.98 | 6.62 | −0.02 | −0.03 | 0 | |
| E484 | −0.08 | 3.15 | −3.35 | 0.01 | 0 | 0 | |
| E484Q | 0.08 | −0.05 | 0.06 | −0.02 | 0 | 0 | |
| Omicron L452 RBD–L452R JMB2002 | −2.19 | −0.21 | −0.1 | −1.94 | 0 | 0 | |
| −0.76 | −0.14 | 1.0 | −1.54 | 0.01 | 0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, D.; Kumar, M.; Sharma, P.; Mohan, T.; Prakash, A.; Kumari, R.; Kaur, P. Effect of Double Mutation (L452R and E484Q) on the Binding Affinity of Monoclonal Antibodies (mAbs) against the RBD—A Target for Vaccine Development. Vaccines 2023, 11, 23. https://doi.org/10.3390/vaccines11010023
Gupta D, Kumar M, Sharma P, Mohan T, Prakash A, Kumari R, Kaur P. Effect of Double Mutation (L452R and E484Q) on the Binding Affinity of Monoclonal Antibodies (mAbs) against the RBD—A Target for Vaccine Development. Vaccines. 2023; 11(1):23. https://doi.org/10.3390/vaccines11010023
Chicago/Turabian StyleGupta, Deepali, Mukesh Kumar, Priyanka Sharma, Trishala Mohan, Amresh Prakash, Renu Kumari, and Punit Kaur. 2023. "Effect of Double Mutation (L452R and E484Q) on the Binding Affinity of Monoclonal Antibodies (mAbs) against the RBD—A Target for Vaccine Development" Vaccines 11, no. 1: 23. https://doi.org/10.3390/vaccines11010023