
Re-Evaluating Human Cytomegalovirus Vaccine Design: Prediction of T Cell Epitopes
Abstract
:1. Introduction
2. Methods
2.1. Overview
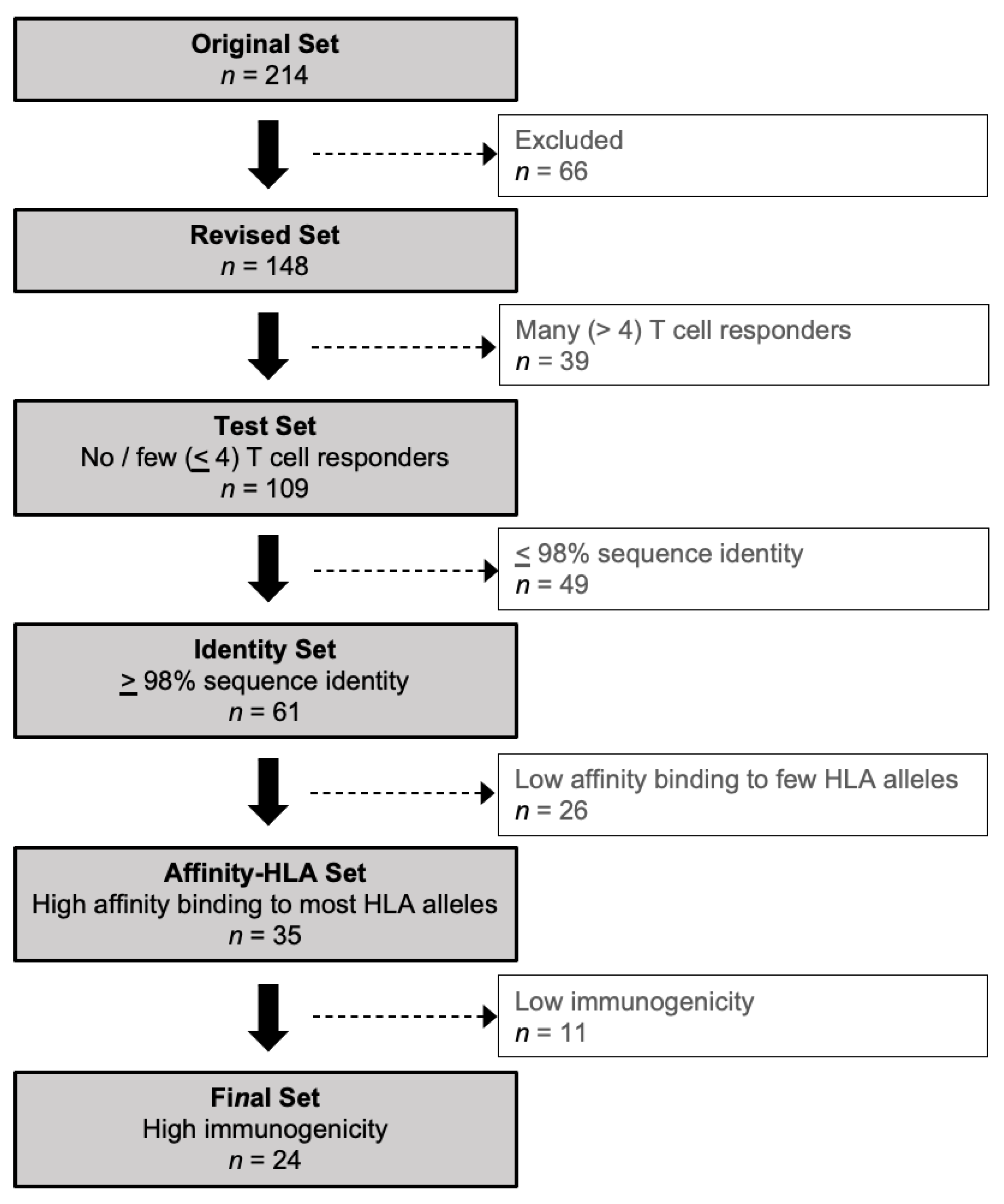
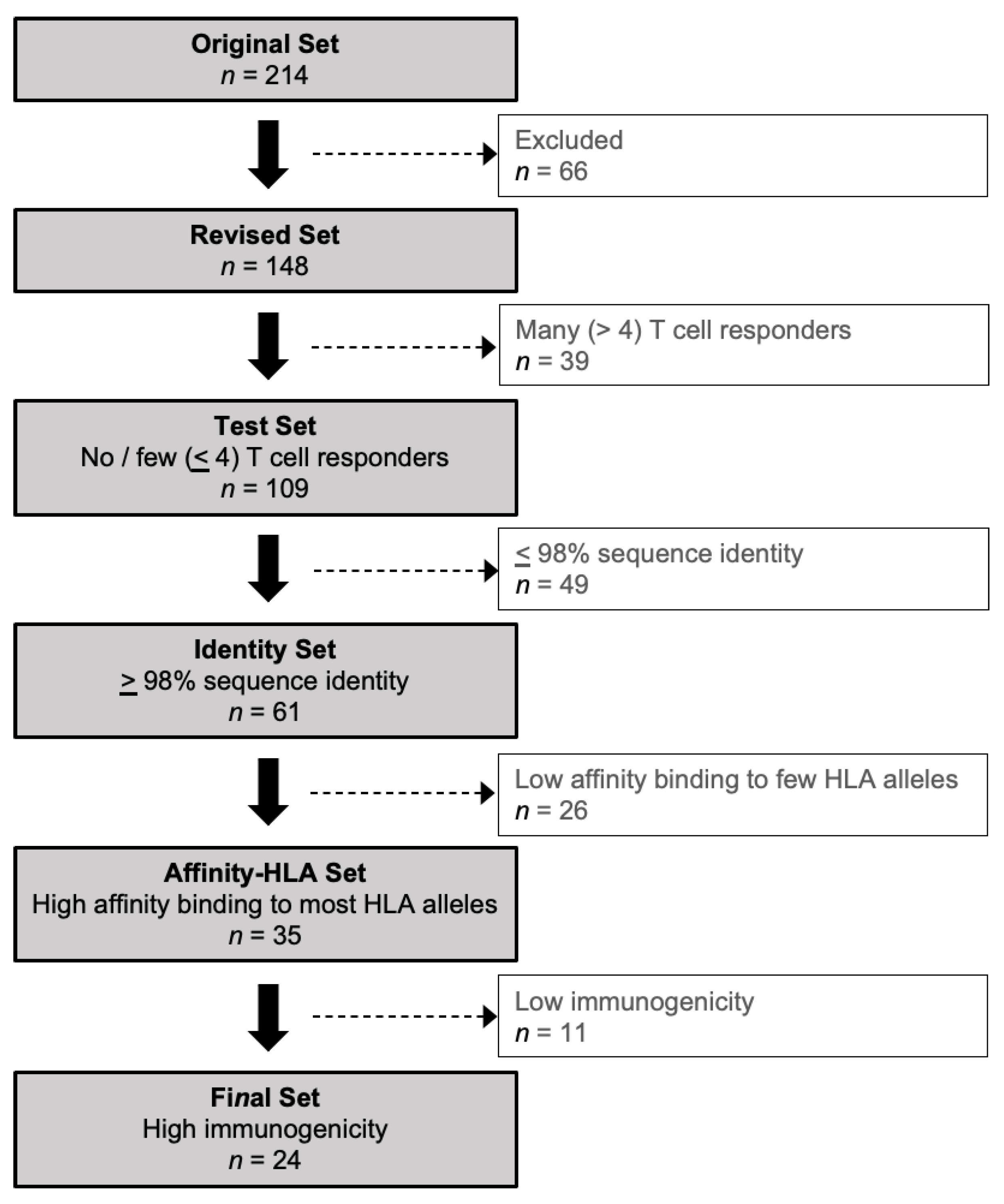
2.2. Creating a Revised Set of HCMV Proteins
2.3. T Cell Responses to the Revised Set of HCMV Proteins
2.4. Protein Sequence Identity
2.5. MHC Class I-Peptide Binding Affinity
2.6. Immunogenicity Prediction
3. Results
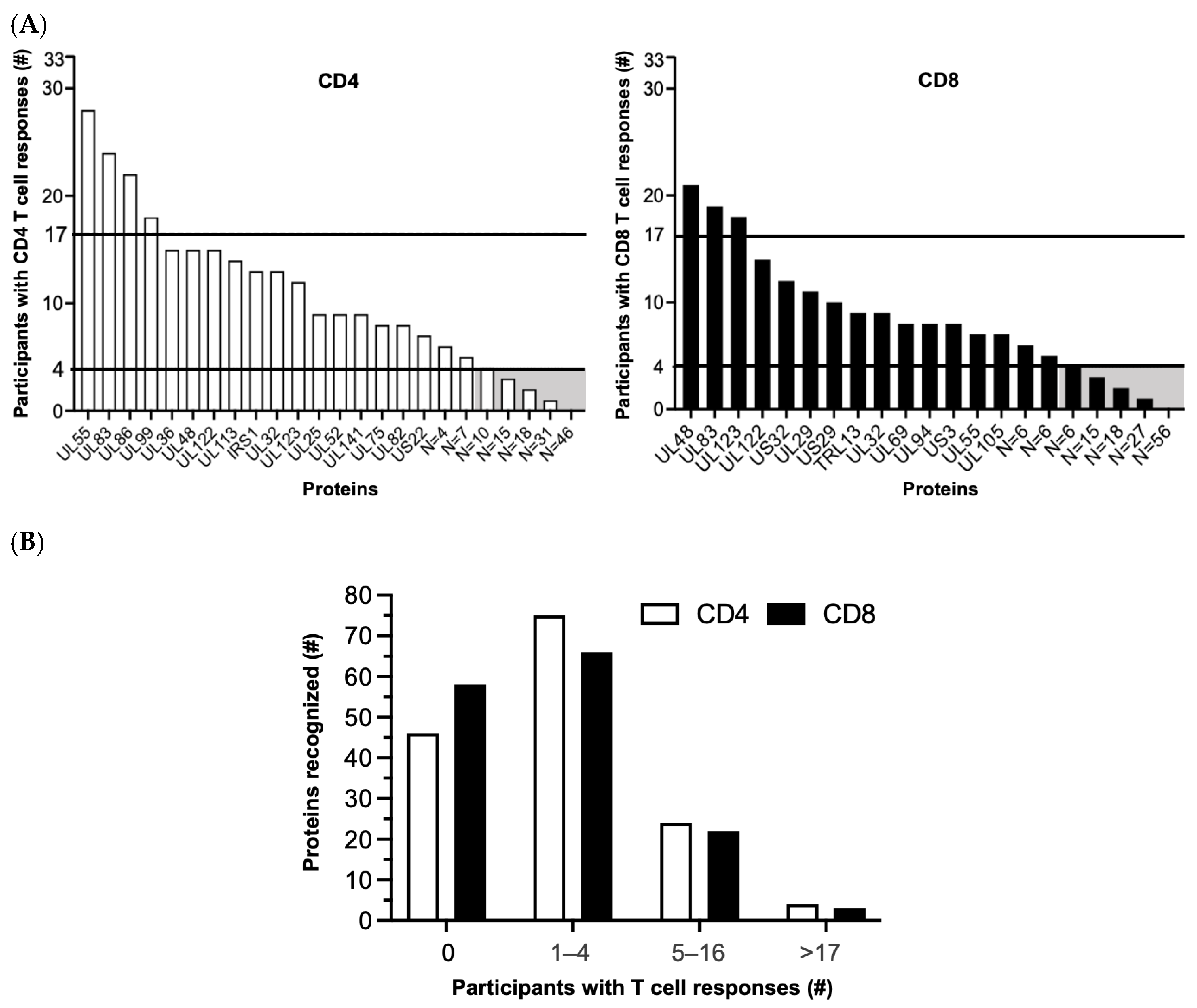
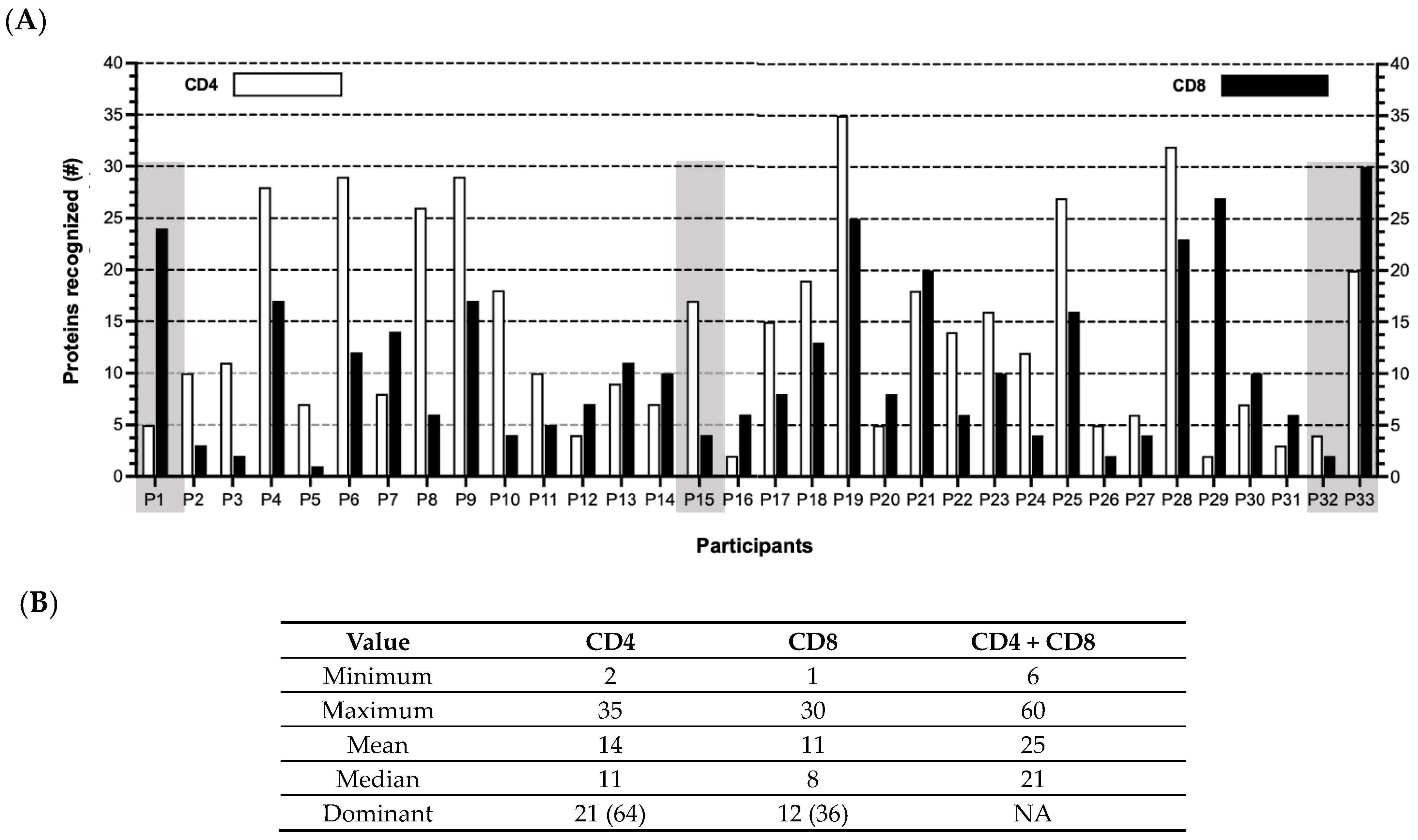
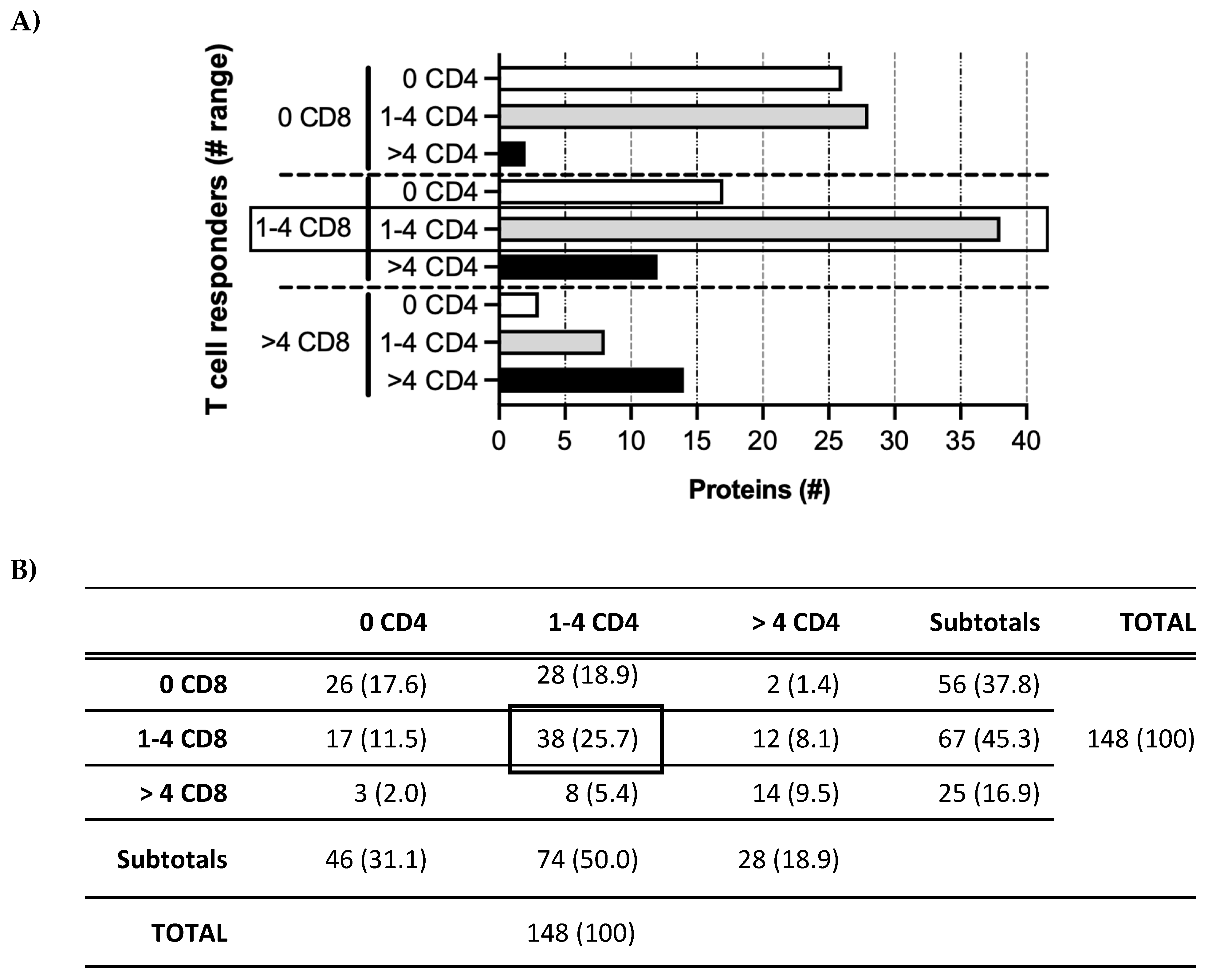
3.1. Patterns of T Cell Responses to HCMV Proteins
3.2. Amino Acid Sequence Identity in HCMV Proteins with No or Few T Cell Responders
3.3. Predicted Epitopes in Conserved HCMV Proteins with No or Few T Cell Responders
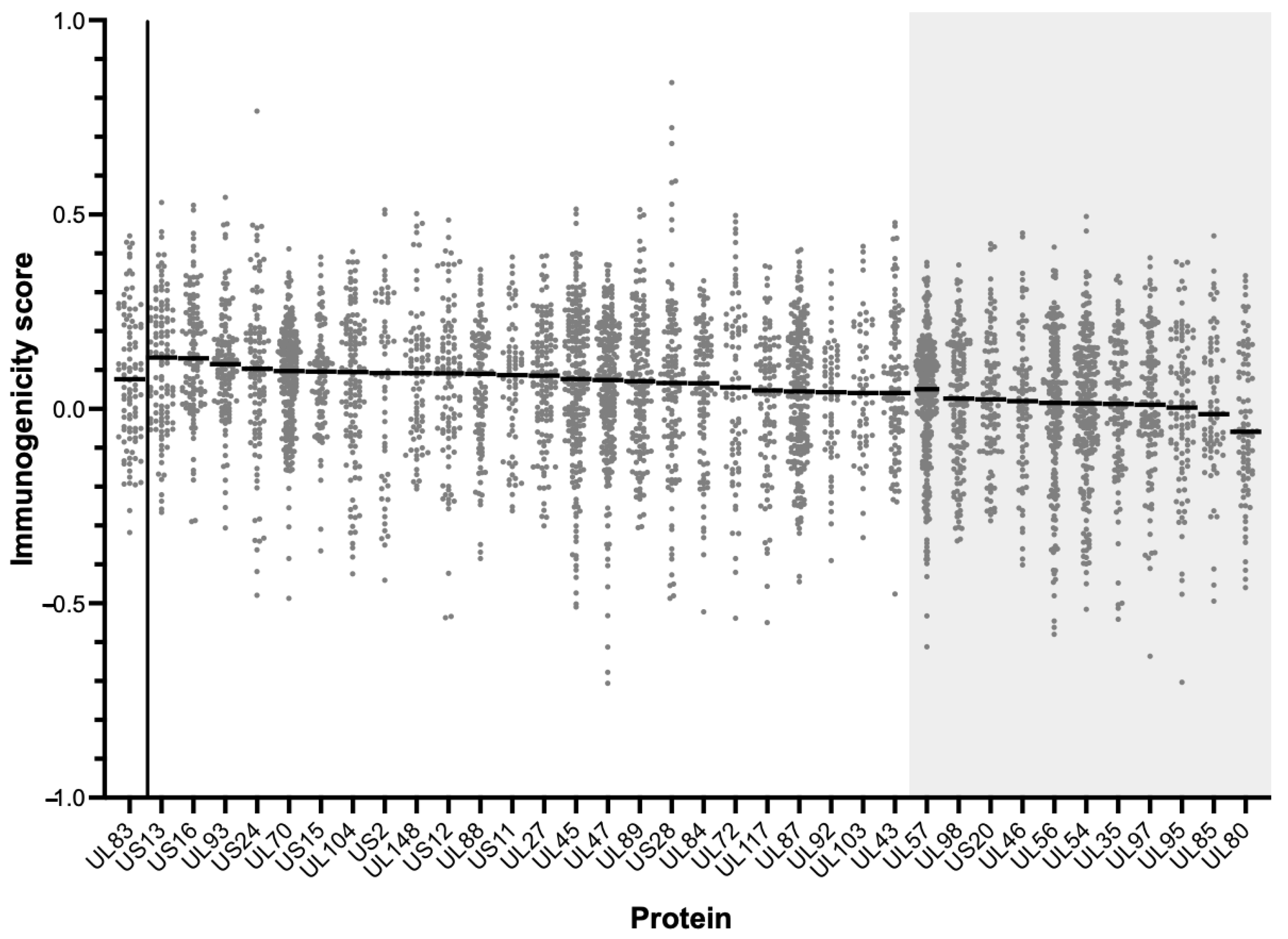
3.4. Predicted Immunogenicity of Peptides in the Affinity-HLA Set
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanshaw, J.B. Congenital cytomegalovirus infection: A fifteen year perspective. J. Infect. Dis. 1971, 123, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- Boppana, S.B.; Britt, W.J. Recent Approaches and Strategies in the Generation of Anti-human Cytomegalovirus Vaccines. Methods Mol. Biol. 2021, 2244, 403–463. [Google Scholar] [CrossRef]
- Mussi-Pinhata, M.M.; Yamamoto, A.Y. Natural history of congenital cytomegalovirus infection in highly seropositive populations. J. Infect. Dis. 2020, 221 (Suppl. S1), S15–S22. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://ourworldindata.org/grapher/births-and-deaths-projected-to-2100. (accessed on 2 May 2023).
- Stebbins, R.C.; Noppert, G.A.; Aiello, A.E.; Cordoba, E.; Ward, J.B.; Feinstein, L. Persistent socioeconomic and racial and ethnic dis-parities in pathogen burden in the United States, 1999–2014. Epidemiol. Infect. 2019, 147, e301. [Google Scholar] [CrossRef]
- Lantos, P.M.; Hoffman, K.; Permar, S.R.; Jackson, P.; Hughes, B.L.; Kind, A.; Swamy, G. Neighborhood Disadvantage is Associated with High Cytomegalovirus Seroprevalence in Pregnancy. J. Racial Ethn. Health Disparities 2018, 5, 782–786. [Google Scholar] [CrossRef]
- Lantos, P.M.; Hoffman, K.; Permar, S.R.; Jackson, P.; Hughes, B.L.; Swamy, G.K. Geographic Disparities in Cytomegalovirus Infection During Pregnancy. J. Pediatr. Infect. Dis. Soc. 2017, 6, e55–e61. [Google Scholar] [CrossRef]
- Lantos, P.M.; Maradiaga-Panayotti, G.; Barber, X.; Raynor, E.; Tucci, D.; Hoffman, K.; Permar, S.R.; Jackson, P.; Hughes, B.L.; Kind, A.; et al. Geographic and Racial Disparities in Infant Hearing Loss. Otolaryngol.-Head Neck Surg. 2018, 159, 1051–1057. [Google Scholar] [CrossRef]
- Lantos, P.M.; Permar, S.R.; Hoffman, K.; Swamy, G.K. The Excess Burden of Cytomegalovirus in African American Communities: A Geospatial Analysis. Open Forum. Infect. Dis. 2015, 2, ofv180. [Google Scholar] [CrossRef]
- Dowd, J.B.; Aiello, A.E.; Alley, D.E. Socioeconomic disparities in the seroprevalence of cytomegalovirus infection in the US popu-lation: NHANES III. Epidemiol. Infect. 2009, 137, 58–65. [Google Scholar] [CrossRef]
- Dowd, J.B.; Zajacova, A.; Aiello, A. Early origins of health disparities: Burden of infection, health, and socioeconomic status in U.S. children. Soc. Sci. Med. 2009, 68, 699–707. [Google Scholar] [CrossRef]
- Bresson, J.; Clavequin, M.; Mazeron, M.; Mengelle, C.; Scieux, C.; Segondy, M.; Houhou, N. Risk of cytomegalovirus transmission by cryopreserved semen: A study of 635 semen samples from 231 donors. Hum. Reprod. 2003, 18, 1881–1886. [Google Scholar] [CrossRef]
- Diafouka, F.; Foulongne, V.; Hauhouot-Attoungbre, M.-L.; Monnet, D.; Segondy, M. Cytomegalovirus DNA in semen of men seeking fertility evaluation in Abidjan, Côte d’Ivoire. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Moretti, E.; Figura, N.; Campagna, M.S.; Iacoponi, F.; Gonnelli, S.; Collodel, G. Infectious Burden and Semen Parameters. Urology 2017, 100, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, N.G.; Yamamoto, A.Y.; Duarte, G.; Aragon, D.C.; Fowler, K.B.; Boppana, S.; Britt, W.J.; Mussi-Pinhata, M.M. Cytomegalovirus (CMV) Shedding in Se-ropositive Pregnant Women from a High Seroprevalence Population: “The Brazilian Cytomegalovirus Hearing and Maternal Secondary Infection Study” (BraCHS). Clin. Infect. Dis. 2018, 67, 743–750. [Google Scholar] [CrossRef]
- Cannon, M.J.; Stowell, J.D.; Clark, R.; Dollard, P.R.; Johnson, D.; Mask, K.; Stover, C.; Wu, K.; Amin, M.; Hendley, W.; et al. Repeated measures study of weekly and daily cyto-megalovirus shedding patterns in saliva and urine of healthy cytomegalovirus-seropositive children. BMC Infect. Dis. 2014, 14, 569. [Google Scholar] [CrossRef]
- Hamprecht, K.; Maschmann, J.; Jahn, G.; Poets, C.F.; Goelz, R. Cytomegalovirus transmission to preterm infants during lactation. J. Clin. Virol. 2008, 41, 198–205. [Google Scholar] [CrossRef]
- Hotsubo, T.; Nagata, N.; Shimada, M.; Yoshida, K.; Fujinaga, K.; Chiba, S. Detection of Human Cytomegalovirus DNA in Breast Milk by Means of Polymerase Chain Reaction. Microbiol. Immunol. 1994, 38, 809–811. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, X.; Song, Q.; Wang, H.; Yu, H.; Zhang, Y.; Qiao, E.; Xue, W.; Li, X.; Zhuang, S.; et al. Cytomegalovirus Shedding in Healthy Seropositive Female College Students: A 6-Month Longitudinal Study. J. Infect. Dis. 2017, 217, 1069–1073. [Google Scholar] [CrossRef]
- Meier, J.; Lienicke, U.; Tschirch, E.; Krüger, D.H.; Wauer, R.R.; Prösch, S. Human Cytomegalovirus Reactivation during Lactation and Mother-to-Child Transmission in Preterm Infants. J. Clin. Microbiol. 2005, 43, 1318–1324. [Google Scholar] [CrossRef]
- Stowell, J.D.; Mask, K.; Amin, M.; Clark, R.; Levis, D.; Hendley, W.; Lanzieri, T.M.; Dollard, S.C.; Cannon, M.J. Cross-sectional study of cytomegalovirus shedding and immunological markers among seropositive children and their mothers. BMC Infect. Dis. 2014, 14, 568. [Google Scholar] [CrossRef] [PubMed]
- White, J.L.; Patel, E.U.; Abraham, A.G.; Grabowski, M.K.; Arav-Boger, R.; Avery, R.K.; Quinn, T.C.; Tobian, A.A.R. Prevalence, Magnitude, and Genotype Distribution of Urinary Cytomegalovirus (CMV) Shedding Among CMV-Seropositive Children and Adolescents in the United States. Open Forum Infect. Dis. 2019, 6, ofz272. [Google Scholar] [CrossRef]
- Zanghellini, F.; Boppana, S.B.; Emery, V.C.; Griffiths, P.D.; Pass, R.F. Asymptomatic Primary Cytomegalovirus Infection: Virologic and Immunologic Features. J. Infect. Dis. 1999, 180, 702–707. [Google Scholar] [CrossRef]
- Amin, M.M.; Bialek, S.R.; Dollard, S.C.; Wang, C. Urinary Cytomegalovirus Shedding in the United States: The National Health and Nutrition Examination Surveys, 1999–2004. Clin. Infect. Dis. 2018, 67, 587–592. [Google Scholar] [CrossRef]
- Britt, W.J. Human Cytomegalovirus Infection in Women with Preexisting Immunity: Sources of Infection and Mechanisms of Infection in the Presence of Antiviral Immunity. J. Infect. Dis. 2020, 221 (Suppl. 1), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [PubMed]
- A Plotkin, S. Preventing Infection by Human Cytomegalovirus. J. Infect. Dis. 2020, 221 (Suppl. 1), S123–S127. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2019, 37, 7437–7442. [Google Scholar] [CrossRef]
- Hehnly, C.; Ssentongo, P.; Bebell, L.M.; Burgoine, K.; Bazira, J.; Fronterre, C.; Kumbakumba, E.; Mulondo, R.; Mbabazi-Kabachelor, E.; Morton, S.U.; et al. Cytomegalovirus infections in infants in Uganda: Newborn-mother pairs, neonates with sepsis, and infants with hydrocephalus. Int. J. Infect. Dis. 2022, 118, 24–33. [Google Scholar] [CrossRef]
- Cannon, M.J. Congenital cytomegalovirus (CMV) epidemiology and awareness. J. Clin. Virol. 2009, 46 (Suppl. 4), S6–S10. [Google Scholar] [CrossRef]
- Ssentongo, P.; Hehnly, C.; Birungi, P.; Roach, M.A.; Spady, J.; Fronterre, C.; Wang, M.; Murray-Kolb, L.E.; Al-Shaar, L.; Chinchilli, V.M.; et al. Congenital Cytomegalovirus Infection Burden and Epidemiologic Risk Factors in Countries With Universal Screening: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e2120736. [Google Scholar] [CrossRef]
- Hyde, T.B.; Schmid, D.S.; Cannon, M.J. Cytomegalovirus seroconversion rates and risk factors: Implications for congenital CMV. Rev. Med. Virol. 2010, 20, 311–326. [Google Scholar] [CrossRef]
- Ross, S.A.; Arora, N.; Novak, Z.; Fowler, K.B.; Britt, W.J.; Boppana, S.B. Cytomegalovirus Reinfections in Healthy Seroimmune Women. J. Infect. Dis. 2010, 201, 386–389. [Google Scholar] [CrossRef]
- Coppola, T.; Mangold, J.F.; Cantrell, S.; Permar, S.R. Impact of Maternal Immunity on Congenital Cytomegalovirus Birth Prevalence and Infant Outcomes: A Systematic Review. Vaccines 2019, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Controversies in the natural history of congenital human cytomegalovirus infection: The paradox of infection and disease in offspring of women with immunity prior to pregnancy. Med. Microbiol. Immunol. 2015, 204, 263–271. [Google Scholar] [CrossRef]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Sinha, A.; Fowler, K.B.; Mladsi, D.; Barnett, C.; Samant, S.; Gibson, L. A framework for assessing the lifetime economic burden of congenital cytomegalovirus in the United States. Cost Eff. Resour. Alloc. 2019, 17, 21. [Google Scholar] [CrossRef]
- Weil, C.; Wang, W.; Marks, M.A.; Bilavsky, E.; Sinha, A.; Chodick, G.; Goodman, E. Health Care Resource Utilization and Economic Burden Associated With Congenital Cytomegalovirus Infection: A Longitudinal Analysis of Data From Clinical Practice at a Large Health Care Provider in Israel. Clin. Ther. 2022, 44, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Smithers-Sheedy, H.; Khandaker, G.; Raynes-Greenow, C.; Flack, L.; Britton, P.N.; McIntyre, S.; Badawi, N.; Burgner, D.; Galea, C.; Jones, C.A. The long-term burden of con-genital cytomegalovirus: Hospitalisation and mortality in a population-based matched cohort study. Eur. J. Paediatr. Neurol. 2022, 37, 82–86. [Google Scholar] [CrossRef]
- Fang, Y.; Doyle, M.F.; Chen, J.; Mez, J.; Satizabal, C.L.; Alosco, M.L.; Qiu, W.Q.; Lunetta, K.L.; Murabito, J.M. Circulating immune cell phenotypes are associated with age, sex, CMV, and smoking status in the Framingham Heart Study offspring participants. Aging 2023, 15, 3939–3966. [Google Scholar] [CrossRef]
- Barry, P.A.; Deere, J.D.; Yue, Y.; Chang, W.W.; Schmidt, K.A.; Wussow, F.; Chiuppesi, F.; Diamond, D.J.; Sparger, E.E.; Walter, M.R.; et al. Cytomegalovirus-vectored vaccines for HIV and other pathogens. AIDS 2020, 34, 335–349. [Google Scholar] [CrossRef]
- Mocarski, E.S., Jr. Immunomodulation by cytomegaloviruses: Manipulative strategies beyond evasion. Trends Microbiol. 2002, 10, 332–339. [Google Scholar] [CrossRef]
- Jackson, J.W.; Sparer, T. There Is Always Another Way! Cytomegalovirus’ Multifaceted Dissemination Schemes. Viruses 2018, 10, 383. [Google Scholar] [CrossRef]
- Miller-Kittrell, M.; E Sparer, T. Feeling manipulated: Cytomegalovirus immune manipulation. Virol. J. 2009, 6, 4. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef]
- Britt, W. Virus entry into host, establishment of infection, spread in host, mechanisms of tissue damage. In Human Herpesviruses: Biology, Therapy and Immunoprophy-Laxis; Arvin, A., Campadelli-Fiume, G., Moore, P., Mocarski, E., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 737–764. [Google Scholar]
- A Plotkin, S.; Wang, D.; Oualim, A.; Diamond, D.J.; Kotton, C.N.; Mossman, S.; Carfi, A.; Anderson, D.; Dormitzer, P.R. The Status of Vaccine Development Against the Human Cytomegalovirus. J. Infect. Dis. 2020, 221 (Suppl. 1), S113–S122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Freed, D.C.; Li, L.; Tang, A.; Li, F.; Murray, E.M.; Adler, S.P.; McVoy, M.A.; Rupp, R.E.; Barrett, D.; et al. A Replication-Defective Human Cytomegalovirus Vaccine Elicits Hu-moral Immune Responses Analogous to Those with Natural Infection. J. Virol. 2019, 93, e00747-19. [Google Scholar] [CrossRef]
- Aldoss, I.; La Rosa, C.; Baden, L.R.; Longmate, J.; Ariza-Heredia, E.J.; Rida, W.N.; Lingaraju, C.R.; Zhou, Q.; Martinez, J.; Kaltcheva, T.; et al. Poxvirus Vectored Cytomegalovirus Vaccine to Prevent Cytomegalovirus Viremia in Transplant Recipients: A Phase 2, Randomized Clinical Trial. Ann. Intern. Med. 2020, 172, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Adler, S.P.; Manganello, A.M.; Lee, R.; McVoy, M.A.; Nixon, D.E.; Plotkin, S.; Mocarski, E.; Cox, J.H.; Fast, P.E.; Nesterenko, P.A.; et al. A Phase 1 Study of 4 Live, Recombinant Human Cytomegalovirus Towne/Toledo Chimera Vaccines in Cytomegalovirus-Seronegative Men. J. Infect. Dis. 2016, 214, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; Mcneal, M.M.; et al. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: A randomized clinical trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef]
- Elek, S.D.; Stern, H. Development of a vaccine against mental retardation caused by cytomegalovirus infection in utero. Lancet 1974, 1, 1–5. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cy-tomegalovirus. J. Gen. Virol. 2004, 85 Pt 5, 1301–1312. [Google Scholar] [CrossRef]
- Paul, S.; Sidney, J.; Sette, A.; Peters, B. TepiTool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr. Protoc. Immunol. 2016, 114, 18.19.1–18.19.24. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Weiskopf, D.; Angelo, M.A.; Sidney, J.; Peters, B.; Sette, A. HLA Class I Alleles Are Associated with Peptide-Binding Repertoires of Different Size, Affinity, and Immunogenicity. J. Immunol. 2013, 191, 5831–5839. [Google Scholar] [CrossRef]
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 1994, 153, 5586–5592. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef]
- Martini, S.; Nielsen, M.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource Program 2003–2018: Reflections and outlook. Immunogenetics 2020, 72, 57–76. [Google Scholar] [CrossRef]
- Chauhan, V.; Singh, M.P. Immuno-informatics approach to design a multi-epitope vaccine to combat cytomegalovirus infection. Eur. J. Pharm. Sci. 2020, 147, 105279. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Ojha, R.; Dipti, K.; Kumar, R.; Prajapati, V.K. Immunoselective algorithm to devise multi-epitope subunit vaccine fighting against human cytomegalovirus infection. Infect. Genet. Evol. 2020, 82, 104282. [Google Scholar] [CrossRef] [PubMed]
- Quinzo, M.J.; Lafuente, E.M.; Zuluaga, P.; Flower, D.R.; Reche, P.A. Computational assembly of a human Cytomegalovirus vaccine upon experimental epitope legacy. BMC Bioinform. 2019, 20, 476. [Google Scholar] [CrossRef]
- Bakkari, M.A. Targeted Protein-Specific Multi-Epitope-Based Vaccine Designing against Human Cytomegalovirus by Using Immunoinformatics Approaches. Vaccines 2023, 11, 203. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.S.; Sethi, G.; Ramadas, K. Design of multi-epitope based vaccine against Mycobacterium tuberculosis: A subtractive proteomics and reverse vaccinology based immunoinformatics approach. J. Biomol. Struct. Dyn. 2023, Online ahead of print. 1–19. [Google Scholar] [CrossRef]
- Lichtman, A. Finding TB vaccine antigens: Follow the TCRs. Sci. Immunol. 2023, 8, eadg8281. [Google Scholar] [CrossRef] [PubMed]
- Arif, S.; Akhter, M.; Khaliq, A.; Akhtar, M.W. Fusion peptide constructs from antigens of M. tuberculosis producing high T-cell mediated immune response. PLoS ONE 2022, 17, e0271126. [Google Scholar] [CrossRef]
- Di Salvatore, V.; Russo, G.; Pappalardo, F. Reverse Vaccinology for Influenza A Virus: From Genome Sequencing to Vaccine Design. Methods Mol. Biol. 2023, 2673, 401–410. [Google Scholar] [CrossRef]
- Dasari, V.; McNeil, L.K.; Beckett, K.; Solomon, M.; Ambalathingal, G.; Le Thuy, T.; Panikkar, A.; Smith, C.; Steinbuck, M.P.; Jakubowski, A.; et al. Lymph node targeted multi-epitope subunit vaccine promotes effective immunity to EBV in HLA-expressing mice. Nat. Commun. 2023, 14, 4371. [Google Scholar] [CrossRef]
- Dhanwani, R.; Dhanda, S.K.; Pham, J.; Williams, G.P.; Sidney, J.; Grifoni, A.; Picarda, G.; Arlehamn, C.S.L.; Sette, A.; Benedict, C.A. Profiling Human Cytomegalovirus-Specific T Cell Responses Reveals Novel Immunogenic Open Reading Frames. J. Virol. 2021, 95, e0094021. [Google Scholar] [CrossRef]
- Jackson, S.E.; Sedikides, G.X.; Romashova, V.; Okecha, G.; Remmerswaal, E.B.M.; Bemelman, F.J.; Sinclair, J.H.; Wills, M.R. IL-10-Secreting CD8(+) T Cells Specific for Human Cytomegalovirus (HCMV): Generation, Maintenance and Phenotype. Pathogens 2022, 11, 1530. [Google Scholar] [PubMed]
- Di Rosa, F.; Pabst, R. The bone marrow: A nest for migratory memory T cells. Trends Immunol. 2005, 26, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Giam, K.; Croft, N.P.; Akutsu, T.; Smith, A.I.; Li, J.; Rossjohn, J.; et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Briefings Bioinform. 2020, 21, 1119–1135. [Google Scholar] [CrossRef]
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omaïs, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient Origin and Gene Mosaicism of the Progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005, 1, e5. [Google Scholar] [CrossRef]
- Ernst, J.D. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe 2018, 24, 34–42. [Google Scholar] [CrossRef]
- Coscolla, M.; Copin, R.; Sutherland, J.; Gehre, F.; de Jong, B.; Owolabi, O.; Mbayo, G.; Giardina, F.; Ernst, J.D.; Gagneux, S. M. tuberculosis T Cell Epitope Analysis Reveals Paucity of Antigenic Variation and Identifies Rare Variable TB Antigens. Cell Host Microbe 2015, 18, 538–548. [Google Scholar] [CrossRef]
- Lee, M.S.J.; Coban, C. Unforeseen pathologies caused by malaria. Int. Immunol. 2018, 30, 121–129. [Google Scholar] [CrossRef]
- Taher, H.; Mahyari, E.; Kreklywich, C.; Uebelhoer, L.S.; McArdle, M.R.; Moström, M.J.; Bhusari, A.; Nekorchuk, M.E.X.; Whitmer, T.; Scheef, E.A.; et al. In vitro and in vivo characterization of a recombinant rhesus cytomegalovirus containing a complete genome. PLoS Pathog. 2020, 16, e1008666. [Google Scholar] [CrossRef]
- Hansen, S.G.; Strelow, L.I.; Franchi, D.C.; Anders, D.G.; Wong, S.W. Complete sequence and genomic analysis of rhesus cytomegalo-virus. J. Virol. 2003, 77, 6620–6636. [Google Scholar]
- Rivailler, P.; Kaur, A.; Johnson, R.P.; Wang, F. Genomic Sequence of Rhesus Cytomegalovirus 180.92: Insights into the Coding Potential of Rhesus Cytomegalovirus. J. Virol. 2006, 80, 4179–4182. [Google Scholar] [CrossRef]
- Yue, Y.; Kaur, A.; Zhou, S.S.; Barry, P.A. Characterization and immunological analysis of the rhesus cytomegalovirus homologue (Rh112) of the human cytomegalovirus UL83 lower matrix phosphoprotein (pp65). J. Gen. Virol. 2006, 87 Pt 4, 777–787. [Google Scholar] [CrossRef]
- Malouli, D.; Hansen, S.G.; Nakayasu, E.S.; Marshall, E.E.; Hughes, C.M.; Ventura, A.B.; Gilbride, R.M.; Lewis, M.S.; Xu, G.; Kreklywich, C.; et al. Cytomegalovirus pp65 limits dissemination but is dispensable for persistence. J. Clin. Investig. 2014, 124, 1928–1944. [Google Scholar] [CrossRef] [PubMed]
- Verweij, M.C.; Hansen, S.G.; Iyer, R.; John, N.; Malouli, D.; Morrow, D.; Scholz, I.; Womack, J.; Abdulhaqq, S.; Gilbride, R.M.; et al. Modulation of MHC-E transport by viral decoy ligands is required for RhCMV/SIV vaccine efficacy. Science 2021, 372, eabe9233. [Google Scholar] [CrossRef] [PubMed]
- Manley, T.J.; Luy, L.; Jones, T.; Boeckh, M.; Mutimer, H.; Riddell, S.R. Immune evasion proteins of human cytomegalovirus do not prevent a diverse CD8+ cytotoxic T-cell response in natural infection. Blood 2004, 104, 1075–1082. [Google Scholar] [CrossRef]
- Hansen, S.G.; Powers, C.J.; Richards, R.; Ventura, A.B.; Ford, J.C.; Siess, D.; Axthelm, M.K.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J.; et al. Evasion of CD8 + T Cells Is Critical for Superinfection by Cytomegalovirus. Science 2010, 328, 102–106. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Revised Set n = 148 Total | Test Set n = 109 Total | Identity Set n = 61 Total | Affinity-HLA Set n = 35 Total | Final Set n = 24 Total | |||
|---|---|---|---|---|---|---|---|
| HCMV Protein | GenBank or Swiss Prot ID | CD8 (#) | CD4 (#) | Sequence Identity (Median %) | Peptides with IC50 ≤ 50 nM (#) | Restricting HLA Alleles (#) | Immunogenicity Score (Median) |
| UL 27 | CAA35426 | 0 | 1 | 98.7 | 186 | 23 | 0.0860 |
| UL 43 | CAA74075 | 2 | 4 | 99.1 | 151 | 21 | 0.0410 |
| UL 45 | CAA35404 | 4 | 3 | 99.5 | 319 | 26 | 0.0770 |
| UL 47 | CAA35406 | 2 | 1 | 99.7 | 332 | 26 | 0.0740 |
| UL 48 | CAA35407 | 21 | 15 | 99.3 | 542 | 26 | 0.0810 |
| UL 70 | CAA35386 | 0 | 2 | 99.6 | 341 | 25 | 0.0970 |
| UL 72 | CAA35387 | 0 | 0 | 99.0 | 104 | 23 | 0.0550 |
| UL 83 (pp65) | CAA35357 | 19 | 24 | 99.5 | 137 | 22 | 0.0760 |
| UL 84 | CAA35358 | 2 | 0 | 98.5 | 130 | 23 | 0.0660 |
| UL 87 | CAA35361 | 3 | 4 | 99.6 | 328 | 23 | 0.0450 |
| UL 88 | CAA35362 | 1 | 0 | 99.8 | 148 | 24 | 0.0910 |
| UL 89 | CAA35363 | 0 | 3 | 99.4 | 242 | 24 | 0.0710 |
| UL 92 | CAA35366 | 0 | 0 | 99.5 | 103 | 24 | 0.0430 |
| UL 93 | CAA35367 | 0 | 0 | 98.7 | 172 | 21 | 0.1150 |
| UL103 | CAA35339 | 2 | 0 | 99.6 | 93 | 21 | 0.0410 |
| UL104 | CAA35341 | 0 | 0 | 99.7 | 170 | 24 | 0.0950 |
| UL117 | CAA35319 | 1 | 0 | 99.3 | 121 | 22 | 0.0470 |
| UL123 (IE1) | CAA35325 | 18 | 12 | 97.2 | 115 | 21 | −0.108 |
| UL148Tol | AAA85887.1 | 0 | 3 | 98.7 | 122 | 22 | 0.0920 |
| US 2 | CAA35313 | 2 | 2 | 99.5 | 88 | 22 | 0.0920 |
| US 11 | CAA35278 | 1 | 0 | 98.6 | 105 | 23 | 0.0870 |
| US 12 | CAA35279 | 3 | 3 | 98.9 | 162 | 21 | 0.0910 |
| US 13 | CAA35280 | 0 | 0 | 99.6 | 169 | 21 | 0.1320 |
| US 15 | CAA35282 | 0 | 2 | 99.6 | 148 | 21 | 0.0960 |
| US 16 | CAA35283 | 2 | 3 | 99.4 | 179 | 21 | 0.1300 |
| US 24 | CAA35291 | 3 | 3 | 99.6 | 166 | 24 | 0.1030 |
| US 28 | P09704 | 1 | 0 | 98.9 | 208 | 26 | 0.0670 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barry, P.A.; Iyer, S.S.; Gibson, L. Re-Evaluating Human Cytomegalovirus Vaccine Design: Prediction of T Cell Epitopes. Vaccines 2023, 11, 1629. https://doi.org/10.3390/vaccines11111629
Barry PA, Iyer SS, Gibson L. Re-Evaluating Human Cytomegalovirus Vaccine Design: Prediction of T Cell Epitopes. Vaccines. 2023; 11(11):1629. https://doi.org/10.3390/vaccines11111629
Chicago/Turabian StyleBarry, Peter A., Smita S. Iyer, and Laura Gibson. 2023. "Re-Evaluating Human Cytomegalovirus Vaccine Design: Prediction of T Cell Epitopes" Vaccines 11, no. 11: 1629. https://doi.org/10.3390/vaccines11111629
APA StyleBarry, P. A., Iyer, S. S., & Gibson, L. (2023). Re-Evaluating Human Cytomegalovirus Vaccine Design: Prediction of T Cell Epitopes. Vaccines, 11(11), 1629. https://doi.org/10.3390/vaccines11111629