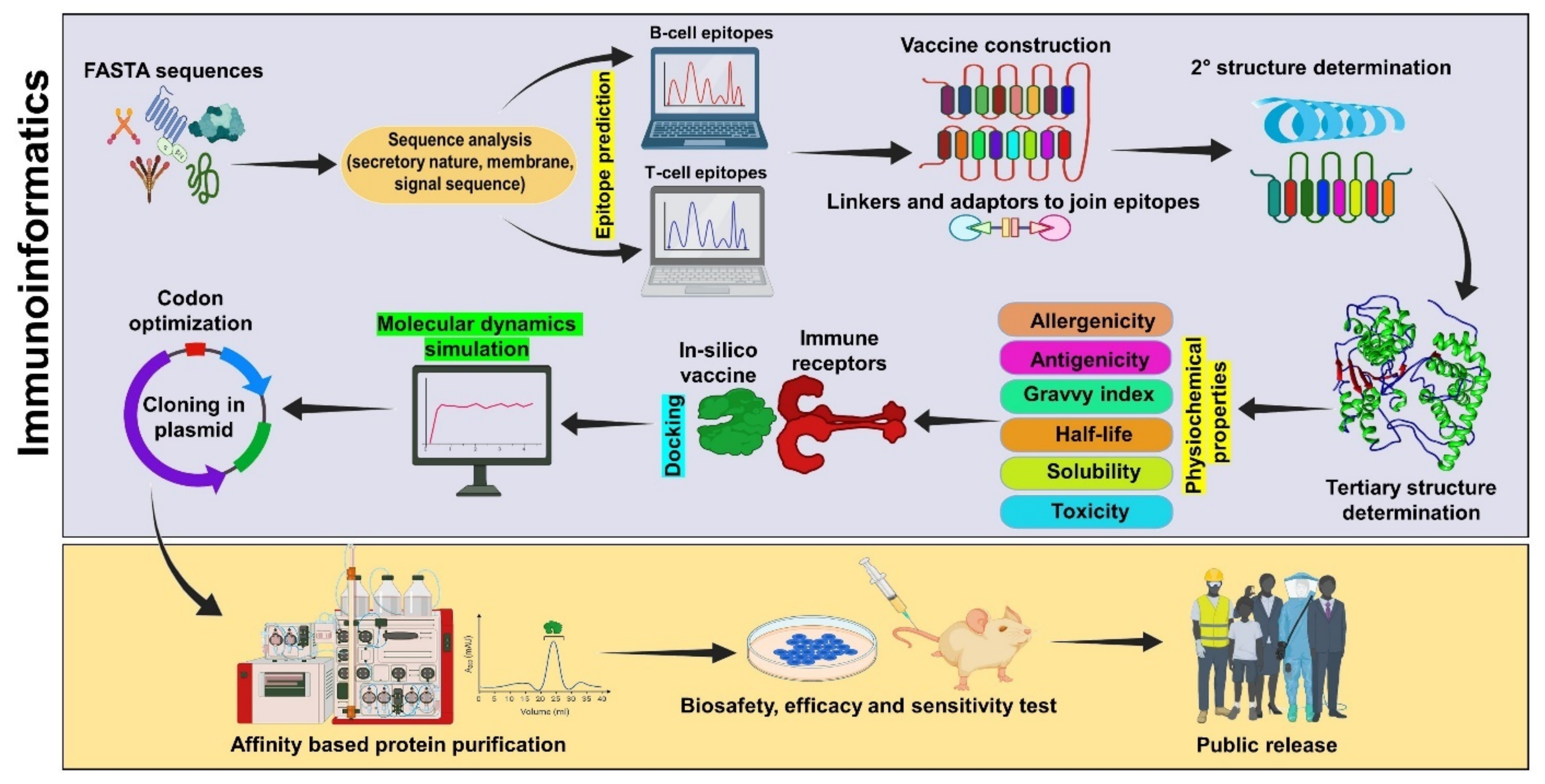
Immunoinformatics Approaches for Vaccine Design: A Fast and Secure Strategy for Successful Vaccine Development


{kind=link}
Author Contributions
Conflicts of Interest
References
- Black, S.; Bloom, D.E.; Kaslow, D.C.; Pecetta, S.; Rappuoli, R. Transforming vaccine development. Semin. Immunol. 2020, 50, 101413. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Usmani, S.S.; Agrawal, P.; Nagpal, G.; Gautam, A.; Raghava, G.P.S. Novel in silico tools for designing peptide-based subunit vaccines and immunotherapeutics. Brief. Bioinform. 2017, 18, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Arora, N.; Keshri, A.K.; Kaur, R.; Rawat, S.S.; Prasad, A. Immunoinformatic Approaches for Vaccine Designing for Pathogens with Unclear Pathogenesis. Methods Mol. Biol. 2022, 2412, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Joon, S.; Singla, R.K.; Shen, B. Vaccines and Immunoinformatics for Vaccine Design. Adv. Exp. Med. Biol. 2022, 1368, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Galanis, K.A.; Nastou, K.C.; Papandreou, N.C.; Petichakis, G.N.; Pigis, D.G.; Iconomidou, V.A. Linear B-Cell Epitope Prediction for In Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface. Int. J. Mol. Sci. 2021, 22, 3210. [Google Scholar] [CrossRef]
- Akbari, E.; Kardani, K.; Namvar, A.; Ajdary, S.; Ardakani, E.M.; Khalaj, V.; Bolhassani, A. In silico design and in vitro expression of novel multiepitope DNA constructs based on HIV-1 proteins and Hsp70 T-cell epitopes. Biotechnol. Lett. 2021, 43, 1513–1550. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Devi, Y.D.; Goswami, H.B.; Konwar, S.; Doley, C.; Dolley, A.; Devi, A.; Chongtham, C.; Dowerah, D.; Biswa, V.; Jamir, L.; et al. Immunoinformatics mapping of potential epitopes in SARS-CoV-2 structural proteins. PLoS ONE 2021, 16, e0258645. [Google Scholar] [CrossRef]
- Khalid, K.; Irum, S.; Ullah, S.R.; Andleeb, S. In-Silico Vaccine Design Based on a Novel Vaccine Candidate Against Infections Caused by Acinetobacter baumannii. Int. J. Pept. Res. Ther. 2022, 28, 16. [Google Scholar] [CrossRef]
- Ding, M.D.; Wang, H.N.; Cao, H.P.; Fan, W.Q.; Ma, B.C.; Xu, P.W.; Zhang, A.Y.; Yang, X. Development of a multi-epitope antigen of S protein-based ELISA for antibodies detection against infectious bronchitis virus. Biosci. Biotechnol. Biochem. 2015, 79, 1287–1295. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.B.; Niu, S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. 2021, 11, 1249. [Google Scholar] [CrossRef]
- Kaur, R.; Arora, N.; Jamakhani, M.A.; Malik, S.; Kumar, P.; Anjum, F.; Tripathi, S.; Mishra, A.; Prasad, A. Development of multi-epitope chimeric vaccine against Taenia solium by exploring its proteome: An in silico approach. Expert Rev. Vaccines 2020, 19, 105–114. [Google Scholar] [CrossRef]
- Kaur, R.; Arora, N.; Rawat, S.S.; Keshri, A.K.; Singh, N.; Show, S.K.; Kumar, P.; Mishra, A.; Prasad, A. Immunoinformatics driven construction of multi-epitope vaccine candidate against Ascaris lumbricoides using its entire immunogenic epitopes. Expert Rev. Vaccines 2021, 20, 1637–1649. [Google Scholar] [CrossRef]
- Schaap-Johansen, A.L.; Vujović, M.; Borch, A.; Hadrup, S.R.; Marcatili, P. T Cell Epitope Prediction and Its Application to Immunotherapy. Front. Immunol. 2021, 12, 712488. [Google Scholar] [CrossRef]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, S.N.H.; Jain, A.; Haq, E.; Mehbodniya, A.; Webber, J. Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review. Pathogens 2022, 11, 146. [Google Scholar] [CrossRef]
- Zhao, T.; Cheng, L.; Zang, T.; Hu, Y. Peptide-Major Histocompatibility Complex Class I Binding Prediction Based on Deep Learning With Novel Feature. Front. Genet. 2019, 10, 1191. [Google Scholar] [CrossRef] [Green Version]
- Laimer, J.; Lackner, P. MHCII3D-Robust Structure Based Prediction of MHC II Binding Peptides. Int. J. Mol. Sci. 2020, 22, 12. [Google Scholar] [CrossRef]
- Degoot, A.M.; Chirove, F.; Ndifon, W. Trans-Allelic Model for Prediction of Peptide:MHC-II Interactions. Front. Immunol. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Lin, L.; Ting, S.; Yufei, H.; Wendong, L.; Yubo, F.; Jing, Z. Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2. Virus Res. 2020, 288, 198082. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirohi, P.R.; Gupta, J.; Somvanshi, P.; Prajapati, V.K.; Grover, A. Multiple epitope-based vaccine prediction against SARS-CoV-2 spike glycoprotein. J. Biomol. Struct. Dyn. 2022, 40, 3347. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Rampogu, S.; Lee, G.; Hong, J.C.; Lee, K.W. Exploring the Binding Interaction of Raf Kinase Inhibitory Protein With the N-Terminal of C-Raf Through Molecular Docking and Molecular Dynamics Simulation. Front. Mol. Biosci. 2021, 8, 655035. [Google Scholar] [CrossRef]
- Hasanzadeh, S.; Habibi, M.; Shokrgozar, M.A.; Ahangari Cohan, R.; Ahmadi, K.; Asadi Karam, M.R.; Bouzari, S. In silico analysis and in vivo assessment of a novel epitope-based vaccine candidate against uropathogenic Escherichia coli. Sci. Rep. 2020, 10, 16258. [Google Scholar] [CrossRef]
- Shehata, M.M.; Mahmoud, S.H.; Tarek, M.; Al-Karmalawy, A.A.; Mahmoud, A.; Mostafa, A.; Elhefnawi, M.M.; Ali, M.A. In Silico and In Vivo Evaluation of SARS-CoV-2 Predicted Epitopes-Based Candidate Vaccine. Molecules 2021, 26, 6182. [Google Scholar] [CrossRef]
- Shabani, S.H.; Kardani, K.; Milani, A.; Bolhassani, A. In Silico and in Vivo Analysis of HIV-1 Rev Regulatory Protein for Evaluation of a Multiepitope-based Vaccine Candidate. Immunol. Investig. 2022, 51, 1–28. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [Green Version]
- Oli, A.N.; Obialor, W.O.; Ifeanyichukwu, M.O.; Odimegwu, D.C.; Okoyeh, J.N.; Emechebe, G.O.; Adejumo, S.A.; Ibeanu, G.C. Immunoinformatics and Vaccine Development: An Overview. ImmunoTargets Ther. 2020, 9, 13–30. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rawat, S.S.; Keshri, A.K.; Kaur, R.; Prasad, A. Immunoinformatics Approaches for Vaccine Design: A Fast and Secure Strategy for Successful Vaccine Development. Vaccines 2023, 11, 221. https://doi.org/10.3390/vaccines11020221
Rawat SS, Keshri AK, Kaur R, Prasad A. Immunoinformatics Approaches for Vaccine Design: A Fast and Secure Strategy for Successful Vaccine Development. Vaccines. 2023; 11(2):221. https://doi.org/10.3390/vaccines11020221
Chicago/Turabian StyleRawat, Suraj Singh, Anand Kumar Keshri, Rimanpreet Kaur, and Amit Prasad. 2023. "Immunoinformatics Approaches for Vaccine Design: A Fast and Secure Strategy for Successful Vaccine Development" Vaccines 11, no. 2: 221. https://doi.org/10.3390/vaccines11020221