


Neutrophil Extracellular Traps and NLRP3 Inflammasome: A Disturbing Duo in Atherosclerosis, Inflammation and Atherothrombosis

 ,
,  , and
, and 
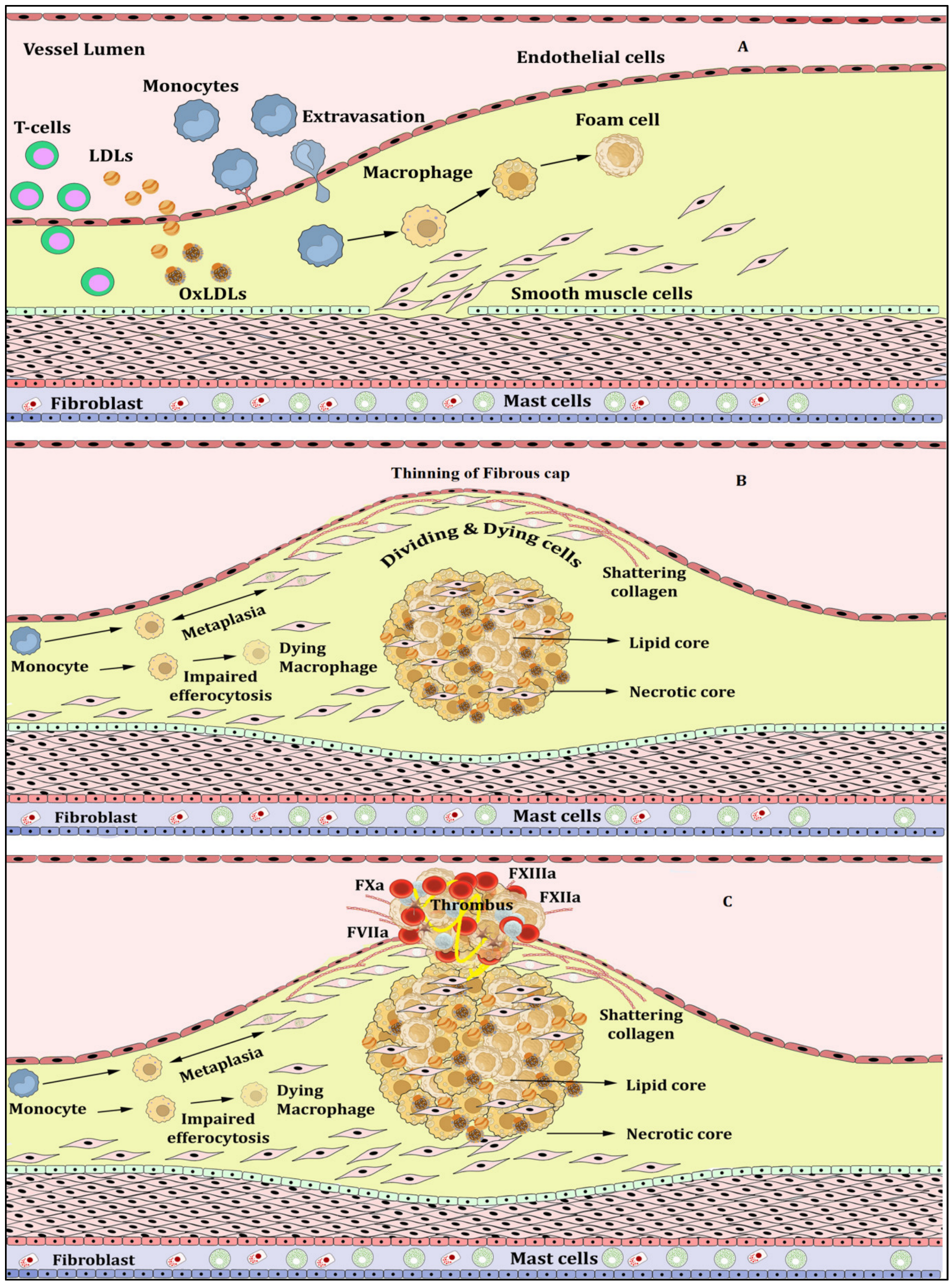
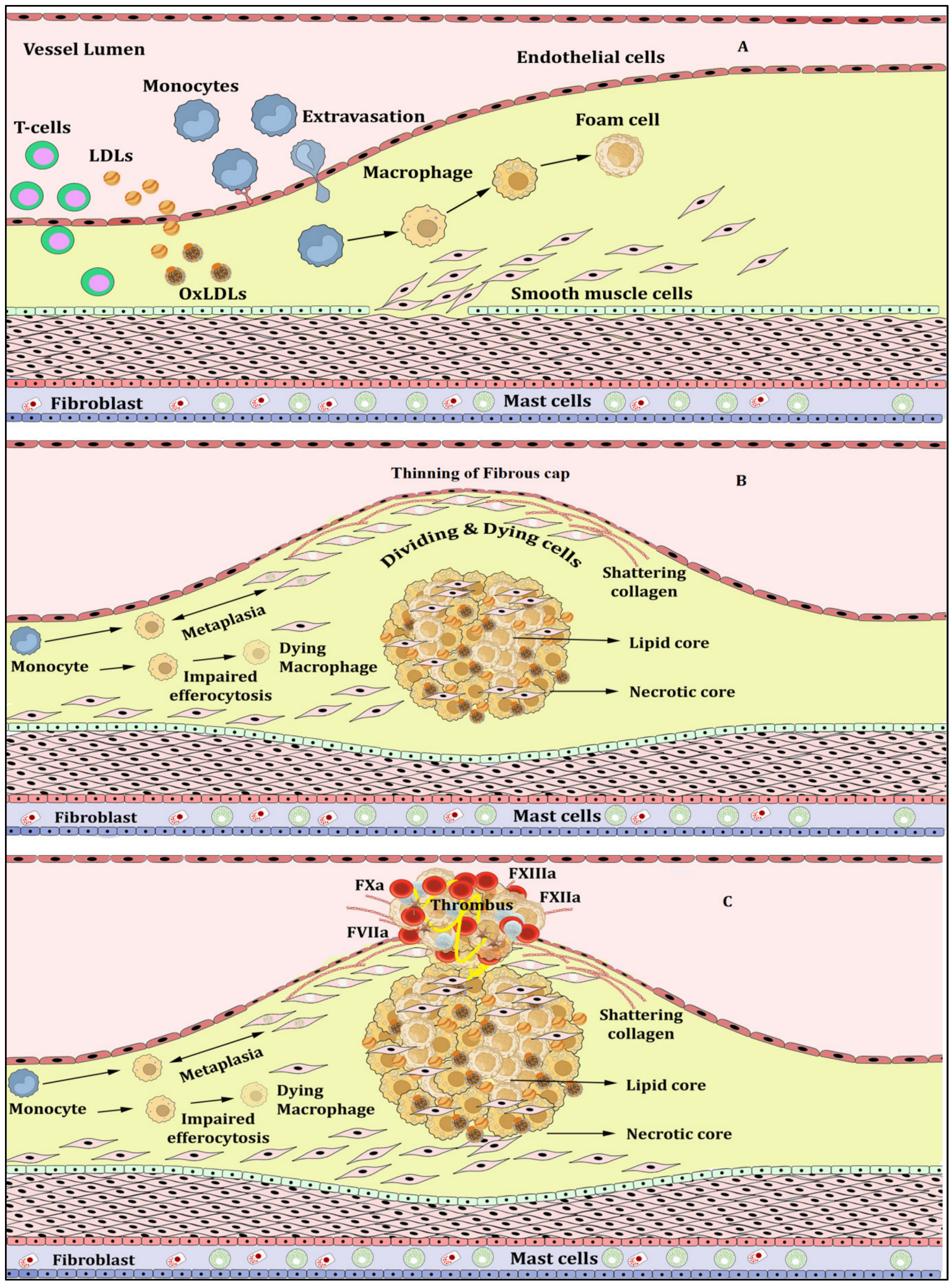{kind=link}
{kind=link}
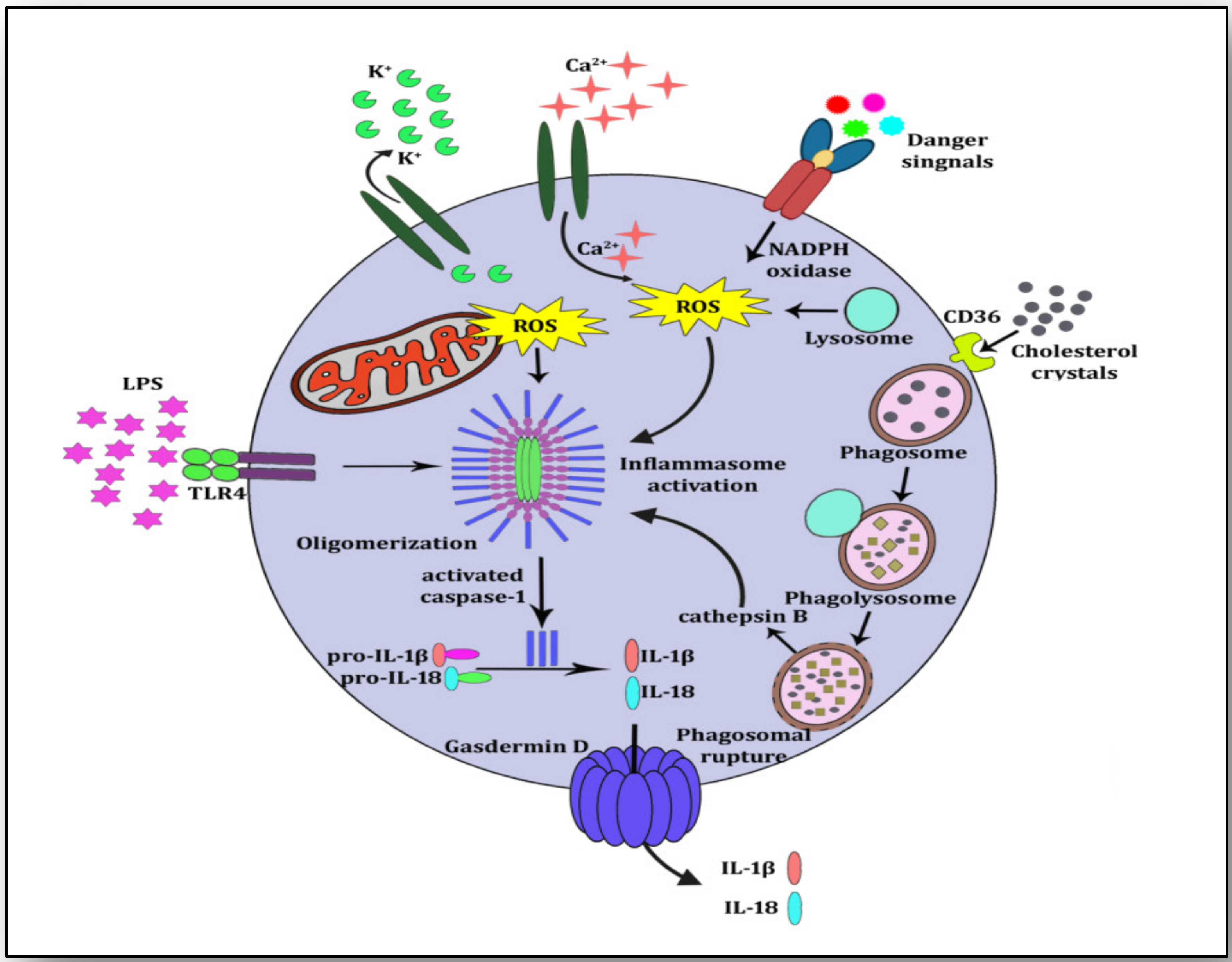{kind=link}
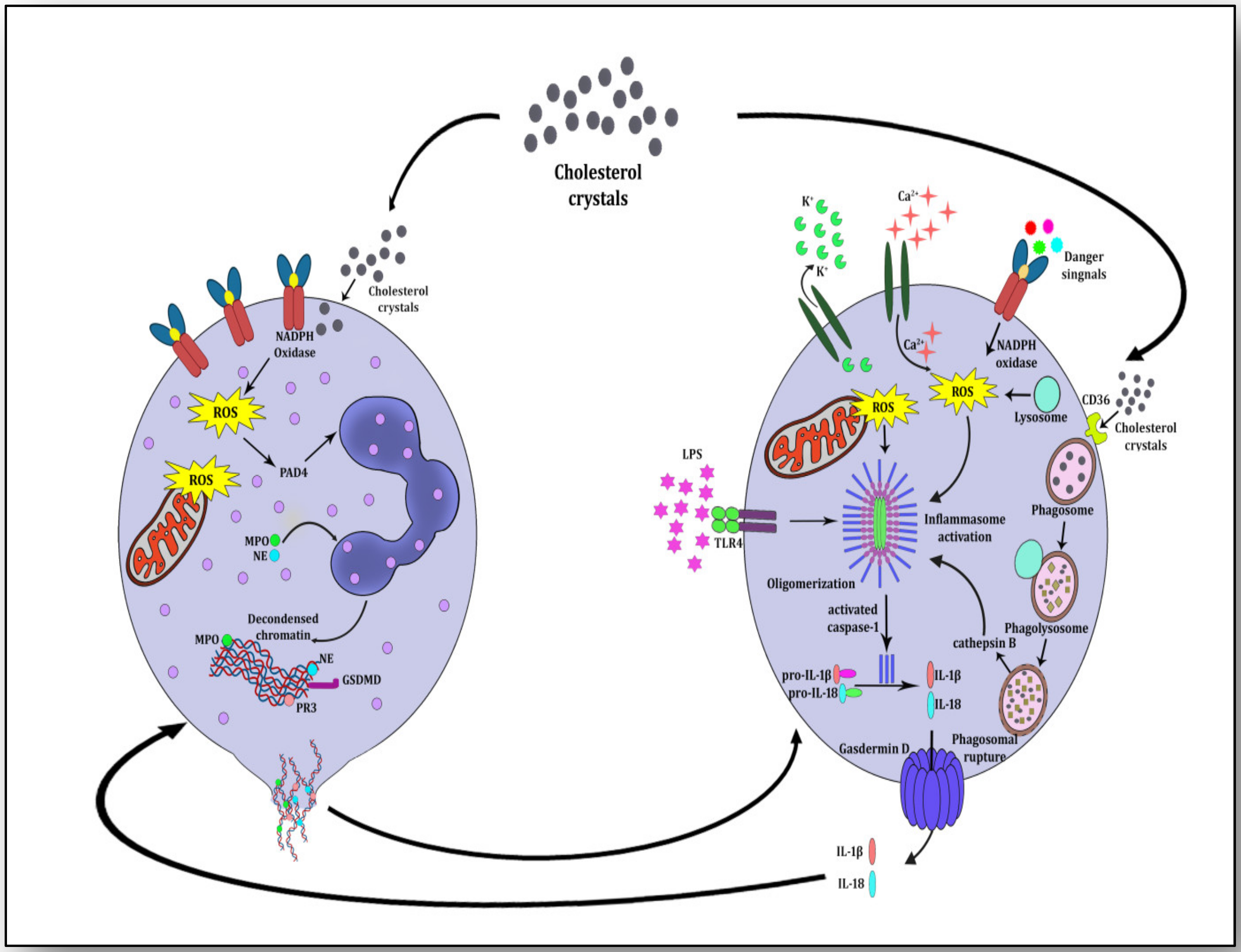{kind=link}
Abstract
:1. Introduction
Atherosclerosis, Inflammation and Atherothrombosis
2. NETs, NETosis and Atherosclerosis
3. NLRP3 Inflammasome Activation and Atherosclerosis
4. NETosis-NLRP3 Inflammasome Activation Link: A Maleficent Crosstalk
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle factors and high-risk atherosclerosis: Pathways and mechanisms beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell. Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Dabagh, M.; Jalali, P.; Tarbell, J.M. The transport of LDL across the deformable arterial wall: The effect of endothelial cell turnover and intimal deformation under hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H983–H996. [Google Scholar] [CrossRef]
- Noble, M.I.M.; Drake-Holland, A.J.; Vink, H. Hypothesis: Arterial glycocalyx dysfunction is the first step in the atherothrombotic process. QJM 2008, 101, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl. Acad. Sci. USA 2020, 117, 7326–7337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines induced neutrophil extracellular traps formation: Implication for the inflammatory disease condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.-X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: How to proceed? Nat. Rev. Cardiol. 2022, 19, 522–542. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Damascena, H.L.; Silveira, W.A.A.; Castro, M.S.; Fontes, W. Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate). Cells 2022, 11, 2889. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell. Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthopoulou, A.; Gkaliagkousi, E.; Lazaridis, A.; Arelaki, S.; Pateinakis, P.; Ntinopoulou, M.; Mitsios, A.; Antoniadou, C.; Argyriou, C.; Georgiadis, G.S.; et al. Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension. JCI Insight 2021, 6, e148668. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.-H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef] [Green Version]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschonas, I.C.; Tselepis, A.D. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis 2019, 288, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Zou, X.; Wang, D.; Liu, Y.; Zhang, Z.; Guo, J.; Lu, R.; Huang, W.; Wang, S.; Li, Z.; et al. IRGM/Irgm1 deficiency inhibits neutrophil-platelet interactions and thrombosis in experimental atherosclerosis and arterial injury. Biomed. Pharmacother. 2023, 158, 114152. [Google Scholar] [CrossRef]
- Dou, H.; Kotini, A.; Liu, W.; Fidler, T.; Endo-Umeda, K.; Sun, X.; Olszewska, M.; Xiao, T.; Abramowicz, S.; Yalcinkaya, M.; et al. Oxidized Phospholipids Promote NETosis and Arterial Thrombosis in LNK(SH2B3) Deficiency. Circulation 2021, 144, 1940–1954. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Mahley, R.W.; Huang, Y.; Weisgraber, K.H. Putting cholesterol in its place: ApoE and reverse cholesterol transport. J. Clin. Invest. 2006, 116, 1226–1229. [Google Scholar] [CrossRef]
- Allam, G.; Abdel-Moneim, A.; Gaber, A.M. The pleiotropic role of interleukin-17 in atherosclerosis. Biomed. Pharmacother. 2018, 106, 1412–1418. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Yang, X.; Li, Y.; Wang, X.; Tan, S.; Chen, F. LPS enhances platelets aggregation via TLR4, which is related to mitochondria damage caused by intracellular ROS, but not extracellular ROS. Cell. Immunol. 2018, 328, 86–92. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S.; Lappegård, K.T.; Brekke, O.-L.; Lambris, J.D.; Damås, J.K.; et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef] [Green Version]
- Nadra, I.; Mason, J.C.; Philippidis, P.; Florey, O.; Smythe, C.D.W.; McCarthy, G.M.; Landis, R.C.; Haskard, D.O. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: A vicious cycle of inflammation and arterial calcification? Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Pazár, B.; Ea, H.-K.; Narayan, S.; Kolly, L.; Bagnoud, N.; Chobaz, V.; Roger, T.; Lioté, F.; So, A.; Busso, N. Basic calcium phosphate crystals induce monocyte/macrophage IL-1β secretion through the NLRP3 inflammasome in vitro. J. Immunol. 2011, 186, 2495–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Liu, J.; Pang, X.; Wang, S.; Zhao, J.; Zhang, X.; Feng, L. Palmitic acid exerts pro-inflammatory effects on vascular smooth muscle cells by inducing the expression of C-reactive protein, inducible nitric oxide synthase and tumor necrosis factor-α. Int. J. Mol. Med. 2014, 34, 1706–1712. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.-S. Caspase-11 non-canonical inflammasome: A critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 2017, 152, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Lim, G.B. IL-1 signalling in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 200. [Google Scholar] [CrossRef] [PubMed]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Hettwer, J.; Hinterdobler, J.; Miritsch, B.; Deutsch, M.-A.; Li, X.; Mauersberger, C.; Moggio, A.; Braster, Q.; Gram, H.; Robertson, A.A.B.; et al. Interleukin-1β suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc. Res. 2022, 118, 2778–2791. [Google Scholar] [CrossRef]
- Ren, X.-S.; Tong, Y.; Ling, L.; Chen, D.; Sun, H.-J.; Zhou, H.; Qi, X.-H.; Chen, Q.; Li, Y.-H.; Kang, Y.-M.; et al. NLRP3 Gene Deletion Attenuates Angiotensin II-Induced Phenotypic Transformation of Vascular Smooth Muscle Cells and Vascular Remodeling. Cell. Physiol. Biochem. 2017, 44, 2269–2280. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L. Interleukin-18: A potent pro-inflammatory cytokine in atherosclerosis. Cardiovasc. Res. 2012, 96, 172–175; discussion 176–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.-G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.V.; Kaul, D.; Kumar, M.J.M.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E-/- mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020, 41, 2153–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; St Hilaire, C.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Ogura, Y.; Sutterwala, F.S.; Flavell, R.A. The inflammasome: First line of the immune response to cell stress. Cell 2006, 126, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzer, P.; Negro, R.; Fukui, S.; di Meglio, L.; Aymonnier, K.; Chu, L.; Cherpokova, D.; Gutch, S.; Sorvillo, N.; Shi, L.; et al. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front. Immunol. 2021, 12, 683803. [Google Scholar] [CrossRef]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, T.-L.; Chen, Y.-M.; Tang, K.-T.; Chen, P.-K.; Liu, H.-J.; Chen, D.-Y. MicroRNA-223 inhibits neutrophil extracellular traps formation through regulating calcium influx and small extracellular vesicles transmission. Sci. Rep. 2021, 11, 15676. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Yang, P.; Gao, M.; Yu, T.; Shi, Y.; Zhang, M.; Yao, M.; Liu, Y.; Zhang, X. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin. Sci. 2019, 133, 565–582. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.; Kumar, N.; Singh, M.; Kaur, M.; Singh, G.; Narang, A.; Kanwal, A.; Sharma, K.; Singh, B.; Napoli, M.D.; et al. Neutrophil Extracellular Traps and NLRP3 Inflammasome: A Disturbing Duo in Atherosclerosis, Inflammation and Atherothrombosis. Vaccines 2023, 11, 261. https://doi.org/10.3390/vaccines11020261
Singh P, Kumar N, Singh M, Kaur M, Singh G, Narang A, Kanwal A, Sharma K, Singh B, Napoli MD, et al. Neutrophil Extracellular Traps and NLRP3 Inflammasome: A Disturbing Duo in Atherosclerosis, Inflammation and Atherothrombosis. Vaccines. 2023; 11(2):261. https://doi.org/10.3390/vaccines11020261
Chicago/Turabian StyleSingh, Puneetpal, Nitin Kumar, Monica Singh, Manminder Kaur, Gurjinderpal Singh, Amit Narang, Abhinav Kanwal, Kirti Sharma, Baani Singh, Mario Di Napoli, and et al. 2023. "Neutrophil Extracellular Traps and NLRP3 Inflammasome: A Disturbing Duo in Atherosclerosis, Inflammation and Atherothrombosis" Vaccines 11, no. 2: 261. https://doi.org/10.3390/vaccines11020261
APA StyleSingh, P., Kumar, N., Singh, M., Kaur, M., Singh, G., Narang, A., Kanwal, A., Sharma, K., Singh, B., Napoli, M. D., & Mastana, S. (2023). Neutrophil Extracellular Traps and NLRP3 Inflammasome: A Disturbing Duo in Atherosclerosis, Inflammation and Atherothrombosis. Vaccines, 11(2), 261. https://doi.org/10.3390/vaccines11020261