1. Introduction
Immunotherapy (the use of immunological factors or an immune stimulator/modifier to treat or prevent diseases) is one of the most promising approaches for the treatment of diseases including inflammations and cancers. For cancers, the immunotherapeutic agents harness or boost the immune system to work more efficiently, making it more specific and/or more able to recognize and attack the targets [
1]. Immunotherapy can stand alone or be performed alongside, after, or prior to conventional cancer treatments (surgery, radiotherapy, and chemotherapy), i.e., it can serve as an adjuvant, neoadjuvant, or induction therapy [
2,
3,
4]. In many instances, immunotherapy can work for cancers when other treatment measures do not or cannot work, such as in unresectable or radio- and/or chemo-resistant cancers [
5].
Nowadays, there are several types of immunotherapies that have been practiced that suit different cancers including the use of adoptive immune cells, i.e., tumor-infiltrating lymphocytes [
6], engineered CAR-T/Natural Killer (NK) cells [
7,
8], and T cell receptor (TCR)-engineered T cells [
9]; cancer vaccines (either for the treatment of existing cancer or to prevent cancer development) [
10,
11]; immunomodulators, i.e., cytokines, Bacillus Calmette–Guérin (BCG), and biological response modifiers (e.g., Imiquimod, Lenalidomide) [
12,
13,
14,
15]; immune system agonists [
16,
17]; and monoclonal antibodies (mAbs) [
18,
19]. Usually, the immune modifiers, immune system agonists, and monoclonal antibodies do not kill cancer cells directly but work by enhancing and/or helping the immune system to better find cancers in the body (marking cancerous cells) for the immune system to attack them.
Tumorigenesis involves cancer immunoediting, which is a continual process consisting of three phases [
20,
21,
22]. The first phase involves the initially transformed body cell that, if not eliminated by the host immune surveillance of natural killer (NK) cells, can manage to form a nascent tumor; this will be followed by the second phase, a relatively prolonged equilibrium phase in which the tumor outgrowth is immunologically restrained but not eliminated; and, finally, the third phase involves immunological sculpting of the tumor and establishment of a tumor microenvironment (TME) that favors tumor metabolism and growth, leading to tumor progression/metastasis and clinical manifestations [
22,
23,
24]. The TME is a complex milieu consisting of vasculature (blood and lymph vessels), fibroblasts, mesenchymal stem cells (MSCs), adipocytes, neuroendocrine (NE) cells, immune cells (myeloid cells, lymphocytes), extracellular matrix, and vesicles as well as physical and chemical factors that contribute to low extracellular pH (acidosis), hypoxia, and elevated interstitial fluid pressure [
25]. The immune cells in the TME play different roles at different stages of tumor development, i.e., tumor suppression in the early stage by some cell types, but tumor growth promotion at the later stage by others [
20,
21,
22,
25]. In the early stage of cancer, NK cells, CD8
+ cytotoxic T lymphocytes (CTL), Th1 cells, and antigen-presenting cells (APCs), like dendritic cells (DCs) and M1 macrophages, are effective in suppressing tumor growth. Cytotoxic T cells and NK cells induce apoptosis, necrosis, and growth arrest of the cancerous cells by releasing IFN-γ, perforin, granzymes, etc. The apoptotic cancer cells and released components are phagocytosed/endocytosed by APCs, which process and present the antigenic peptides to lymphocytes in the lymphoid tissues for adaptive immune responses against the tumor. However, in the later phase, tumors can evade the immune defenses and progress by creating an immunosuppressive TME containing M2 tumor-associated macrophages (TAMs), CD4
+ Th2 cells, and, especially, regulatory T cells (Tregs) [
25]. Tregs in the TME attenuate/inhibit effector T cells/NK cells and DCs by producing a myriad of immunosuppressive factors (both soluble and cell-bound) including immunosuppressive cytokines (like IL-10, TGF-β, and IL-35); high levels of cell-surface IL-2R (CD25) that deprive the microenvironment of IL-2, which is important for the proliferation and survival of effector T cells, esp. CD8
+; surface-exposed ectonucleotidases (CD39 and CD73) that generate adenosine for impairment of effector cell functions; and, most of all, surface expressed immune-checkpoint molecules (e.g., PD1, CTLA-4, and LAG-3) that bind to respective receptors/ligands (PD-L1, CD86, and MHC-II, respectively) on the APCs, causing the refractoriness of the effector cells [
26]. The blockage of immune-checkpoint signaling pathways in the effector T cells has shown promising results in cancer immunotherapy [
27,
28,
29]. Monoclonal antibodies targeting immune checkpoints, e.g., Nivolumab for PD-1, Avelumab for PD-L1, Ipilimumab for CTLA-4, Relatlimab for LAG-3, or combined treatment with these have been approved for the immunotherapy of many cancers [
30].
Refractory effector T cells in the TME can be revitalized to regain their anti-tumor activities, i.e., the ability to restrain tumor growth, by stimulating T cell costimulatory receptors, such as those belonging to the tumor necrosis factor receptor superfamily (TNFRSF), i.e., CD40, 4-1BB, and OX40 [
31,
32]. Ligation of OX40 (TNFRSF4, CD134) on the effector T cell surface by OX40 agonists causes the receptor to oligomerize (cluster) and stimulates intracellular signaling, which enhances anti-tumor immunity leading to therapeutic effects in cancers [
33,
34,
35]. Resting T cells do not constitutively express OX40 on the cell surface but transiently upregulate it upon being activated [
31]. The interaction between OX40 on T cells and OX40L (CD252) expressed on antigen-presenting cells, such as DCs, macrophages, and B cells, enhances T cell expansion, trafficking, cytokine release and survival, and the generation of long-lived memory T cells [
31,
32]. Monoclonal antibodies that trigger OX40 signaling have been developed and tested in preclinical trials and in patients with advanced solid tumors; these were found to be well-tolerated and effective [
36,
37]. The treatment of tumor-bearing hosts with OX40 agonistic antibodies (using a variety of antibody formats) resulted in tumor regression [
38,
39,
40]. The mechanisms of those OX40 agonistic antibodies against tumors involved the promotion of antigen-specific effector T cell expansion and survival in the TME and macrophage activation, as well as the reverse immune suppression of Tregs [
38,
41]. Regulatory T cells constitutively express OX40. OX40 signaling controls Treg proliferation and their suppressive activities [
42,
43]. OX40 agonists alone or in combination with immune-checkpoint blockers have high therapeutic potential for patients with advanced malignancies. Despite numerous preclinical and clinical trials of several OX40 agonists, none of these have reached clinical use [
44]. The lack of sufficient potency of the OX40 antibodies in a therapeutic setting may be due to their inefficiency or low potency in properly clustering the OX40 molecules on the T cell surface in vivo [
44]. The OX40 agonist-OX40 complexes must be able to cause clustering of the spatially distant OX40 molecules on the cell surface to enable their intracytoplasmic OX40 tails to generate a hexameric TRAF complex for downstream signaling and activation of the NK-κB and NFAT transcription factors, and this requires the optimal positioning of the complex on the cell surface [
44]. The lack of this intrinsic property renders the OX40 agonists ineffective in therapy [
44]. In this study, engineered human OX40 agonistic antibodies that target the human OX40 ectodomain in the form of bivalent human single-chain variable fragments (HuscFvs) linked to the human IgG1-Fc portion (fusion antibodies) were generated. The production, characterization, and evaluation of these novel OX40 agonistic antibodies in T cell stimulation to enhance anti-tumor activities form the core of this article.
2. Materials and Methods
2.1. Ethics Statement
Experiments involving human samples were approved by the Institutional Review Board of the Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand (IRB Si 651/2018).
2.2. Cells and Culture Media
Human embryonic kidney (HEK) 293T cells and Jurkat T cells (a leukemic cell line isolated from peripheral blood of a patient with acute T lymphocytic leukemia) were from the American Type Culture Collection (ATCC; Manassas, VA, USA). HEK293E suspension cells were from IBA Lifesciences (Göttingen, Germany). The ovarian cancer cell line, SK-OV-3, was from Dr. Somponnat Sampattavanich, Department of Pharmacology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok. Human peripheral blood mononuclear cells (PBMCs) were isolated from blood of healthy donors. HEK293T and SK-OV-3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone; Cytiva, Marlborough, MA, USA), 100 units/mL penicillin, 100 mg/mL streptomycin, and 2 mM L-glutamine (Gibco, Thermo Fisher Scientific). HEK293E suspension cells were cultured in CDM4HEK293 serum-free medium (Cytiva, Marlborough, MA, USA) supplemented with 100 units/mL penicillin, 100 mg/mL streptomycin, and 4 mM L-glutamine (Gibco, Thermo Fisher Scientific). Jurkat cells were cultured in RPMI-1640 (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS (HyClone), 100 units/mL penicillin, 100 mg/mL streptomycin, and 2 mM L-glutamine (Gibco, Thermo Fisher Scientific) (complete RPMI-1640). T cells, monocytes, and dendritic cells (DCs) were cultured in AIM-V (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS (HyClone), 100 units/mL penicillin, 100 mg/mL streptomycin, and 2 mM L-glutamine (Gibco, Thermo Fisher Scientific) (complete AIM-V). Human PBMCs were cultured in either complete RPMI-1640 or complete AIM-V.
2.3. Recombinant Ectodomain of Human OX40
A recombinant human OX40 ectodomain (EcOX40) with inherent OX40 ligand binding activity (Abcam, Cambridge, UK) was used as the antigenic bait in phage panning to fish out HuscFv-displaying phage clones from a previously constructed HuscFv phage display library [
45] for the production of HuscFvs to human EcOX40.
A recombinant human glycosylated EcOX40 produced by transformed HEK293E suspension cells in our laboratory was used as an antigen for checking the binding of antibodies to human EcOX40. An amplicon of the EcOX40 coding sequence was ligated to a mammalian expression vector (pDSG, IBA Lifesciences) and the recombinant vector was transformed into HEK293E suspension cells. The cells were grown and the soluble EcOX40 protein was harvested from the cell culture supernatant.
2.4. Preparation of Mammalian Cells That Overexpress Human OX40 (HEK-OX40 Cells)
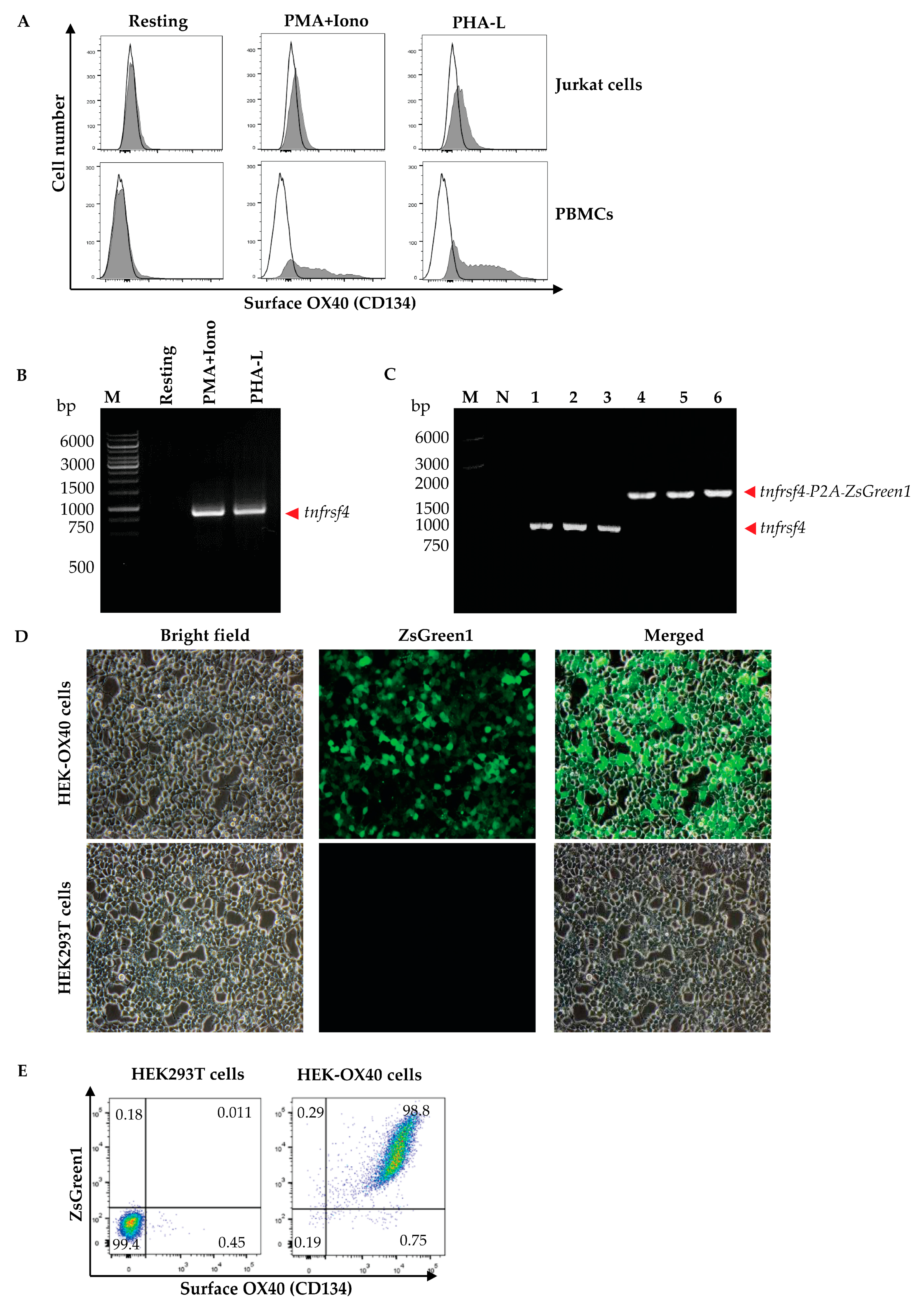
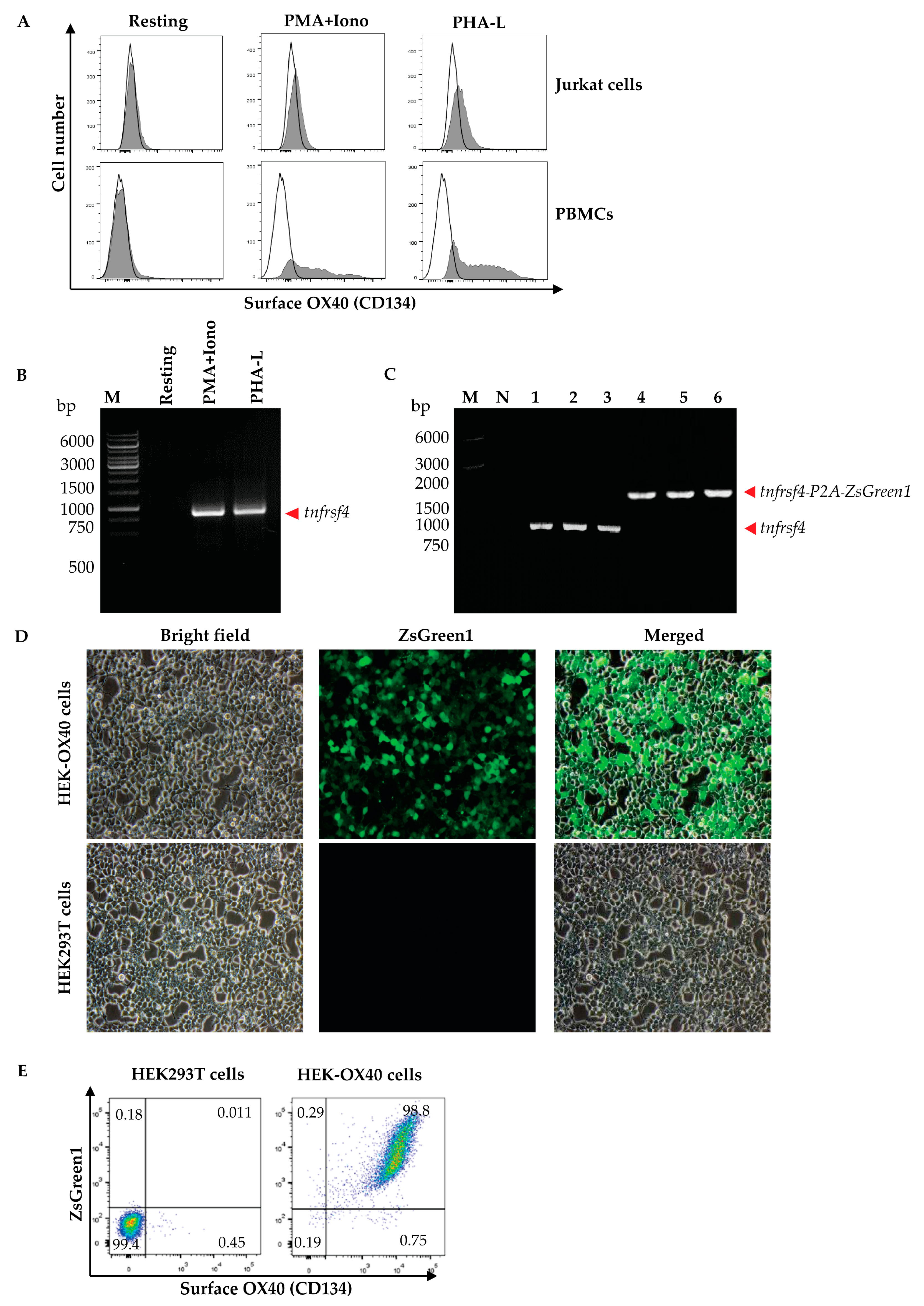
Peripheral blood mononuclear cells (PBMCs) were isolated from 10 mL of heparinized whole blood from a healthy subject by gradient centrifugation using Ficoll Hypaque (Histopaque-1077, Sigma-Aldrich, St. Louis, MO, USA). The isolated PBMCs (1 × 106 cells) and Jurkat cells were cultured in complete RPMI-1640 medium (Gibco, Thermo Fisher Scientific) containing mitogens, i.e., 0.5 μg/mL of phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, USA) and 1 μM of ionomycin (Iono, Sigma-Aldrich, USA) or 1 μg/mL phytohemagglutinin-L (PHA-L, Biochrom, Cambridge, UK) at 37 °C in a 5% CO2 atmosphere for 3 days. The activated cells and respective resting cells (no mitogen activated counterparts) were then stained with anti-OX40-Alexa Fluor 647 to check for OX40 expression by flow cytometry. The activated PBMCs were found to express OX40 more than the Jurkat cells; therefore, the PBMCs were used as a source of the OX40 coding gene (tnfrsf4).
Human PBMCs (1 × 106 cells) were cultured in complete RPMI-1640 medium (Gibco, Thermo Fisher Scientific) containing 0.5 μg/mL of PMA (Sigma-Aldrich) and 1 μM of Iono (Sigma-Aldrich) or 1 μg/mL PHA-L (Biochrom) at 37 °C in a 5% CO2 atmosphere for 6 h. Total RNA was extracted from the mitogen-activated cells by using Trizol reagent (Invitrogen, Waltham, MA, USA) and reversely transcribed into complementary DNA (cDNA) by using the RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific). The cDNA was used as the template in a polymerase chain reaction (PCR) for the amplification of the full-length human OX40 gene (human tnfrsf4) by using specific primers with 5′ XhoI and EcoRI restriction sites, i.e., forward primer: 5′-TACTATCTCGAGGCCACCATGTGCGTGGGGGCTCG-3′ and reverse primer: 5′-TATCATGAATTCGCGATCTTGGCCAGGGTGGAGTG-3′, from the GenBank database (NM_003327). The nucleotide sequence of the amplified PCR product was verified using Sanger sequencing (1st Base DNA sequencing service, Selangor, Malaysia) before ligation into the pJET1.2 cloning vector (Thermo Fisher Scientific) using T4 ligase (New England BioLabs, Ipswich, MA, USA). The recombinant vector was transformed into JM109 Escherichia coli. The preparation was spread onto Luria-Bertani (LB) agar containing 100 µg/mL ampicillin (LB-A agar plate) and incubated at 37 °C overnight. The plasmid-transformed E. coli colonies (clones) that grew on the agar were screened for the target gene, i.e., full-length human tnfrsf4 by direct colony PCR using forward and reverse pJET1.2 primers, and the amplicon was sequenced (1st Base).
For preparing mammalian cells that overexpressed full-length human OX40, bi-cistronic expression vector that expressed full-length human OX40 and the Zoanthus green fluorescent protein (ZsGreen1) was prepared. A commercially synthesized MCS-P2A nucleotide sequence and the pLVX-puro vector (Clontech, Mountain View, CA, USA) were similarly digested with XhoI and XbaI restriction endonucleases (Thermo Fisher Scientific). The cut MCS-P2A was ligated with the cut pLVX-puro vector using T4 ligase. The recombinant vector was transformed into DH5α E. coli. The transformed bacteria were spread on the selective LB-A agar plate and incubated at 37 °C overnight. The E. coli clones carrying the recombinant MCS-P2A-pLVX-puro plasmid vector were screened by direct colony PCR using forward and reverse MSCV primers. The recombinant plasmids were extracted from the DH5α E. coli and verified by DNA sequencing (1st Base).
A DNA fragment coding for the Zoanthus green fluorescent protein (ZsGreen1) (Genscript, Piscataway, NJ, USA) and the MCS-P2A-pLVX-puro vector were similarly digested with NotI and XbaI restriction enzymes. The cut ZsGreen1 fragment was ligated with the cut MCS-P2A-pLVX-puro vector using T4 ligase and transformed into DH5α E. coli. The transformed bacteria were spread onto the LB-A agar plate. After incubation at 37 °C overnight, bacterial colonies were screened by direct colony PCR as above for the clones that harbored the MCS-P2A-ZsGreen1-pLVX-puro vector using the forward and reverse CMV primers. The verified MCS-P2A-ZsGreen1-pLVX-puro vector and the tnfrsf4-pJET1.2 were cut similarly with XhoI and EcoRI enzymes (Thermo Fisher Scientific), ligated using T4 ligase, and transformed into DH5α E. coli. The E. coli clones carrying the tnfrsf4-P2A-ZsGreen1-pLVX-puro vector were screened by direct colony PCR using the forward and reverse MSCV primers. The tnfrsf4-P2A-ZsGreen1-pLVX-puro vector was extracted from the E. coli clone by using the EndoFree Maxi Plasmid kit (Tiangen Biotech, Beijing, China).
Human embryonic kidney (HEK293T) cells were cultured in 10% tetracycline-free-FBS-supplemented DMEM before their use in lentivirus preparation. The HEK293T cells were co-transfected with the tnfrsf4-P2A-ZsGreen1-pLVX-puro vector and Lenti-X packaging single shots (VSV-G) (Clontech), and the cells were incubated at 37 °C in a 5% CO2 incubator for 3 days. The culture supernatant was tested for the presence of lentivirus particles by adding Lenti-X GoStix (Clontech). A positive Lenti-X GoStix result implied that there were more than 5 × 105 infectious units (IFU)/mL of lentivirus particles in the culture supernatant. The lentivirus particles in the culture supernatant were concentrated by using Lenti-X concentrator (Clontech) and checked for quantity and infectivity. For this experiment, the HEK293T cells (3 × 104 cells) were added to the wells of a 96-well-tissue culture plate (Corning, Steuben County, NY, USA) and the plate was kept at 37 °C in a 5% CO2 incubator overnight. A five-fold serially diluted lentivirus preparation (50 μL) was added to individual wells containing the HEK293T cells and incubated further for 4 h before the addition of 10% FBS supplemented-DMEM (150 μL/well) and was then kept in the incubator for 3 more days. The cells from all wells were collected separately and the expression of the ZsGreen1 protein was determined (excitation/emission at 493/505 nm) via flow cytometry. The percentage of ZsGreen1-positive cells was used for calculating the number of lentivirus infectious units or total transducing units (TU)/mL.
HEK293T cells (1 × 104 cells/well of a 96-well tissue culture plate maintained at 37 °C in a 5% CO2 incubator overnight) were added together with the lentivirus (MOI 10; by mixing the lentivirus with 10% FBS-supplemented DMEM in a total volume of 50 μL). The infected HEK293T cells were incubated for 4 h, followed by the addition of 10% FBS-supplemented DMEM (150 μL/well) and the cells were incubated further for 3 more days. The cells were observed for ZsGreen1 protein by using an inverted fluorescent microscope. A positive result indicated that the cells were infected with the lentivirus and that the transfected DNA was integrated into the chromosomes. Thereafter, 2 μg/mL puromycin (Invivogen, San Diego, CA, USA) was added to the cultured cells. The spent medium was replaced with fresh medium every 2–3 days. On days 7–10, non-infected cells were dead. The viable cells were cultured further in 10% FBS-supplemented DMEM. The expression of human OX40 on the cell surface was determined. The cells were stained with an Alexa Fluor 647-conjugated anti-human OX40/CD134 antibody (clone Ber-act35, Biolegend, San Diego, CA, USA) and analyzed via flow cytometry (BD LSRFortessa, BD biosciences, San Jose, CA, USA) in comparison with non-infected HEK293T cells (negative control). The HEK293T cells expressing human OX40 were designated “HEK-OX40 cells”.
2.5. Production of HuscFvs That Bound to Human EcOX40 by Using Phage Display Technology
The HuscFv phage display library used in this study was constructed previously [
45,
46]. In this phage library, complete phage particles display HuscFvs as fusion partners of the phage coat P3 protein and contain the respective HuscFv genes (
huscfvs) in their genomes. The diversity of the HuscFvs of this library was ~2.6 × 10
8 [
45]. After one cycle of the library propagation in TG1
E. coli, ~2.6 × 10
12 cfu/mL of complete phage particles were obtained [
45].
For the selection of HuscFvs displaying phage clones that bound to human EcOX40, recombinant human EcOX40 (Abcam) and HEK-OX40 cells were used as the antigenic baits in the phage panning to fish out the EcOX40-bound HuscFv-displaying phages from the HuscFv phage display library. For panning with the recombinant human EcOX40, the recombinant human EcOX40 and unrelated proteins, including bovine serum albumin (BSA, Calbiochem, San Diego, CA, USA), and skim milk (HiMedia, Maharashtra, India) were applied to separate wells in an EIA/RIA microplate (Corning) (2 μg protein in 100 μL of phosphate buffered saline, pH 7.4 (PBS)) and the plate was kept at 4 °C overnight. After discarding the fluid in all the wells, the coated wells were washed three times with PBS containing 0.05% polysorbate (Tween)-20 (PBST; 200 μL PBST/well each time). Then, each well was blocked with 200 μL of 3% BSA in PBS and the plate was incubated at 37 °C for 1 h. After discarding the excess BSA and washing the wells with PBST, the HuscFv phage display library (50 μL) was added to the skim milk-coated well and incubated at 37 °C for 1 h. The supernatant was then moved to the BSA-coated well and incubated for 1 h before the fluid containing skim milk and the BSA-subtracted phage library were transferred to the human EcOX40-coated well and kept at 37 °C for 1 h. The fluid was discarded and the well was washed thoroughly with PBST. Log-phase-grown HB2151 E. coli (200 μL) was added to the well containing the EcOX40-bound phages and kept at 37 °C for 15 min to allow the phages to infect the E. coli. The phage transformed preparation was diluted appropriately with LB broth and spread onto 2× YT-ampicillin agar (2YT-A) plates containing 2% glucose (2YT-AG agar) and the plates were incubated at 37 °C overnight.
For the selection of HuscFv-displaying phage clones that bound to human EcOX40 expressed on the surface of the HEK-OX40 cells, a cell-based phage panning was performed. The skim milk and BSA-subtracted HuscFv phage display library was prepared as above. The subtracted library (100 μL) was mixed with 1 × 106 HEK293T cells in a microcentrifuge tube and kept at 4 °C on a see-saw rocker for 1 h. The cells were sedimented by centrifugation at 10,000× g for 30 s. The supernatant containing the HEK293T cell-unbound phage was transferred to another centrifuge tube containing 1 × 106 HEK-OX40 cells. After rocking them at 4 °C for 1 h, the cells were collected by centrifugation as above and washed ten times with 1% BSA in PBS containing 0.02% NaN3, followed by washing with cold PBS five times. Log-phase-grown HB2151 E. coli (200 μL) were then added to the washed cells and the preparation was kept at 37 °C for 15 min. The phage-transformed HB2151 E. coli preparation was diluted with LB broth, plated onto the 2× YT-AG agar plates, and the plates were incubated at 37 °C overnight.
The phage-infected HB2151
E. coli clones from the phage panning with recombinant human EcOX40 and HEK-OX40 cells that grew on the 2× YT-AG agar plates were screened by direct colony PCR for the genes coding for HuscFvs (
huscfvs) by using R1 and R2 primers specific for the pCANTAB5E phagemids [
45]. The
huscfv-positive
E. coli clones were grown individually in 2× YT-A broth at 37 °C with shaking aeration (250 rpm) until the optical density (OD) at 600 nm reached about 0.3–0.5. The bacterial cultures were centrifuged (4000×
g, 4 °C, 15 min), the supernatants were discarded, and the bacteria in each pellet were resuspended in fresh 2× YT-A broth containing 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubated further at 30 °C, 250 rpm for 4 h. The bacterial cells of each culture were collected by centrifugation, resuspended in 1 mL PBS, and homogenized by sonication. Bacterial homogenates were centrifuged (12,000×
g, 4 °C, 15 min). Then, the bacterial lysates (supernatants) were collected and the presence of E-tagged-HuscFvs in the lysates was determined by Western blot analysis using an anti-E-tag antibody (1:5000, Abcam, Cambridge, UK) to probe the HuscFvs in the SDS-PAGE-separated bacterial lysates.
2.6. Characterization of the HuscFvs
Binding of the HuscFvs to human EcOX40 was tested using an indirect ELISA and flow cytometry. For the indirect ELISA, the recombinant human EcOX40 (Abcam) and BSA (control antigen) were dissolved in PBS (5 μg/mL), and 100 μL aliquots were added to separate wells of an EIA/RIA plate (Corning). The plate was kept at 4 °C overnight. Then, all wells were washed three times with PBS before blocking each well with 200 μL of 5% skim milk in PBS at 37 °C for 1 h. The blocking solution was removed, the wells were washed with PBS, and 100 μL of the E. coli lysate containing HuscFvs was added to the wells coated with EcOX40 and BSA. The original HB2151 E. coli lysate (HB) was included in the assay as the background binding control (no HuscFv). The plate was kept at 4 °C overnight, and the wells were washed with PBS containing 0.05% Tween-20 (PBST). Rabbit anti-E-tag antibody (1:5000, Abcam) was added to each well (100 μL/well) and the plate was kept at 37 °C for 3 h. After washing all wells with PBST, horseradish peroxidase (HRP)-conjugated anti-rabbit Ig antibody (1:5000, SouthernBiotech, Birmingham, AL, USA) was added to each well (100 μL/well), kept at 37 °C for 1 h, washed, and then the ABTS substrate (100 μL/well, Seracare, Milford, MA, USA) was added to each well and incubated at room temperature (RT, 25 ± 2 °C) for 1 h. The optical density at 405 nm of the content in each well was determined against the blank (wells added with PBS instead of the E. coli lysate). To test the binding of HuscFvs to the human EcOX40 expressed on the HEK-OX40 cells, E. coli lysates containing HuscFvs were added to HEK293T cells and HEK-OX40 cells contained in separate tubes (5 × 105 cells in 100 μL of 5× FACS buffer, i.e., cold 10% FBS-PBS-0.02% NaN3/tube), and the tubes were placed on a rotator at 4 °C for 1 h. The cells were washed with the FACS buffer three times and resuspended in 25 μL of the same buffer. Rabbit anti-E-tag antibody (1:500, Abcam) was added to the cells in each tube (25 μL/tube) and the tubes were placed on ice for 1 h. The cells were washed three times and resuspended in 12.5 μL FACS buffer. An Alexa Fluor 647-conjugated goat anti-rabbit Ig antibody (12.5 μL, 1:200, Invitrogen) was added to the cells and the tubes were placed on ice for 1 h. The cells were washed with FACS buffer, resuspended in 500 μL of 1% paraformaldehyde in PBS, and subjected to flow cytometric analysis using a flow cytometer (BD LSRFortessa™ Cell Analyzer, BD biosciences).
Complementarity-determining regions (CDRs) and immunoglobulin framework regions (FRs) of the HuscFvs were determined.
Escherichia coli clones that expressed HuscFvs bound to human EcOX40 detected by both indirect ELISA and flow cytometry were grown in 5 mL LB-A broth at 37 °C with 250 rpm shaking aeration overnight. The bacterial cells were harvested by centrifugation (4000×
g, 4 °C, 30 min) and the phagemids they contained were extracted by using a plasmid mini-extraction kit (Favogen, Ping-Tung, Taiwan). The isolated phagemid DNAs were Sanger-sequenced (1st Base). The nucleotide sequences were subjected to the CLC Main Workbench 8 program (Qiagen, Hilden, Germany); the nucleotide sequences of
huscfvs of individual
E. coli clones were deduced; and the CDRs and FRs of both VH and VL domains were analyzed using the IMGT/V-QUEST analysis tool, the International Immunogenetics Information, IMGT Systems (
https://academic.oup.com/nar/article/50/D1/D1262/6455007?login=false, accessed on 1 November 2019) [
47]. The
huscfv sequences of all the
E. coli clones were multiply aligned by using Clustal Omega [
48] and compared.
2.7. Homology Modeling and Intermolecular Docking to Predict the OX40 Ectodomain Sites Bound by the HuscFvs
The three-dimensional (3D) structures of the HuscFvs were modeled by subjecting the HuscFv amino acid sequences to the online I-TASSER server [
49]. The geometric and physical qualities of the I-TASSER-predicted 3D structures were improved by making them become closer to their native state using the ModRefinder [
50] and the Fragment-Guided Molecular Dynamics (FG-MD) [
51] systems, respectively. For the human EcOX40 homology model, the protein crystal structure PDB 2HEV [
52] which was built from amino acids 29-214 of human OX40 (NP_003318.1) was used as the template for the I-TASSER modeling of the full-length EcOX40 protein. The human EcOX40 model derived by I-TASSER was also refined by the ModRefinder and FG-MD systems. For intermolecular docking, the modeled HuscFvs were docked against the modeled human EcOX40 by using the ClusPro Protein Docking server [
53]. The largest cluster size with minimal local energy and a near-native state of the protein conformations was chosen from about 30 models derived from the ClusPro for each docking. The protein structural models and the molecular interactions were built and visualized by using the Pymol software (The PyMOL Molecular Graphics System, Version 1.3r1edu, Schrodinger, LLC, New York, NY, USA) and BioVia (Discovery studio). Interactive residues and chemical bonds between the HuscFvs and the human EcOX40 were determined.
2.8. Large-Scaled Production of 6× His-Tagged-HuscFvs to EcOX40
The huscfvs coding for HuscFvs to EcOX40 were subcloned from pCANTAB5E phagemids into the pET23b+ expression vector and transformed into DH5α competent E. coli. Bacterial colonies carrying the recombinant 6× his-huscfv-pET23b+ plasmids were screened by direct colony PCR, and positive clones were cultured in 10 mL of LB broth containing 100 μg/mL ampicillin (LB-A) broth. The plasmids were extracted for DNA sequencing. The verified recombinant plasmids were transformed into competent BL21 (DE3) E. coli expression hosts. One positive transformant of each original clone was grown in LB-A broth at 37 °C with shaking aeration (250 rpm) overnight. The overnight culture (100 μL) was added to 200 mL of fresh Lennox broth containing 100 μg/mL ampicillin (LN-A) and incubated with shaking at 250 rpm until the OD at 600 nm reached 0.3–0.5. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to the culture (0.1 mM final concentration) and the culture was incubated further for 5 h. The bacterial cells collected from each culture after centrifugation (4500× g, 4 °C, 15 min) were resuspended in 1 mL PBS and homogenized by sonication (30% amplitude) in an ice bath for 1 min. The clear supernatants were collected after centrifugation (12,000× g, 4 °C, 20 min). The HuscFvs in the supernatants were purified by using the TALON® Metal Affinity resin (Clontech Lab, Mountain View, CA, USA (presently Takara Bio USA)) and eluted with 150 mM imidazole solution into 1 mL fractions. The purified 6× His-tagged HuscFvs to EcOX40 were subjected to SDS-PAGE and Coomassie Brilliant Blue G250 (CBB) staining and Western blot analysis for verification.
2.9. Production of Human Bivalent HuscFv-Fcγ Antibodies to EcOX40
DNA sequences coding for HuscFvs (
huscfvs) of the selected
E. coli clones were codon-optimized for their expression in mammalian cells. The
huscfvs were analyzed for their codon adaptation index (CAI) and the nucleotides were changed by using the vector NTI software (Thermo Fisher Scientific) and the vector builder [
54]. The codon-optimized
huscfv sequences with
SacI and
BglII endonuclease restriction sites (for subsequent cloning) were synthesized (IDT, Coralville, IA, USA).
The huIgG1 Fc2-pINFUSE mammalian expression vector (Invivogen) was transformed into JM109 E. coli by heat-shock transformation; the transformed bacteria were spread onto an LB-Zeocin agar plate, and the plate was incubated at 37 °C overnight. Bacterial colonies carrying the recombinant huIgG1 Fc2-pINFUSE plasmids were screened by direct colony PCR using the HTL5UTR forward and EBV-RP reverse oligonucleotide primers. The plasmids from the PCR-positive E. coli clones were extracted using a plasmid extraction kit (GeneAid, New Taipei, Taiwan). The bacterial clones with the verified plasmids were grown individually in LB-Zeocin broth at 37 °C for 16 h and the plasmids were extracted from the bacteria. The codon-optimized huscfvs were digested with the SacI and BglII restriction enzymes and ligated to the similarly cut huIgG1 Fc2-pINFUSE plasmids vectors using T4 ligase. The recombinant plasmids were transformed into JM109 E. coli, and the transformed E. coli clones harboring plasmids with the inserted huscfvs were screened again via direct colony PCR as above. Positive clones were grown for plasmid extraction using a plasmid extraction kit (Favogen) and sequenced (1st Base). Escherichia coli clones with verified plasmids were grown and their plasmids were extracted and purified by using the EndoFree Maxi Plasmid kit (Tiangen Biotech)
For the production of human bivalent HuscFv-Fcγ antibodies (designated fusion antibodies), HEK293E suspension cells in IBA solution (IBA suspensions life sciences, Göttingen, Germany) were cultured in MEXi-CM serum-free medium (IBA) in Erlenmeyer culture flasks (Corning, Glendale, AZ, USA) at 37 °C in a 5% CO2 atmosphere with shaking aeration (125 rpm). The MEXi-CM serum-supplemented culture medium was gradually replaced with CDM4HEK293 serum-free medium (Cytiva) until the cells could grow well in the latter alone, which took 3–4 weeks. Log-phase-grown HEK293E cells were suspended in SFM4Transfx-293 transfection medium (Cytiva) (1 × 106 cells/mL); the cells were incubated overnight as above, and they were ready for transfection the next day. For the cell transfection, the huscfv-huIgG1 Fc2-pINFUSE vector was mixed with a transfection reagent (FectoPro, PolyPlus transfection, Illkirch, France). The ratio of plasmid DNA/transfection reagent/booster reagent was 1:1.5:1; the mixture was added to the HEK293E suspension cells, and the preparation was incubated as above for 4 h. The CDM4HEK293 medium was added, and the cells were incubated further for 7–10 days or until 60–75% of the cells were dead. The cell-spent medium containing the fusion antibodies was harvested. The fusion antibodies were purified by mixing the cell culture supernatant containing the fusion antibodies with 10× IgG binding buffer (200 mM sodium phosphate buffer, pH 7.0), filtered through a 0.45-μm membrane, and the preparations were passed through protein G column chromatography (Cytiva). Fractions (1 mL) were collected (ÄKTAprime, Cytiva) and the presence of the fusion antibodies was determined by SDS-PAGE and CBB staining.
2.10. Effective Concentration 50 (EC50) and Binding of the HuscFvs and Bivalent HuscFv-Fcγ Antibodies (Fusion Antibodies) to Human OX40
An indirect ELISA was performed to determine the effective concentration 50 (EC50) of the fusion antibodies (bivalent HuscFv-Fcγ) for purified EcOX40 from HEK293E suspension cells.
Binding of the fusion antibodies to OX40 on activated human T cells was performed using flow cytometry.
Co-immunoprecipitations using HEK-OX40 cells and flow cytometric analysis were performed to demonstrate binding of the HuscFvs and fusion antibodies to full-length human OX40.
For the indirect ELISA, purified EcOX40 from the culture supernatant of HEK293E suspension cells was dissolved in PBS (5 μg/mL). Next, 100 μL aliquots were added to each well of the EIA/RIA plate (Corning) and the plate was kept at 4 °C overnight. The coated wells were washed with PBS, and each well was blocked with 200 μL of 5% skim milk in PBS at 37 °C for 1 h. After washing, various concentrations of fusion antibodies (0.3125, 0.625, 1.25, 2.5, 5, 10, 20 and 40 μg/mL) in 100 μL PBS were added separately to the EcOX40-coated wells, kept at 37 °C for 3 h, washed with PBST, and 100 μL of horseradish peroxidase (HRP)-conjugated anti-human IgG Fc antibody (1:5000, Invitrogen) was added to each well. The plate was incubated at 37 °C for 1 h, washed, and 100 μL of the ABTS substrate (Seracare, Milford, MA, USA) was added to each well. The optical density at 405 nm of the content in each well was determined against blank (wells added with buffer instead of fusion antibodies) using the Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, Santa Clara, CA, USA). The effective concentration 50 (EC50) of the fusion antibodies was calculated.
The binding of the fusion antibodies to human EcOX40 on human T cells was also determined by flow cytometric analysis. Human T cells were isolated from PBMCs by magnetic-based negative selection using the EasySepTM human T cell enrichment kit (Stemcell technologies, Vancouver, BC, Canada). The T cells were cultured with 0.5 μg/mL of PHA-L mitogen for 3 days to stimulate OX40 expression. Activated T cells were collected, washed with PBS, resuspended at 1 × 105 cells in 25 μL of 20% FBS-PBS-0.02% NaN3 (blocking buffer), and placed on ice for 30 min. After that, 25 μL of 40 μg/mL of fusion antibodies were added to the cells, placed on ice for 30 min, and then washed with 2% FBS-PBS-0.02% NaN3 (FACS buffer) three times. The cells were counter-stained with mouse anti-OX40-Alexa Fluor 647 conjugate and anti-human IgG Fc-Brilliant violet 405 conjugate and placed on ice for 30 min. The stained cells were then washed with FACS buffer, fixed with 1% paraformaldehyde in PBS, and assessed for EcOX40 binding of the fusion antibodies by flow cytometry (BD LSRFortessa, BD biosciences).
Binding of the HuscFvs/fusion antibodies to human OX40 was determined via a co-immunoprecipitation assay. The HEK-OX40 cells (5 × 106 cells) that expressed human OX40 were suspended in 500 μL of PBS, 2 μg of HuscFvs (6× His-tagged-HuscFvs) or fusion antibodies were added, and the cells were placed on a rocker at 4 °C for 3 h. The cells were washed with cold PBS five times. The cell pellets were then lysed with 500 μL of 1% Triton-X 100-Tris lysis buffer containing a protease inhibitor cocktail (Sigma-Aldrich) on ice for 1 h and centrifuged (15,000× g, 4 °C, 30 min). The clear cell lysate was collected, mixed with 2 μg of anti-6× His antibody and placed on a rocker at 4 °C for 3 h. Two microliters of protein G magnetic beads (Genscript) were added into the mixture and placed on the rocker at 4 °C for 1 h. The mixture-containing tube was placed on a magnetic stand and the beads were washed thoroughly with 1 mL of cold 1% Triton-X 100 Tris lysis buffer. The proteins were eluted from the beads by adding reducing sample buffer, boiled for 5 min, and subjected to SDS-PAGE and Western blotting. Proteins coprecipitated with OX40 were detected by using the rabbit anti-human OX40 antibody (clone BLR042F, 1:5000, Abcam) in the Western blot analysis.
2.11. NF-κB Assay for Determining the Ability of HuscFvs to EcOX40 to Stimulate OX40 Signaling
Preliminary experiments were performed by using the NF-κB assay to select HuscFvs that have OX40-stimulatory potential.
HEK-OX40 cells were seeded into a well of a 6-well-culture plate (5 × 105 cells/well) and incubated at 37 °C in a 5% CO2 incubator overnight. The lipofectamine™ 3000 transfection reagent (Thermo Fisher Scientific) was prepared: in one test tube, 1 μL of P3000TM, 25 μL of Opti-MEM™I reduced serum, and 1 μg of pNL3.2.NF-κB-RE[NlucP/NF-κB-RE/Hygro] vector (Promega, Madison, WI, USA) were added and mixed; in another test tube, 1.5 μL of Lipo3000 was mixed with 23.5 μL of Opti-MEM™ I reduced serum. Thereafter, the contents of the two test tubes were mixed well, and kept at 25 °C for 15 min. The mixture was gently added dropwise to the HEK-OX40 cells in the well and the cells were incubated at 37 °C in a humidified CO2 incubator for 72 h. The HEK-OX40 cells that contained the pNL3.2.NF-κB-RE[NlucP/NF-κB-RE/Hygro] plasmids were designated “HEK-OX40 reporter cells”.
For the NF-κB assay, HEK-OX40 reporter cells were seeded into wells of a 96-well white plate (15,000 cells/well) and incubated at 37 °C in a 5% CO2 incubator overnight. Various concentrations of purified 6× His-tagged HuscFvs (5, 10, 20 and 40 μg/mL) were added to the HEK-OX40 reporter cells. Mouse anti-His antibody (at half the concentration of the added 6× His-tagged HuscFvs) was added into each well to cross-link the HuscFvs that bound to EcOX40 on the cells to induce OX40 ligation/clustering. The HEK-OX40 reporter cells incubated with mouse anti-His antibody (Biolegend) served as the background control (no HuscFvs/crosslinked HuscFvs); HEK-OX40 reporter cells incubated with 2 ng/mL of human TNF-α served as the positive control for the NF-κB reporter system. After 6 h of treatments, 100 μL of the Nano-Glo substrate (Promega) was added into each well, and luciferase activity was measured by using the BioTek Synergy H1 Multimode Reader (BioTek, Santa Clara, CA, USA).
2.12. Preparation of Primary T Cells
Human PBMCs were isolated from the buffy coat of heparinized whole blood after Ficoll-Hypaque (Histopaque-1077; Sigma-Aldrich) density-gradient centrifugation. The PBMCs were washed twice with Dulbecco’s phosphate-buffered saline (DPBS), counted, and resuspended in 2% FBS-1 mM EDTA-DPBS (sorting medium) at 5 × 107 cells/mL. T cells were isolated from the PBMCs by magnetic-based negative selection using the EasySepTM human T cell enrichment kit (Stemcell technologies) according to the manufacturer’s protocol. The purity of the T cells (>95%) was checked by staining with anti-CD3-APC/Cy7 (clone SK7, Biolegend). The T cells were then resuspended in 10% FBS-AIM-V medium (Life Technologies, Thermo Fisher Scientific, Carlsbad, CA, USA).
2.13. T Cell Proliferation Assay
T cells (1 × 106 cells/mL in DPBS) were labeled with 5 μM of CellTrace Violet (Life Technologies) and incubated at 37 °C for 20 min. Medium containing 2% FBS was added (five times the original volume of the staining medium) and centrifuged once to remove free dye. The cell pellet was resuspended in 10% FBS-AIM-V medium (Life Technologies) at 1 × 106 cells/mL, and 100 μL aliquots of the cells were added into wells precoated with 100 μL of anti-CD3 antibody (0.5 μg/mL, clone UCHT-1, Biolegend) and fusion antibodies to OX40 (0.25, 0.5, 1.0 and 2.0 μg/mL); soluble anti-CD28 (1 μg/mL; clone CD28.2, Biolegend) was added to the wells containing T cells. T cells in culture medium alone served as the non-stimulated cell control. The cells were then kept at 37 °C in a CO2 incubator for 5 days. They were then harvested and the reduction of CellTrace Violet (indicating cell proliferation) was assessed via flow cytometry (BD FACSymphony A1, BD biosciences).
2.14. Detection of Intracellular Cytokines and Granzyme B
Human T cells in 10% FBS-AIM-V medium (1 × 106 cells/mL) were stimulated with immobilized anti-CD3 antibody and fusion antibodies and soluble anti-CD28 as above for 2 days. The cytokine blocker Brefeldin A (Biolegend) was added to the cells in each well 12 h prior to measuring the intracellular cytokines and granzyme B. The total T cells (CD3+ cells) were also stained for CD4+ and CD8+ subpopulations. In brief, 1 × 106 cells were resuspended in 50 μL of 10% human AB serum-FACS buffer and placed on ice for 30 min. The cells were stained with a cocktail of antibodies (Biolegend) against CD3-APC/Cy7 (clone SK7), CD4-Alexa Fluor 488 (clone SK3), and CD8-PE (clone SK1) and placed on ice for 30 min. After washing, the stained cells (CD3+ T cell and CD4+ and CD8+ T subpopulations) were then fixed with 4% paraformaldehyde-DPBS at RT for 15 min, washed twice with DPBS, and incubated in intracellular staining permeabilization wash buffer (Biolegend) for 20 min at RT. Cytokine/granzyme B-specific antibodies (Biolegend), i.e., anti-IL-2-Brilliant Violet 421 (clone MQ1-17H12), anti-IFN-γ-Brilliant Violet 421 (clone 4S.B3), anti-TNF-α-Brilliant Violet 421 (clone Mab11) or anti-granzyme B-Alexa Fluor 647 (clone QA16A02) were added to the permeabilized cells and incubated at RT for 30 min. The stained cells were washed with intracellular staining permeabilization wash buffer and the presence of intracellular cytokines/granzyme B was assessed by flow cytometry (BD FACSymphony A1, BD biosciences).
2.15. Determination of the Enhancing Effect of the Fusion Antibodies on T Cell Survival
To determine the effect of fusion antibodies on T cell survival, T cells (1 × 106 cells/mL of 10% FBS-AIM-V medium) that had been activated with immobilized anti-CD3 antibody and fusion antibodies in the presence of soluble anti-CD28 were cultured at 37 °C in CO2 incubator for 5 days. The cells were harvested, washed twice with DPBS, then stained with 0.5 µM ethidium homodimer D-1 (EthD-1) (Invitrogen) and 400 nM Apotracker green (Biolegend). Percentages of living cells, total dead cells, necrotic cells, and early/late apoptotic cells were assessed via flow cytometry (BD FACSymphony A1, BD biosciences) and compared with the CD3-CD28-activated T cells in the medium alone (no immobilized fusion antibodies)
2.16. Preparation of Monocyte-Derived Dendritic Cells (MoDCs)
Human PBMCs were suspended in sorting medium (2% FBS-1 mM EDTA-DPBS) at 5 × 10
7 cells/mL. Monocytes were isolated from the PBMCs by magnetic-based negative selection using the EasySep Human Monocyte Enrichment kit (Stemcell technologies) according to the manufacturer’s protocol. The purity of the monocytes was checked by staining them with anti-CD14-fluorescein isothiocyanate (FITC) (clone QA19A47, Biolegend). The purity of CD14-positive cells was higher than 90%. Monocytes were then resuspended in 10% FBS-AIM-V medium (Life Technologies) at 1 × 10
6 cells/mL. The monocytes (500 μL) were cultured in 24-well flat-bottomed tissue culture plates. For the differentiation of monocytes into moDCs, the cell culture medium was supplemented with 800 units/mL of recombinant human GM-CSF and 400 units/mL of recombinant human IL-4 (Stemcell technologies) (differentiation medium). Cell-spent differentiation medium was replaced with 50% fresh differentiation medium on day 3 and cultured further for 2 more days. Thereafter, the cells were cultured in maturation medium (medium supplemented with 400 ng/mL CD40L (Biolegend) and 100 units/mL TNF-α (Stemcell technologies)) for 2 days [
55,
56]. The cells were harvested and checked for expression of the immunophenotypic markers, CD14 and CD83, by staining with anti-CD14-FITC and anti-CD83-Alexa Fluor 647 (clone HB15e, Biolegend) and analysis via flow cytometry (BD FACSymphony A1, BD biosciences).
2.17. Preparation of Tumor Cell Lysate
The ovarian cancer cells, SK-OV-3, from a confluent culture were harvested and washed twice with PBS. The cells were resuspended to a density of 1 × 10
7 cells/mL in sterile PBS and kept in a water bath at 42 °C for 1 h and 37 °C for 2 h. Then, they were lysed by rapid freezing in liquid nitrogen and thawing in a 37 °C water bath for 4–5 cycles [
57]. The preparation was centrifuged (10,000×
g, 4°C, 30 min), and the clear lysates were collected and passed through a 0.2 μm filter. The protein concentration of the lysate was determined using the BCA protein assay (Bio-Rad, Munich, Germany).
2.18. Preparation of Tumor-Lysate-Pulsed DCs and Tumor-Antigen-Primed T Cells
Monocyte-derived DCs (1 × 105 cells) in 1 mL 10% FBS-AIM medium were added to the ovarian cancer cell lysate (100 μg/mL) and incubated at 37 °C in aCO2 incubator for 12 h. MHC class-I partially matched human T cells were added to the tumor-lysate-pulsed DCs at a ratio of 5:1, and the cells were cultured for 7 days. On days 3 and 5, 50% of the spent culture medium was replaced with fresh medium containing low doses of 10 units/mL IL-2 (Stemcell technologies) and 5 ng/mL IL-7 (Stemcell technologies). On day 5, the cells were observed under an inverted microscope, and sample was taken to check for the expression of OX40 by staining with an antibody cocktail containing anti-CD3-APC/Cy7, anti-CD4-Alexa Fluor 488, anti-CD8-PE/Dazzle 594, and anti-OX40-Alexa Fluor 647, followed by analysis via flow cytometry. On day 7, the tumor-antigen-primed T cells were harvested, washed twice with PBS, and resuspended in fresh 10% FBS-AIM-V medium (1 × 106 cells/mL).
2.19. Cytotoxicity Assay
After stimulation with the tumor-lysate-pulsed DCs, the tumor-antigen-primed T cells were additionally stimulated by incubating with anti-EcOX40 fusion antibodies for 24 h. The calcein-release assay was used for determining the ovarian tumor cell lysis caused by the fusion-antibody-stimulated tumor-antigen-primed T cells in comparison with the tumor-antigen-primed T cells without fusion antibody stimulation (control tumor antigen primed-T cells). Ovarian cancer cells were labeled with 1 μM of calcein-AM (Invitrogen), and 1 × 10
4 labeled cells were seeded into individual wells of a black 96-wellplate for 12 h. The fusion-antibody-stimulated tumor-antigen-primed T cells and control cells were added separately to the calcein-labeled ovarian cancer cells at target/effector (T:E) ratios of 1:5, 1:10, and 1:20. The cells of all treatments were cultured at 37 °C in CO
2 incubator for 24 h. After incubation, 100 μL of the supernatant from each well (triplicate wells for each treatment) was collected, and the fluorescence intensity of the released calcein was measured using the BioTek Synergy H1 Multimode Reader (BioTek). Two independent and reproducible experiments were performed using T cells of two blood donors whose MHC class-I partially matched that of the ovarian cancer cells. Specific lysis of the cancer cells was calculated as follows:
2.20. Statistical Analysis
Data obtained from independent experiments were analyzed using Kruskal–Wallis and Dunn’s multiple comparison tests using GraphPad Prism, version 10 (GraphPad Software Inc., San Diego, CA, USA). Two-tailed p values of less than 0.05 were considered significant (*, p < 0.05; **, p < 0.01).
4. Discussion
An investigation into the role of the immune system in fighting against tumors was embarked on in the late 18th century [
58,
59]. During the early stages of tumor initiation, naïve T cells are primed and activated by tumor immunogenic antigens in the draining lymph nodes from where tumor-specific T cells recirculate back to destroy the cancer. Advanced cancers with highly infiltrated T cells into the TME have a good prognosis, i.e., enhanced survival [
60]. Both CD4
+ and CD8
+ T cells are tumor destructors. Th1 cells secrete cytokines (IL-2, TNF-α, and IFN-γ) that stimulate M1 macrophages, NK cells, and CD8
+ T cells to increase the respective anti-tumor activities and prolong cell survival [
61]. Utilizing a cell–cell contact-dependent mechanism, the tumor-antigen-primed CD8
+ T cells (CTLs) directly kill the target cancer cells through specialized exocytosis toward the immunological synapse to deliver (in a paracrine manner) death-inducing granules containing granzymes, perforin, cathepsin C, and granulysin. One CTL (as well as one NK cell) can kill multiple targets via different pathways of cytotoxicity (e.g., caspase-mediated apoptosis, cell membrane disruption, microtubule disruption, formation of GSDM pores in the membrane, and others) in a subsequential manner [
62,
63,
64]. The CTLs also indirectly kill the tumor via the secretion of cytokines, especially IFN-γ [
65]. Despite a multitude of opposing immunological factors (innate and adaptive), tumors manage to escape the host immune control and manifest clinically; they conduct this using several strategies, including the induction of an immunosuppressive TME; expression of immunosuppressive surface proteins, i.e., immune-checkpoint proteins (PD-1, CTLA-4, and LAG-3 that suppress effector cell activities) and ectonucleotidases (CD39/CD73 that convert ATP to immunosuppressive adenosine); downregulation of MHC-class I, FAS, and TRAIL to escape cell killing; secretion of immunosuppressive cytokines and molecules (IL-10, TGF-β, VEGF, idolamine-2,3-dioxygenase, lactate, prostaglandin-E2, galectin 1, and arginase); as well as by the creation of a hypoxic environment [
66].
Insightful information on tumor immune suppressive factors and mechanisms has led to a cancer treatment revolution toward immunotherapy. Among the immunotherapeutic approaches, blocking the immune checkpoints has gained the most success in the treatment of many cancers. Nowadays, several immune-checkpoint blockers have been developed for therapeutic purposes [
67]. Nevertheless, there are limitations in using the immune-checkpoint blockers. While a given blocker may be relatively effective for one cancer, it may fail to yield a beneficial outcome for other cancers [
68]. Immune-checkpoint blocking alone is not sufficient to promote tumor regression in most patients [
69]. In addition, repeated dosing with the checkpoint blockers may cause adverse immune-related reactions and toxicities to several organs/tissues, e.g., endocrine, cardiac, pulmonary, and gastro-intestinal [
67].
Other than immune-checkpoint blockers, the stimulation of T cell costimulatory receptors, particularly tumor necrosis factor receptor (TNFR) superfamily members, e.g., OX40/CD134, 4-1BB, causes T cell proliferation, migration, survival, and activation, which can lead to tumor regression [
70]. Co-receptor stimulators (agonists) can be used in combined immunotherapy with checkpoint blockers [
71] or as an adjunctive, neo adjunctive, or induction therapy with conventional cancer treatments, i.e., surgery, radiation, and chemotherapy [
72]. The proteins of the TNFR superfamily are generally characterized by an extracellular portion composed of several cysteine-rich domains (CRDs) for interacting with their natural ligands. The interaction of cell-surface TNFRSF proteins with their natural ligands leads to receptor homo-trimerization and clustering that triggers downstream signaling, including the activation of the nuclear factor-κB (NF-κB) and nuclear-factor-activated protein (NFAT) transcription factors, which drives immune cell activation, proliferation, and survival. For OX40, the extracellular portion (EcOX40) is composed of four CRDs (CRD1-CRD4) and the stalk. The natural ligand (OX40L) binds CRDs 1, 2, and 3 of the OX trimer to cause receptor clustering and activation of the NF-κB signaling cascade [
73]. Agonistic monoclonal antibodies (mAbs) in a variety of formats that target T cell costimulatory receptors have been developed for cancer therapy [
69]. Nevertheless, none of the OX40 agonists have reached the late phase of clinical trials [
44]. Proper clustering of the naturally expressed, spatially distant OX40 molecules on the T cell surface such that the cytoplasmic tails of OX40 can recruit TRAFs (TRAF2 and TRAF5) to form a hexameric complex is required for adequate downstream signaling for sequential transcription factor and gene activation [
44,
70]. A hexameric OX40L-TRAF2-IgG4Fc fusion protein (MEDI6383) that clustered OX40 molecules by Fc gamma receptors (FcγRs) on the surface of adjacent FcγR-positive cells was found to stimulate and induce NF-κB activity in OX40-expressing T cells, as well as induce Th1 proliferation and cytokine production, and resistance to Treg-mediated suppression [
39]. In this study, we generated novel OX40 agonistic antibodies in the form of bivalent fully human scFvs (HuscFvs) fused to a human IgG1 Fc fragment (fusion antibodies). The fusion antibodies by themselves could stimulate T cell proliferation, survival, and some effector functions, e.g., cytokine production, and they showed enhancement trends for other effector T cell activities like granzyme B production and the lysis of ovarian cancer cells that were used as a target cancer model.
The production of fully human antibodies to human proteins (own species) requires specialized strategies. To generate the effective fusion antibodies to human OX40, we started by producing fully human single-chain antibody fragments (HuscFvs) that bound to the OX40 ectodomain (EcOX40). A phage library that contained phage particles displaying a repertoire of HuscFvs [
45] was used as a tool for the production of the EcOX40-specific HuscFvs. The HuscFv phage display library that we used as a source of human OX40-specific HuscFvs in this study was generated from human immunoglobulin genes isolated from peripheral blood B cells of multiple healthy donors. All families and subfamilies of the genes coding for human VH (
vhs) and VL (
vls) domains were PCR-amplified using 48 pairs of degenerate oligonucleotide primers to
vhs (16 forward and three reverse primers) and 26 pairs of degenerate primers to
vls (13 forward and two reverse primers). The degenerate primers were chosen such that one
vh/vl cDNA template could yield multiple variants after PCR amplification; this was to increase the antibody gene diversity and the possibility of also obtaining gene sequences that encode antibodies (VHs/VLs) to human self-antigens that are absent in the normal peripheral blood B cell pool (like immunoglobulin genes during in-frame V
H-DJ
H rearrangements in pre-B cells and
IGK/IGL gene rearrangements in small pre-B II cells prior to positive selection and allelic exclusion in immature B cells) [
74]. The PCR-amplified
vh and
vl sequences were linked randomly via a polynucleotide linker to yield a repertoire of
vh-linker vl or
huscfvs. The
huscfv repertoire was cloned into the pCANTAB5E phagemid downstream of the phage gene
p3. The recombinant phagemids were introduced into competent TG1
E. coli and the phage-transformed
E. coli were grown and co-infected with a M13KO7 helper phage. The complete phage particles displaying the human scFvs (HuscFvs) as fusion partners of the P3 protein and containing the respective HuscFv genes (
huscfvs) in the phage genomes were recovered from the
E. coli culture supernatant (i.e., the HuscFv phage display library was obtained) [
45]. To select out phage clones displaying HuscFvs to the OX40 ectodomain (EcOX40), a commercialized recombinant EcOX40 that bound to OX40L and HEK-OX40 cells was used as a panning antigen. A total of 33 phage-transformed
E. coli clones produced HuscFvs that bound to the recombinant EcOX40 via indirect ELISA, but among them, HuscFvs of 12 clones bound also to the surface-exposed OX40 on HEK-OX40 cells. These results verified that the HuscFv phage display library does contain phage particles that display HuscFvs to human self-antigens. The same library was used successfully for selecting out phage clones that displayed HuscFvs to the human PIM2 kinase [
46]. Among the 12
E. coli clones that produced EcOX40-bound HuscFvs (by both indirect ELISA and HEK-OX40 cells), many clones were sibling clones; therefore, only seven clones (C2, D11, F8, C36, C41, C61, and C78) were studied further.
Computerized simulation was used to predict the region of the EcOX40 protein that is bound by the HuscFvs to select the HuscFvs with OX40-stimulation potential. Satisfactory 3D structures of the HuscFvs of individual
E. coli clones were built (i.e., the HuscFv residues in the most favored regions of the Ramachandran plots were higher than 90%) (
Table S2)) and used in the intermolecular docking with the full-length EcOX40 3D homology model [acquired by I-TASSER modeling using the crystal structure of PDB 2HEV [
52] that was built from amino acids 29-214 of human OX40 (NP_003318.1)). It was found that HuscFvs of six clones (clones C2, D11, F8, C36, C41, and C78) interacted with the cysteine-rich domain-2 (CRD2) of EcOX40, which has been shown to function in OX40 stimulation [
44], while the HuscFv of clone C62 formed a contact interface with CRD4 and the stalk of EcOX40, which do not participate in OX40 stimulation. Thus, HuscFvs of clones C2, D11, F8, C36, C41, and C78 were selected for further experiments.
The genes coding for HuscFvs (
huscfvs) of clones C2, D11, F8, C36, C41, and C78 were subcloned from phagemids into protein expression plasmid vectors for large-scale production of the HuscFvs. High yields of HuscFvs were obtained from the sibling clones of C2, D11, C36, and C41 after subcloning. HuscFvC2, HuscFvD11, HuscFvC36, and HuscFvC41 were subjected to primary testing for their ability to stimulate OX40 signaling by using HEK-OX40 reporter cells prior to performing experiments that used freshly isolated human blood cells. For this experiment, the monomeric HuscFvs that bound to OX40 on the surface of HEK-OX40 reporter cells were crosslinked by an anti-His antibody. HuscFvC2 and HuscFvC41 could increase the luciferase activity of the reported cells indicating that they could stimulate downstream OX40 signaling to activate the NF-κB transcription factor. Therefore, HuscFvC2 and HuscFvC41 were used for preparing bivalent HuscFv-IgG1 Fc (Fcγ) fusion proteins, designated C2Fc and C41Fc fusion antibodies, respectively. Before using these further, the antigenic specificity of the fusion antibodies had to be verified; these were found to retain the EcOX40-binding capability of the original HuscFvs as tested by both co-immunoprecipitation with OX40 in the HEK-OX40 lysate and with EcOX40 on the HEK-OX40 cells, as well as on stimulated human T cells by flow cytometry. The EC50 values of the C2Fc and the C41Fc were also monitored, and they were in a nanomolar range (81.53 and 236.96 nM, respectively), which may require affinity improvement [
75,
76], if higher affinity is needed for in vivo use.
Naturally, naïve T cells require at least two signals to sustain T cell activation. The first activation signal is from the TCR-CD3 complex, and the second signal is from the interaction of the T cell CD28 with a B7 ligand (CD80) expressed on antigen-presenting cells. OX40 is expressed only on the simulated (effector) T cells. Thus, in our in vitro experiments for testing the T cell activating activities of the fusion antibodies, peripheral blood T cells isolated from the healthy donors had to be stimulated with anti-CD3 (first signal) and anti-CD28 (second signal) to induce OX40 expression on the T cell surface before the fusion antibodies were added to the cells; then, the expected outcomes of different experiments were observed. The prior stimulation by anti-CD3 and anti-CD28 (not the natural ligands) before adding the fusion antibodies is a limitation of our experiments, as the level of stimulation is not known and uncontrollable; if the cells were activated to the maximal or near-maximal capacity of their response, the additional stimulatory/expected effect after adding the fusion antibodies may not be detected. Fortunately, in most experiments, additional effects after adding the fusion antibodies to the CD3-CD28-stimulated T cells were observed, including an enhancement of T cell proliferation (
Figure 7B–D), an ability to enhance cytokine production (
Figure 8), an enhancement of T cell survival, and a reduction in total T cell death (
Figure 9). Adding C41Fc to the CD3-CD28-activated total T cells and CD4
+ T cells caused a statistically significant increment in granzyme B production; nevertheless, the small scales of granzyme B graphs (y axes of
Figure 8D,H) suggest that the observed increases may not have a biological significance compared to the activated cells without the fusion antibodies. This may be due to the limitation of the in vitro condition of the experiment as stated above, or the amounts (and affinity) of the fusion antibodies may be suboptimal.
It is not possible to mimic the in vivo situation of cancer by the cell-based in vitro assay used in this study. Nevertheless, we successfully generated monocyte-derived DCs (MoDCs). The MoDCs were pulsed by an ovarian cancer-cell lysate and the pulsed DCs were used to stimulate the T cells of healthy donors. The addition of the fusion antibodies to tumor-antigen-primed T cells showed an enhancing trend in mediating the lysis of the cancer cells at an increasing T:E ratio. The relatively low enhancing activity of the fusion antibodies on the tumor-primed T cells might be due to their low target-binding affinity, which requires improvement (e.g., via CDR resurfacing); the number of the effector cells not being adequate (still too few); or because the MHCs of the effector cells (Thai donors) and the target cancer cells (Caucasian female with adenocarcinoma of the ovary) were only partially matched. Unfortunately, higher numbers of effector cells could not be tested due to the limitation in the whole blood volume from individual donors (ethical issue), and completely MHC-matched donors were not available. In real situations, the autologous target effector cell system exists, which might positively impact the capability of the fusion antibodies to enhance effector T cell activity.
Between the two fusion antibodies, C2Fc seemed to perform better than C41Fc in promoting T cell proliferation and survival and reducing T cell death (
Figure 7 and
Figure 9), as well as enhancing cytokine (IL-2, TNF-α and IFN-γ) production (
Figure 8). However, C41Fc seemed to perform better than C2Fc in inducing CD3-CD28-stimulated T cells (total T cell pool and CD4
+ T cells) to produce more granzyme B compared with stimulated cells without the addition of fusion antibodies (
Figure 8). Granzymes are serine proteases that play an important role in tumor cell killing by immune cells (CTL and NK). In addition to granzyme B, humans also have granzymes A, H, K, and M, the roles of which in the immune system are lesser known than that of granzyme B [
76]. The role of granzyme-producing CD4
+ T cells in antitumor immunity has emerged recently [
76]. By using single-cell RNA-sequencing (RNA-seq) technology, it has become clear that several CD4
+ T helper cell subtypes infiltrate into the tumor microenvironment (TME); these are cytolytic CD4
+ T cells that express high levels of granzymes and perforin and may contribute to the anti-cancer response [
77]. The novel multifaceted involvement of CD4
+ T cells in the anti-tumor immune response has been reviewed along with their old paradigm in cancer [
77]. Experiments using a fusion antibody made up of a hybrid HuscFvC2-HuscFvC41 linked to Fcγ to enhance T cell activities against tumors should be carried out.
In conclusion, novel OX40 agonistic antibodies that are fully human proteins in the form of a bivalent HuscFv-Fcγ have been generated. The fusion antibodies enhanced the proliferation, survival, and cytokine production of CD3-CD28-activated T cells. Under the limitation of the in vitro experiments, the fusion antibodies modestly enhanced granzyme B production from the activated T cells and showed trends in enhancing the anti-ovarian cancer activity of tumor-reactive T cells. Further step-by-step testing of fusion antibodies (after affinity improvement) will be a progression toward their use as an adjunctive agent in cancer immunotherapy.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}