
Immunoinformatics Study: Multi-Epitope Based Vaccine Design from SARS-CoV-2 Spike Glycoprotein
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. The Sequences of SARS-CoV-2 Spike Glycoprotein in Indonesia
2.3. CTL Epitope Prediction
2.4. HTL Epitope Prediction
2.5. B-Cell Epitope
2.6. IFN-γ Prediction
2.7. Antigenicity, Allergenicity, Toxicity, and Population Coverage Analysis
2.8. Multiepitope Vaccine Construction, Modeling, and Validation
2.9. Molecular Docking
2.10. Molecular Dynamics
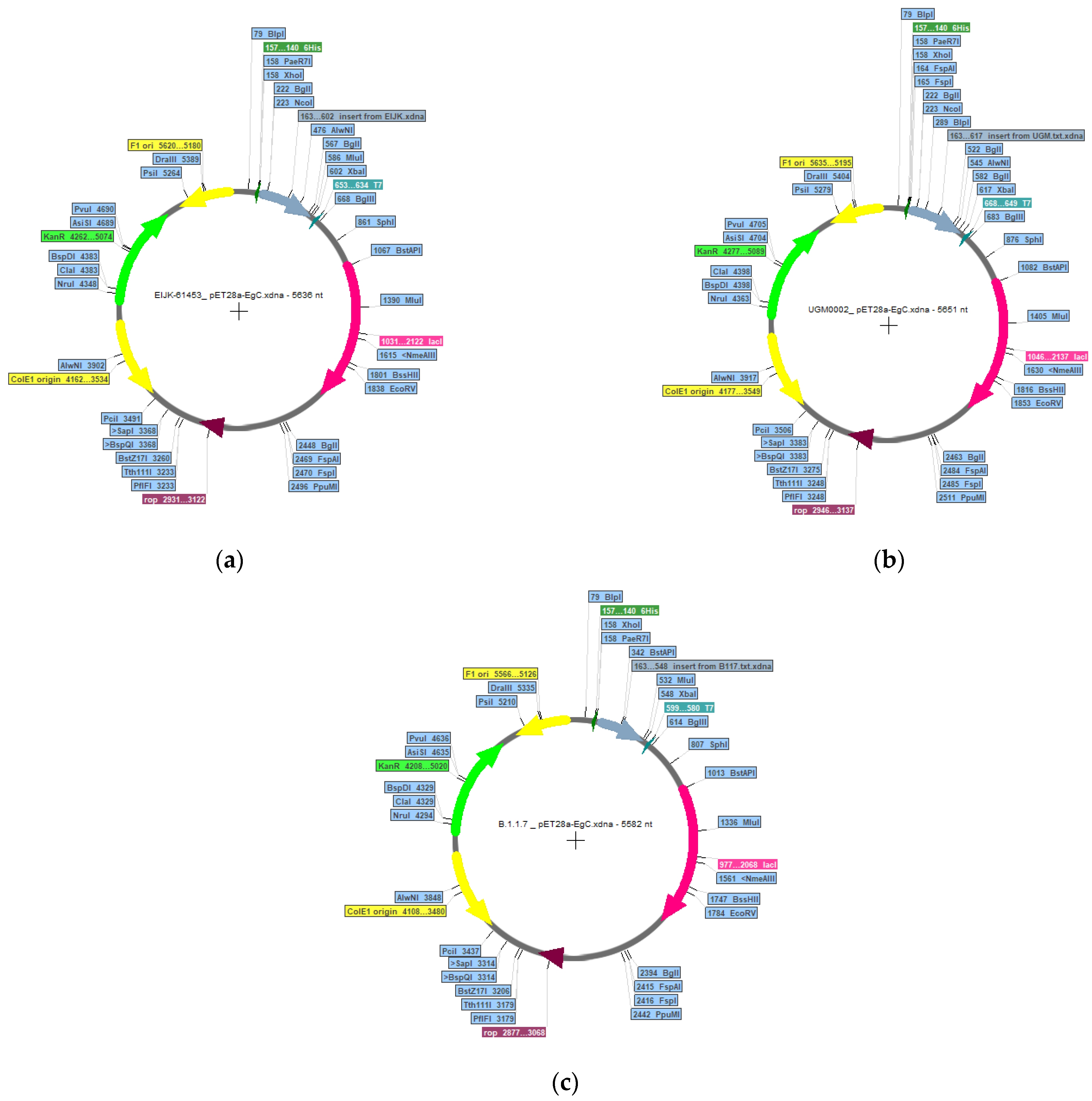
2.11. In Silico Cloning and Immune Simulation
3. Results and Discussion
3.1. Evolutionary Relationship of SARS-CoV-2 in Indonesia and Detection of Its Mutation Site
3.2. Identification and Selection of T-Cell Epitopes
3.3. Identification and Selection of B-Cell Epitopes
3.4. Indonesian Population Coverage from Selected Epitopes
3.5. Epitope Analysis in Human Peptide
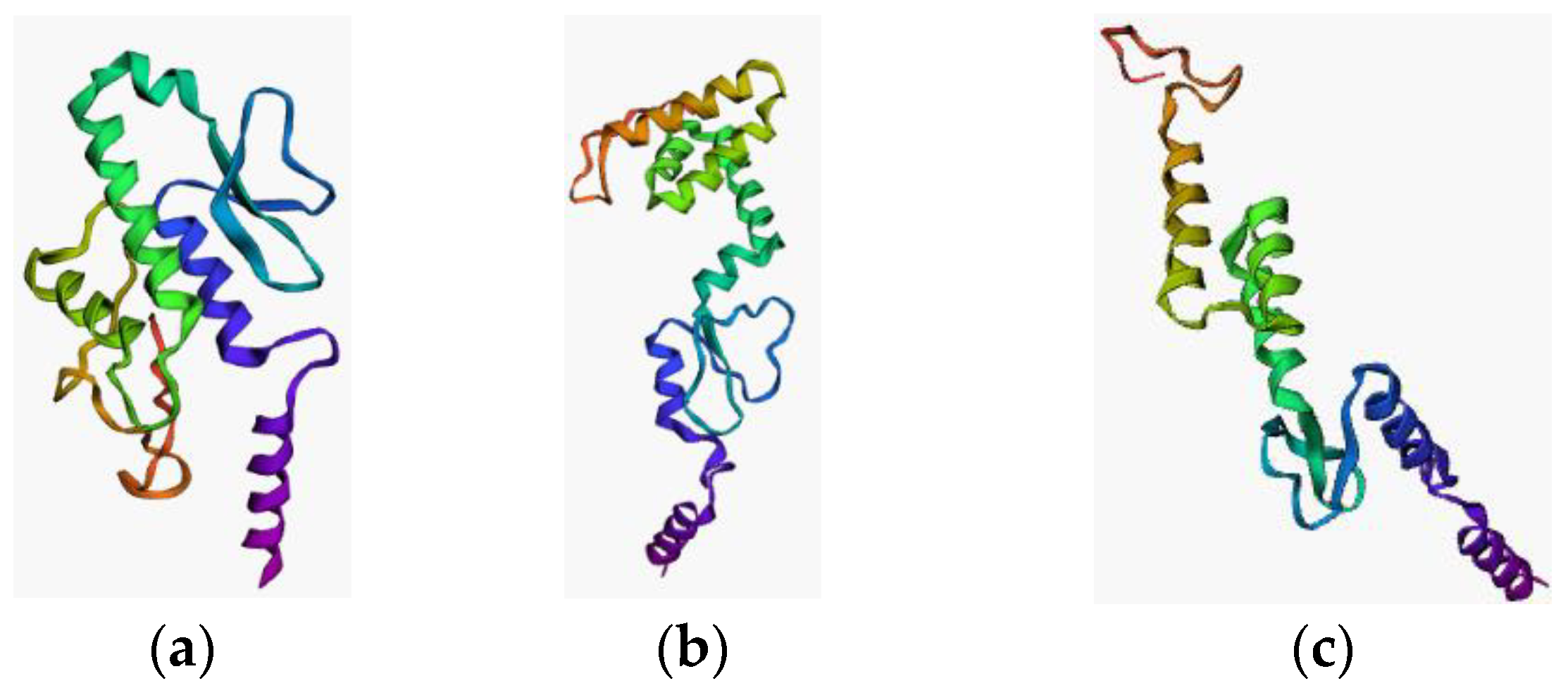
3.6. Vaccine Construction and Its Validation
3.7. Docking Study of the Constructed Vaccine Model against TLR-3
3.8. Docking Study of the Constructed Vaccine Model against TLR-4
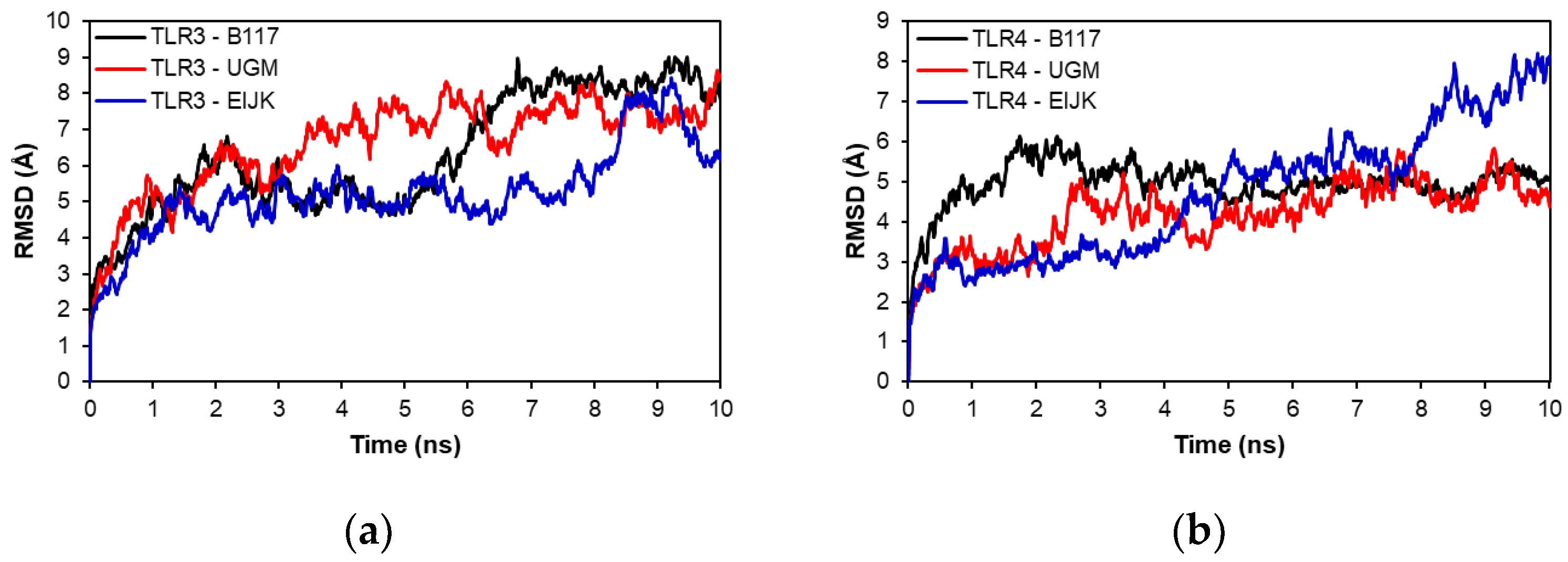
3.9. Molecular Dynamics
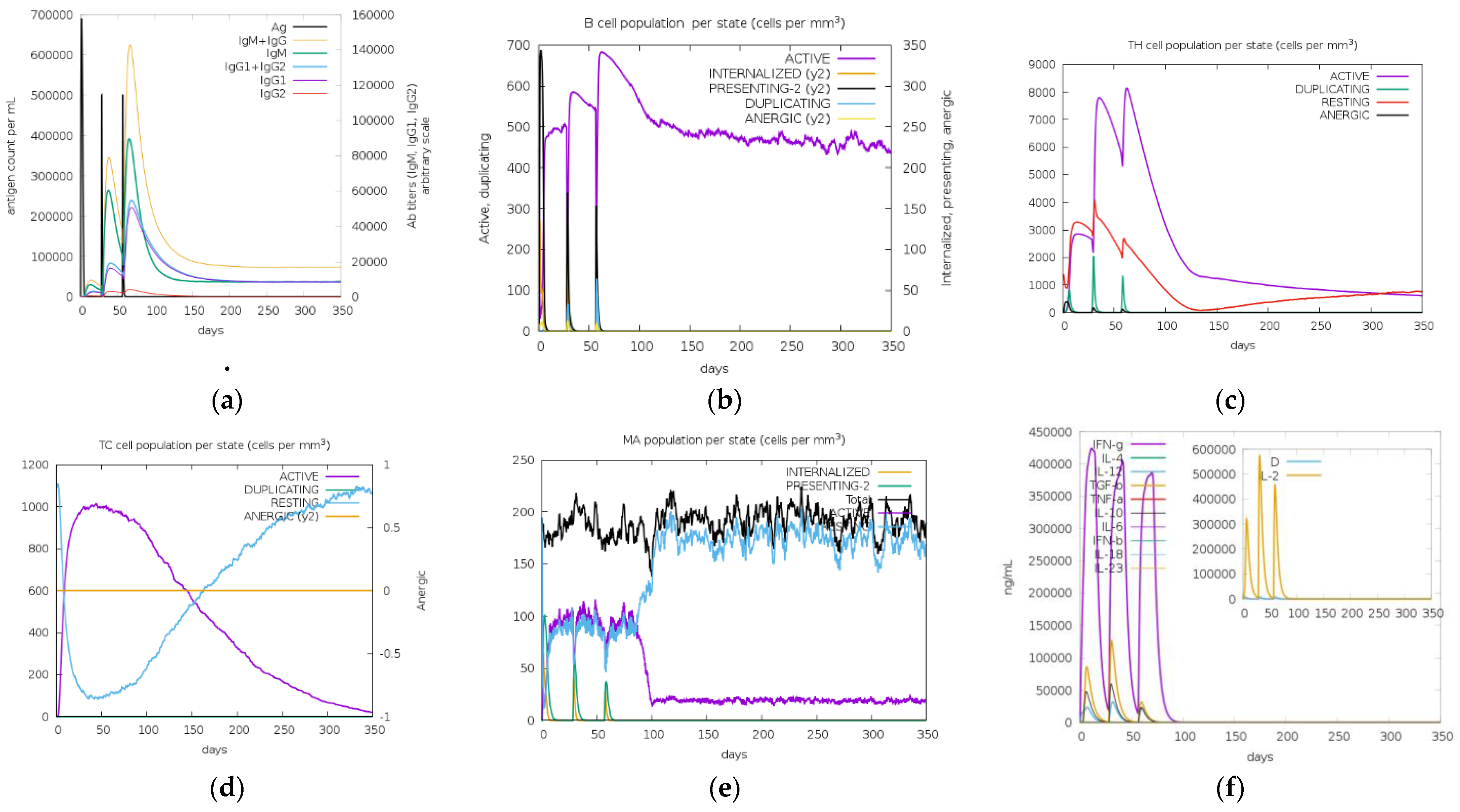
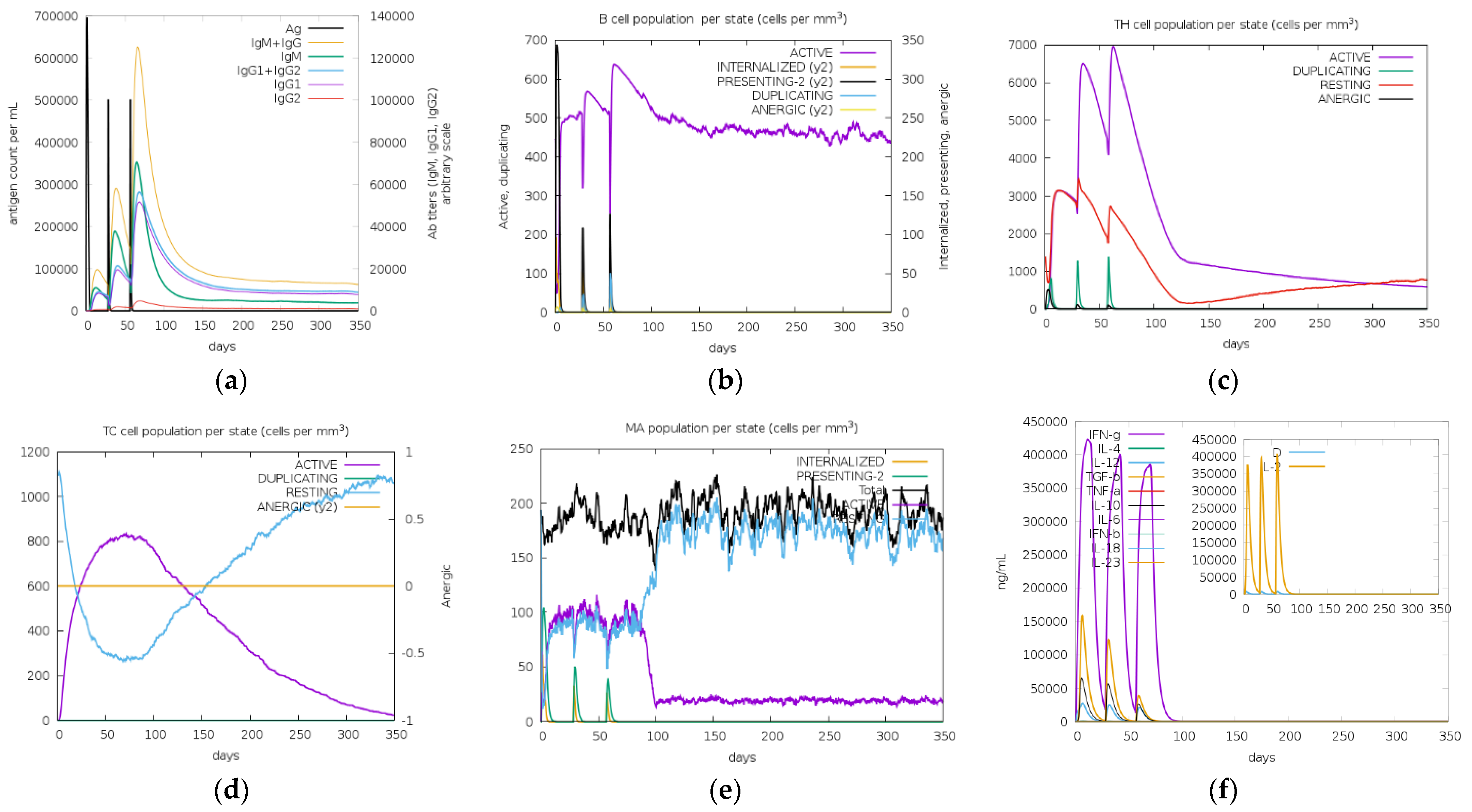
3.10. In Silico Cloning and Immune Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ceraolo, C.; Giorgi, F.M. Genomic variance of the 2019-nCoV coronavirus. J. Med. Virol. 2020, 92, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Pillay, T.S. Gene of the Month: The 2019-NCoV/SARS-CoV-2 Novel Coronavirus Spike Protein. J. Clin. Pathol. 2020, 73, 366–369. [Google Scholar] [PubMed]
- Astuti, I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An Overview of Viral Structure and Host Response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharm. Sin. 2020, 41, 1141–1149. [Google Scholar]
- Li, W.; Li, L.; Sun, T.; He, Y.; Liu, G.; Xiao, Z.; Fan, Y.; Zhang, J. Spike Protein-Based Epitopes Predicted against SARS-CoV-2 through Literature Mining. Med. Nov. Technol. Devices 2020, 8, 100048. [Google Scholar] [CrossRef]
- Ahsan, M.A.; Liu, Y.; Feng, C.; Zhou, Y.; Ma, G.; Bai, Y.; Chen, M. Bioinformatics Resources Facilitate Understanding and Harnessing Clinical Research of SARS-CoV-2. Brief Bioinform. 2021, 22, 714–725. [Google Scholar]
- Bowick, G.C.; McAuley, A.J. Vaccine and Adjuvant Design for Emerging Viruses: Mutations, Deletions, Segments and Signaling. Bioeng. Bugs 2011, 2, 129–135. [Google Scholar] [CrossRef]
- Topuzoğullari, M.; Acar, T.; Arayici, P.P.; Uçar, B.; Uğurel, E.; Abamor, E.; Arasoğlu, T.; Turgut-Balik, D.; Derman, S. An insight into the epitope-based peptide vaccine design strategy and studies against COVID-19. Turk. J. Biol. 2020, 44, 215–227. [Google Scholar] [CrossRef]
- Oreshkova, N.; Molenaar, R.J.; Vreman, S.; Harders, F.; Munnink, B.B.O.; Hakze-van Der Honing, R.W.; Gerhards, N.; Tolsma, P.; Bouwstra, R.; Sikkema, R.S. SARS-CoV-2 Infection in Farmed Minks, The Netherlands, April and May 2020. Eurosurveillance 2020, 25, 2001005. [Google Scholar] [PubMed]
- Zhao, F.; Zai, X.; Zhang, Z.; Xu, J.; Chen, W. Challenges and Developments in Universal Vaccine Design against SARS-CoV-2 Variants. NPJ Vaccines 2022, 7, 167. [Google Scholar] [PubMed]
- Yousaf, M.; Ismail, S.; Ullah, A.; Bibi, S. Immuno-informatics profiling of monkeypox virus cell surface binding protein for designing a next generation multi-valent peptide-based vaccine. Front. Immunol. 2022, 13, 1035924. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2017, 15, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Gustiananda, M.; Sulistyo, B.P.; Agustriawan, D.; Andarini, S. Immunoinformatics Analysis of SARS-CoV-2 ORF1ab Polyproteins to Identify Promiscuous and Highly Conserved T-Cell Epitopes to Formulate Vaccine for Indonesia and the World Population. Vaccines 2021, 9, 1459. [Google Scholar] [PubMed]
- Febrianti, R.A.; Narulita, E. In-Silico Analysis of Recombinant Protein Vaccines Based on the Spike Protein of Indonesian SARS-CoV-2 through a Reverse Vaccinology Approach. J. Taibah Univ. Med. Sci. 2022, 17, 467–478. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data–from Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-Scale Validation of Methods for Cytotoxic T-Lymphocyte Epitope Prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2015, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Galarza, F.F.; McCabe, A.; Dos Santos, E.J.M.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Del Cid-Pavon, G.M.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2019, 48, D783–D788. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- White, R.E.; Manitpisitkul, P. Pharmacokinetic theory of cassette dosing in drug discovery screening. Drug Metab. Dispos. 2001, 29, 957–966. [Google Scholar]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for udp-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (auci/auc) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef]
- Zhao, W.; Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018, 14, e1006457. [Google Scholar] [CrossRef]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.V.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A Server for in Silico Prediction of Allergens. J. Mol. Model 2014, 20, 2278. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Ahammad, I.; Lira, S.S. Designing a Novel MRNA Vaccine against SARS-CoV-2: An Immunoinformatics Approach. Int. J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar]
- Sarkar, B.; Ullah, M.A.; Johora, F.T.; Taniya, M.A.; Araf, Y. Immunoinformatics-Guided Designing of Epitope-Based Subunit Vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology 2020, 225, 151955. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Karplus, P.A. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol. Concepts 2010, 1, 271–283. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Su, P. Stereochemistry of Polypeptide Chain Configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; Askins, J.; Hall, P.R.; Davies, D.R.; Segal, D.M. The dsRNA binding site of human Toll-like receptor 3. Proc. Natl. Acad. Sci. USA 2006, 103, 8792–8797. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.R.; Basu, M.; Swain, B.; Maharana, J.; Dikhit, M.R.; Jayasankar, P.; Samanta, M. Structural insights of rohu TLR3, its binding site analysis with fish reovirus dsRNA, poly I:C and zebrafish TRIF. Int. J. Biol. Macromol. 2012, 51, 531–543. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein–protein complexes. Elife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum New Things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J. PDBsum: Structural summaries of PDB entries. Protein Sci. 2017, 27, 129–134. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Castiglione, F.; Deb, D.; Srivastava, A.P.; Liò, P.; Liso, A. From Infection to Immunity: Understanding the Response to SARS-CoV2 through in-Silico Modeling. Front. Immunol. 2021, 12, 3433. [Google Scholar] [CrossRef]
- Hossain, S.; Hossan, M.I.; Mizan, S.; Moin, A.T.; Yasmin, F.; Akash, A.-S.; Powshi, S.N.; Hasan, A.R.; Chowdhury, A.S. Immunoinformatics approach to designing a multi-epitope vaccine against Saint Louis Encephalitis Virus. Informatics Med. Unlocked 2020, 22, 100500. [Google Scholar] [CrossRef]
- Ding, C.; He, J.; Zhang, X.; Jiang, C.; Sun, Y.; Zhang, Y.; Chen, Q.; He, H.; Li, W.; Xie, J. Crucial Mutations of Spike Protein on SARS-CoV-2 Evolved to Variant Strains Escaping Neutralization of Convalescent Plasmas and RBD-Specific Monoclonal Antibodies. Front. Immunol. 2021, 12, 3231. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N. The Effect of Spike Mutations on SARS-CoV-2 Neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E. Escape from Neutralizing Antibodies by SARS-CoV-2 Spike Protein Variants. Elife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Plasma Antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Andreano, E.; Rappuoli, R. SARS-CoV-2 escaped natural immunity, raising questions about vaccines and therapies. Nat. Med. 2021, 27, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Chesler, D.A.; Reiss, C.S. The role of IFN-γ in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002, 13, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 155–162. [Google Scholar] [CrossRef]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.J.; Soundararajan, V. SARS-CoV-2 Strategically Mimics Proteolytic Activation of Human ENaC. Elife 2020, 9, e58603. [Google Scholar] [CrossRef]
- Angileri, F.; Legare, S.; Gammazza, A.M.; de Macario, E.C.; Macario, A.J.; Cappello, F. Molecular mimicry may explain multi-organ damage in COVID-19. Autoimmun. Rev. 2020, 19, 102591. [Google Scholar] [CrossRef]
- Lucchese, G.; Flöel, A. Molecular Mimicry between SARS-CoV-2 and Respiratory Pacemaker Neurons. Autoimmun. Rev. 2020, 19, 102556. [Google Scholar] [CrossRef]
- Kanduc, D. From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry. Antibodies 2020, 9, 33. [Google Scholar] [CrossRef]
- Sanches, R.C.O.; Tiwari, S.; Ferreira, L.C.G.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.F.; Maia, E.H.B.; Taranto, A.G.; Kato, R.; Azevedo, V.A.C.; et al. Immunoinformatics Design of Multi-Epitope Peptide-Based Vaccine Against Schistosoma mansoni Using Transmembrane Proteins as a Target. Front. Immunol. 2021, 12, 621706. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.H.; Grisham, C.M. Biochemistry; Cengage Learning: Boston, MA, USA, 2016; ISBN 1305886895. [Google Scholar]
- Qamar, M.T.U.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- Donald, J.E.; Kulp, D.W.; DeGrado, W.F. Salt bridges: Geometrically specific, designable interactions. Proteins Struct. Funct. Bioinform. 2010, 79, 898–915. [Google Scholar] [CrossRef]
- Ghosh, T.; Garde, S.; García, A.E. Role of Backbone Hydration and Salt-Bridge Formation in Stability of α-Helix in Solution. Biophys. J. 2003, 85, 3187–3193. [Google Scholar] [CrossRef] [PubMed]
- Meuzelaar, H.; Vreede, J.; Woutersen, S. Influence of Glu/Arg, Asp/Arg, and Glu/Lys Salt Bridges on α-Helical Stability and Folding Kinetics. Biophys. J. 2016, 110, 2328–2341. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M.; Grimsley, G.R. Forces stabilizing proteins. FEBS Lett. 2014, 588, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Tsai, C.J.; Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. Des. Sel. 1997, 10, 999–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Zamane, S.; Alam Nobel, F.; Jebin, R.A.; Amin, M.B.; Somadder, P.D.; Antora, N.J.; Hossain, I.; Islam, M.J.; Ahmed, K.; Moni, M.A. Development of an in silico multi-epitope vaccine against SARS-CoV-2 by précised immune-informatics approaches. Inform. Med. Unlocked 2021, 27, 100781. [Google Scholar] [CrossRef]
- Hu, X.; Chakravarty, S.D.; Ivashkiv, L.B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008, 226, 41–56. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, J.-L. Changing the paradigm of IFN-γ at the interface between innate and adaptive immunity: Macrophage-derived IFN-γ. J. Leukoc. Biol. 2020, 108, 419–426. [Google Scholar] [CrossRef]
- Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol. Adv. 2012, 30, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Analysis | HLA Allele |
|---|---|
| CTL | A*11:01, A*24:02, A*02:01, A*02:03, A*02:06, A*01:01, A*03:01, A*26: 01, A*30:01, A*32:01, B*15:02, B*44:03, B*18:01, B*58:01, B*40:01, B*51:01, B*35:03, B*57:01, B*07:02, and B*15:17 |
| HTL | DRB1*12:02, DRB1*15:02, DRB1*07:01, DRB1*15:01, DRB1*03:01, DRB1*16:02, DRB1*09:01, DRB1*11:01, DRB1*04:05, DRB1*14:04, DRB1*10:01, DRB1*01:01, DRB1*13:02, DRB1*04:03, DRB1*04:02, DRB1*08:03, DRB1*14:01, DRB1*15:03, DRB1*12:01, DRB1*13:01, DRB1*08:02, DRB1*11:04, DPA1*01:03–DPB1*04:01, DPA1*02:01–DPB1*05:01, DPA1*01:03–DPB1*03:01, DPA1*01:03–DPB1*04:02, DPA1*01:03–DPB1*02:01, DPA1*02:01–DPB1*01:01, DPA1*02:01–DPB1*14:01, DQA1*06:01–DQB1*04:02, DQA1*01:01–DQB1*05:01, DQA1*01:02–DQB1*05:01, DQA1*01:02–DQB1*05:02, DQA1*01:02–DQB1*06:02, DQA1*02:01–DQB1*02:02, DQA1*02:01–DQB1*03:01, DQA1*02:01–DQB1*04:02, DQA1*03:01–DQB1*03:01, DQA1*03:01–DQB1*03:02, DQA1*01:03–DQB1*06:03, DQA1*05:01–DQB1*02:01, DQA1*05:01–DQB1*03:01, DQA1*05:01–DQB1*03:02, DQA1*05:01–DQB1*03:03, DQA1*05:01–DQB1*04:02, and DQA1*04:01–DQB1*04:02 |
| 70 | 69 | 213 | 501 | 570 | 614 | 681 | 716 | 982 | 1118 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Wuhan-Hu-1 (wild type) | V | H | V | N | A | D | P | T | S | D |
| EIJK-61453 | V | H | V | N | A | D | P | T | S | D |
| UGM10002 | V | H | A | N | A | G | P | T | S | D |
| B.1.1.7 | - | - | V | Y | D | D | H | I | A | H |
| Sequence ID | CTL Epitope | HLA Allele | Percentile (%) | IC50 (nM) | Antigenicity | Allergenicity | Decision |
|---|---|---|---|---|---|---|---|
| Wuhan-Hu-1 | SPRRARSVA | HLA-B*07:02 | 0.01 | 4.17 | 0.7729 | N | Used |
| KIYSKHTPI | HLA-A*32:01 | 0.02 | 10.98 | 0.7455 | N | Used | |
| HLA-A*02:03 | 0.25 | 9.39 | |||||
| AEIRASANL | HLA-B*40:01 | 0.05 | 11.05 | 0.7082 | N | Used | |
| HLA-B*44:03 | 0.12 | 71.39 | |||||
| QLTPTWRVY | HLA-B*15:02 | 0.03 | 62.69 | 1.2119 | N | Used | |
| WTAGAAAYY | HLA-A*01:01 | 0.02 | 12.27 | 0.6306 | N | Used | |
| HLA-A*26:01 | 0.03 | 11.63 | |||||
| HLA-B*15:17 | 0.04 | 2.6 | |||||
| IAIPTNFTI | HLA-B*51:01 | 0.06 | 302.84 | 0.7052 | N | Used | |
| HLA-B*58:01 | 0.07 | 11.76 | |||||
| HLA-B*15:17 | 0.3 | 11.97 | |||||
| QYIKWPWYI | HLA-A*24:02 | 0.01 | 13.22 | 1.4177 | Y | Not-used | |
| STQDLFLPF | HLA-A*32:01 | 0.04 | 17.27 | 0.6619 | Y | Not-used | |
| HLA-A*26:01 | 0.3 | 437.88 | |||||
| HLA-B*15:17 | 0.5 | 26.56 | |||||
| TLLALHRS | HLA-B*15:02 | 0.05 | 98.39 | 0.7859 | Y | Not-used | |
| YEQYIKWPW | HLA-B*18:01 HLA-B*44:03 | 0.01 0.05 | 4.19 38.77 | 0.869 | Y | Not-used | |
| EIJK-61453 | SPRRARSVA | HLA-B*07:02 | 0.01 | 4.17 | 0.7729 | N | Used |
| KIYSKHTPI | HLA-A*32:01 | 0.02 | 10.98 | 0.7455 | N | ||
| HLA-A*02:03 | 0.25 | 9.39 | N | ||||
| AEIRASANL | HLA-B*40:01 HLA-B*44:03 | 0.05 0.12 | 11.05 71.39 | 0.7082 | N | ||
| QLTPTWRVY | HLA-B*15:02 | 0.03 | 62.69 | 1.2119 | N | ||
| WTAGAAAYY | HLA-A*01:01 | 0.02 | 12.27 | 0.6306 | N | ||
IAIPTNFTI | HLA-A*26:01 HLA-B*15:17 HLA-B*51:01 HLA-B*58:01 HLA-B*15:17 | 0.03 0.04 0.06 0.07 0.3 | 11.63 2.6 302.84 11.76 11.97 | 0.7052 | N | ||
| QYIKWPWYI STQDLFLPF TLLALHRS YEQYIKWPW | HLA-A*24:02 HLA-A*32:01 HLA-A*26:01 HLA-B*15:17 HLA-B*15:02 HLA-B*18:01 HLA-B*44:03 | 0.01 0.04 0.3 0.5 0.05 0.01 0.05 | 13.22 17.27 437.88 26.56 98.39 4.19 38.77 | 1.4177 0.6619 0.7859 0.869 | Y | Not-used | |
| UGM10002 | SPRRARSVA KIYSKHTPI AEIRASANL GVYFASTEK QLTPTWRVY WTAGAAAYY IAIPTNFTI | HLA-B*07:02 HLA-A*32:01 HLA-A*02:03 HLA-B*40:01 HLA-B*44:03 HLA-A*11:01 HLA-A*03:01 HLA-A*30:01 HLA-B*15:02 HLA-A*01:01 HLA-A*26:01 HLA-B*15:17 HLA-B*51:01 HLA-B*58:01 HLA-B*15:17 | 0.01 0.02 0.25 0.05 0.12 0.07 0.12 0.5 0.03 0.02 0.03 0.04 0.06 0.07 0.3 | 4.17 10.98 9.39 11.05 71.39 15.17 23.87 98.99 62.69 12.27 11.63 2.6 302.84 11.76 11.97 | 0.7729 0.7455 0.7082 0.7112 1.2119 0.6306 0.7052 | N | Used |
| QYIKWPWYI STQDLFLPF ETKCTLKSF TLLALHRSY YEQYIKWPW | HLA-A*24:02 HLA-A*32:01 HLA-A*26:01 HLA-B*15:17 HLA-A*26:01 HLA-B*15:02 HLA-B*18:01 HLA-B*44:03 | 0.01 0.04 0.3 0.5 0.06 0.05 0.01 0.05 | 13.22 17.27 437.88 26.56 36.84 98.39 4.19 38.77 | 1.4177 0.6619 0.8720 0.8009 0.8690 | Y | Not-used | |
| B.1.1.7 | KIYSKHTPI | HLA-A*32:01 HLA-A*02:03 | 0.02 0.25 | 10.98 9.39 | 0.7455 | N | Used |
| AEIRASANL | HLA-B*40:01 HLA-B*44:03 | 0.05 0.12 | 11.05 71.39 | 0.7082 | N | Used | |
| WTAGAAAYY | HLA-A*01:01 HLA-A*26:01 | 0.02 0.03 | 12.27 11.63 | 0.6306 | N | Used | |
| GVYFASTEK | HLA-B*15:17 HLA-A*11:01 HLA-A*03:01 | 0.04 0.07 0.12 | 2.6 15.17 23.87 | 0.6506 | N | Used | |
| QLTPTWRVY | HLA-A*30:01 HLA-B*15:02 | 0.5 0.03 | 98.99 62.69 | 1.2119 | N | Used | |
| QSYGFQPTY YEQYIKWPW IAIPINFTI STQDLFLPF ETKCTLKSF TLLALHRSY IPINFTISV | HLA-B*15:17 HLA-B*58:01 HLA-B*15:02 HLA-B*18:01 HLA-B*44:03 HLA-B*58:01 HLA-A*11:01 HLA-B*15:17 HLA-A*32:01 HLA-A*26:01 HLA-B*15:17 HLA-A*26:01 HLA-B*15:02 HLA-B*51:01 HLA-B*07:02 | 0.03 0.17 0.4 0.01 0.05 0.06 0.07 0.5 0.04 0.3 0.5 0.06 0.05 0.06 0.4 | 2.2 30.54 489.26 4.19 38.77 10.26 376.73 30 17.27 437.88 26.56 36.84 98.39 296.2 91.82 | 1.1150 0.8690 1.5131 0.6619 0.8720 0.8009 1.7137 | Y | Not-used |
| Epitope | Seq. Position | Percentile (%) | Interaction with Allele HLA | IC50 (nM) | Antigenicity | Allergenicity | IFN-γ | Decision |
|---|---|---|---|---|---|---|---|---|
| Wuhan Hu-1 | ||||||||
| GINITRFQTLLALHR | 232–246 | 0.02 | HLA-DRB1*04:02 | 96.22 | 0.5582 | N | + | Used |
| 0.03 | HLA-DRB1*15:01 | 10.96 | ||||||
| 0.04 | HLA-DRB1*04:03 | 182.16 | ||||||
| HWFVTQRNFYEPQII CTFEYVSQPFLMDLE | 1101–1115 166–180 | 0.04 0.06 | HLA-DRB1*04:05 HLA-DPA1*01:03- | 19.97 12.76 | 0.5225 0.5700 | − + | ||
| DPB1*04:01 | ||||||||
| RFQTLLALHRSYLTP | 237–251 | 0.07 | HLA-DRB1*14:01 | 66.04 | 0.5470 | − | ||
| IIAYTMSLGAENSV | 692–705 | 0.07 | HLA-DRB1*10:01 | 7.8 | 0.5350 | − | ||
| KTQSLLIVNNATNVV | 113–127 | 0 | HLA-DRB1*13:02 | 6.37 | 0.6303 | Y | Not-used | |
| AAEIRASANLAATKM | 1015–1029 | 0.03 | HLA-DRB1*04:02 | 101.18 | 0.7125 | |||
NCTFEYVSQPFLMDL | 165–179 | 0.07 0.07 | HLA-DRB1*04:03 HLA-DPA1*01:03-DPB1*04:01 | 220.76 12.96 | 0.5206 | |||
| AEIRASANLAATKMS | 1016–1030 | 0.07 | HLA-DRB1*04:02 | 122.53 | 0.8255 | |||
| PINLVRDLPQGFSAL | 209–223 | 0.07 | HLA-DRB1*03:01 | 27.42 | 0.6086 | |||
| EIJK-61453 | ||||||||
| GINITRFQTLLALHR | 232–246 | 0.02 | HLA-DRB1*04:02 | 96.22 | 0.5582 | + | Used | |
| 0.03 | HLA-DRB1*15:01 | 10.96 | ||||||
| 0.04 | HLA-DRB1*04:03 | 182.16 | ||||||
| HWFVTQRNFYEPQII CTFEYVSQPFLMDLE | 1101–1115 166–180 | 0.04 0.06 | HLA-DRB1*04:05 HLA-DPA1*01:03- | 19.97 12.76 | 0.5225 0.5700 | N | − + | |
| DPB1*04:01 | ||||||||
| RFQTLLALHRSYLTP | 237–251 | 0.07 | HLA-DRB1*14:01 | 66.04 | 0.5470 | − | ||
| IIAYTMSLGAENSVA | 692–705 | 0.07 | HLA-DRB1*10:01 | 7.8 | 0.5350 | − | ||
| KTQSLLIVNNATNVV | 113–127 | 0 | HLA-DRB1*13:02 | 6.37 | 0.6303 | Not-used | ||
| AAEIRASANLAATKM NCTFEYVSQPFLMDL | 1015–1029 165–179 | 0.03 0.07 0.07 | “HLA-DRB1*04:02 HLA-DRB1*04:03 HLA-DPA1*01:03- | 101.18 220.76 12.96 | 0.7125 0.5206 | Y | ||
AEIRASANLAATKMS PINLVRDLPQGFSAL | 1016–1030 209–223 | 0.07 0.07 | DPB1*04:01 HLA-DRB1*04:02 HLA-DRB1*03:01 | 122.53 27.42 | 0.8255 0.6086 | |||
| UGM10002 | ||||||||
| GINIFQTLLALHRTR HWFVTQRNFYEPQII CTFEYVSQPFLMDLE NCTFEYVSQPFLMDL | 232–246 1101–1115 166–180 165–179 | 0.02 0.03 0.04 0.04 0.06 0.07 | HLA-DRB1*04:02 HLA-DRB1*15:01 HLA-DRB1*04:03” HLA-DRB1*04:05 HLA-DPA1*01:03- DPB1*04:01 HLA-DPA1*01:03- DPB1*04:01 | 96.22 10.96 182.16 19.97 12.76 12.96 | 0.5582 0.5225 0.5700 0.5206 | N | + − + − | Used |
| KTQSLLIVNNATNVV AAEIRASANLAATKM IIAYTMSLGAENSVA AEIRASANLAATKMS | 113–127 1015–1029 692–705 1016–1030 | 0 0.03 0.07 0.07 0.07 | HLA-DRB1*13:02 “HLA-DRB1*04:02 HLA-DRB1*04:03” HLA-DRB1*10:01 HLA-DRB1*04:02 | 6.37 101.18 220.76 7.8 122.53 | 0.6303 0.7125 0.5426 0.8255 | Y | Not-used | |
| B.1.1.7 | ||||||||
| RAAEIRASANLAATK KHTPINLVRDLPQGF KGIYQTSNFRVQPTE | 1014–1028 206–220 310–324 | 0.08 0.08 0.07 | HLA-DRB1*04:02 HLA-DRB1*04:02 HLA-DPA1*02:01- DPB1*05:01 | 125.96 193.31 1110.1 | 0.5709 0.5644 0.8838 | Non-Al Non-Al Non-Al | + − + | Used |
| TGCVIAWNSNNLDSK EKGIYQTSNFRVQPT VEKGIYQTSNFRVQP AAEIRASANLAATKM NDPFLGVYYHKNNKS CNDPFLGVYYHKNNK | 430–444 309–323 308–322 1015–1029 137–151 136–150 | 0.06 0.07 0.09 0.03 0.07 0.02 0.03 | HLA-DRB1*15:01 HLA-DRB1*15:02 HLA-DRB1*15:02 HLA-DRB1*04:02, HLA-DRB1*04:03 HLA-DPA1*02:01- DPB1*05:01 HLA-DPA1*02:01- DPB1*05:01 | 30.1 84.33 86.62 101.18 220.76 923.7 994.78 | 0.6531 0.9243 0.7959 0.7637 0.8199 0.6472 | Al Al Al Al Al Al | Not-used | |
| Sequence ID | Epitope | Antigenicity | IFN-γ |
|---|---|---|---|
| Wuhan-Hu-1 | NNLDSKVGGNYNY | 0.9437 | + |
| FQPTNG | 0.7429 | + | |
| AYTMSLGAENSVAYSN | 0.6003 | + | |
| GQSKRVDFC | 1.779 | + | |
| SCCKFDEDDSEPVLKGVKL | 0.6085 | + | |
| EIJK-61453 | NNLDSKVGGNYNY | 0.9437 | + |
| FQPTNG | 0.7429 | + | |
| AYTMSLGAENSVAYSN | 0.6003 | + | |
| GQSKRVDFC | 1.779 | + | |
| SCCKFDEDDSEPVLKGVKL | 0.6085 | + | |
| UGM0002 | NNLDSKVGGNYNY | 0.9437 | + |
| SNKKFLPF | 1.3952 | + | |
| VNCTEV | 0.6529 | + | |
| TNTSNQ | 1.6803 | + | |
| LTPTWRVYSTGSNVFQT | 0.5474 | + | |
| GQSKRVDFC | 1.779 | + | |
| B.1.1.7 | GDEVRQIAPGQTGKIA | 1.0202 | + |
| SNKKFLPF | 1.3952 | + | |
| VNCTEV | 1.6803 | + | |
| LGQSKRVDFC | 1.8685 | + | |
| SCCKFDEDDSEPVLKGVK | 0.5409 | + |
| Vaccine Sequences | Score | Explanation |
|---|---|---|
| Wuhan-Hu-1 | ||
| MRIHYLLFALLFLFLVPVPGHGGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKKEAAAKSPRRARSVAKKWTAGAAAYYGPGPGGINITRFQTLLALHRGPGPGCTFEYVSQPFLMDLEGPGPGGQSKRVDFC | Quality Factor ERRAT = 91.73 TM-Score = 0.383 Template = 1KJ6_A Confidence = 97.0% | Using the template protein 1KJ6_A with 97.0% confidence Vaccine length 146 aa |
| EIJK-61453 | ||
| MRIHYLLFALLFLFLVPVPGHGGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKKEAAAKSPRRARSVAKKWTAGAAAYYGPGPGGINITRFQTLLALHRGPGPGCTFEYVSQPFLMDLEGPGPGGQSKRVDFC | Quality Factor ERRAT = 91.73 TM-Score = 0.383 Template = 1KJ6_A Confidence = 97.0% | Using the template protein 1KJ6_A with 97.0% confidence Vaccine length 146 aa |
| UGM0002 | ||
| MRIHYLLFALLFLFLVPVPGHGGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKKEAAAKSPRRARSVAAAYKIYSKHTPIAAYGVYFASTEKAAYWTAGAAAYYGPGPGGINIFQTLLALHRTRGPGPGGQSKRVDFC | Quality Factor ERRAT = 99.27 TM-Score = 0.358 | Using a de novo folding approach Vaccine length 151 aa |
| B.1.1.7 | ||
| MRIHYLLFALLFLFLVPVPGHGGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKKEAAAKKIYSKHTPIAAYWTAGAAAYYGPGPGRAAEIRASANLAATKGPGPGLGQSKRVDFC | Quality Factor ERRAT = 97.32 TM-Score = 0.405 Template = 6VSJ_C Confidence = 100% | Using the 6VSJ_C protein template with 100% confidence Vaccine length 128 aa |
| Vaccine Model | Binding Affinity ΔG (kcal/mol) | RMSD | Interaction with TLR-3 |
|---|---|---|---|
| EIJK-61453 Total Interactions:
| −18.7 | 0.5 ± 0.3 | Hydrogen bond: Vaccine Residues—TLR-3 TYR5-ASP575 (2.65 Å), LEU15-HIS539 (2.70 Å), PRO17-ARG489 (2.86 Å), VAL18-ARG489 (3.10 Å), GLY22-ARG488 (2.60 Å), GLU50-ARG331 (2.67 Å), GLN51-LYS335 (3.00 Å), LYS54-ASN388 (2.65 Å), LYS54-LYS416 (2.78 Å), ARG58-LYS418 (2.70 Å), ARG78-ASN257 (2.73 Å), ARG78-GLN259 (2.89 Å), THR85-TYR307 (2.71 Å), TYR91-LYS416 (2.68 Å), LEU131-GLN618 (2.76 Å), GLU132-LYS619 (2.60 Å), GLY133-GLU570 (3.25 Å), GLN139-ASP536 (2.64 Å) Salt bridges: GLU 50-ARG331 (2.67 Å), LYS 61-ASP366 (3.56 Å), GLU132-LYS619 (2.60 Å) |
| UGM0002 Total Interactions:
| −19.4 | 12.4 ± 0.2 | Hydrogen bond: Vaccine Residues—TLR-3 PHE14-HIS563 (2.69 Å), HIS21-HIS565 (2.73 Å), GLY23-GLU533 (2.72 Å), ASN26-GLU533 (2.73 Å), GLN51-TYR326 (2.77 Å), LYS67-GLU358 (2.88 Å), ALA71-ARG251 (2.88 Å), LYS72-GLU301 (3.24 Å), ILE124-ASN247 (2.75 Å), GLN128-TRP273 (2.85 Å), ARG135-ALA295 (2.78 Å) |
| B.1.1.7 Total Interactions:
| −15.6 | 2.3 ± 0.5 | Hydrogen bond: Vaccine Residues—TLR-3 PHE14-TYR307 (2.95 Å), VAL16-LYS330 (2.61 Å), HIS21-GLU363 (2.58 Å), ARG36-GLU533 (2.92 Å), LYS54-LYS382 (3.11 Å), LYS54-HIS410 (2.94 Å), CYS55-TYR383 (2.90 Å), SER56-TYR462 (3.18 Å), THR57-TYR383 (2.78 Å), ARG64-GLU533 (2.77 Å), LYS72-GLU533 (2.70 Å), TYR84-GLU587 (2.61 Å) Salt bridges: HIS 21-GLU363 (2.58 Å), ARG 36-GLU533 (2.92 Å), ARG 64-GLU533 (2.77 Å), LYS 72-GLU533 (2.70 Å) |
| Vaccine Model | Binding Affinity ΔG (kcal/mol) | RMSD | Interaction with TLR-4 |
|---|---|---|---|
| EIJK-61453 Total Interactions:
| −16.3 | 1.1 ± 0.3 | Hydrogen bond: Vaccine Residues—TLR-4 ARG34-ARG264 (2.79 Å), PRO116-ARG264 (2.77 Å), ARG64-ASN265 (2.68 Å), GLU68-ASN265 (3.04 Å), ARG76-GLU266 (2.76 Å), CYS118-TYR296 (3.23 Å), CYS146-LYS341 (2.98 Å), HIS21-HIS41 (2.84 Å), LEU13-ASN433 (2.84 Å), ALA9-LYS435 (2.73 Å), ARG2-GLU439 (2.60 Å) Salt bridges: LYS72-GLU266 (2.98 Å), ARG2-GLU439 (2.60 Å) |
| UGM0002 Total Interactions:
| −16.1 | 1.0 ± 0.3 | Hydrogen bond: Vaccine Residues—TLR-3 ARG2-GLU270 (2.95 Å), ARG2-ASP294 (2.55 Å), LEU6-TYR296 (2.68 Å), ARG34-PHE387 (2.97 Å), ARG39-LYS388 (3.20 Å), ARG34-LYS399 (3.20 Å), ARG39-GLN436 (2.98 Å), LYS105-GLU439 (3.22 Å), LYS66-GLU439 (2.60 Å), ARG147-ASP490 (2.59 Å) Salt bridges: ARG2-ASP294 (2.55 Å), LYS66-GLU439 (2.60 Å), ARG147-ASP490 (2.59 Å) |
| B.1.1.7 Total Interactions:
| −15.5 | 26.4 ± 0.2 | Hydrogen bond: Vaccine Residues—TLR-3 ALA9-ARG264 (2.79 Å), ARG2-ASN264 (2.68 Å), ARG2-GLU266 (3.04 Å), ARG64-GLU439 (2.60 Å), GLN51-ARG460 (2.85 Å), GLU50-ARG460 (2.72 Å), GLN51-ARG460 (3.12 Å), GLU50-GLN484 (3.19 Å), LYS66-GLU485 (2.66 Å) Salt bridges: ARG2-GLU266 (2.64 Å), ARG64-GLU439 (2.60 Å), GLU50-ARG460 (2.72 Å), LYS66-GLU485 (2.66 Å) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umitaibatin, R.; Harisna, A.H.; Jauhar, M.M.; Syaifie, P.H.; Arda, A.G.; Nugroho, D.W.; Ramadhan, D.; Mardliyati, E.; Shalannanda, W.; Anshori, I. Immunoinformatics Study: Multi-Epitope Based Vaccine Design from SARS-CoV-2 Spike Glycoprotein. Vaccines 2023, 11, 399. https://doi.org/10.3390/vaccines11020399
Umitaibatin R, Harisna AH, Jauhar MM, Syaifie PH, Arda AG, Nugroho DW, Ramadhan D, Mardliyati E, Shalannanda W, Anshori I. Immunoinformatics Study: Multi-Epitope Based Vaccine Design from SARS-CoV-2 Spike Glycoprotein. Vaccines. 2023; 11(2):399. https://doi.org/10.3390/vaccines11020399
Chicago/Turabian StyleUmitaibatin, Ramadhita, Azza Hanif Harisna, Muhammad Miftah Jauhar, Putri Hawa Syaifie, Adzani Gaisani Arda, Dwi Wahyu Nugroho, Donny Ramadhan, Etik Mardliyati, Wervyan Shalannanda, and Isa Anshori. 2023. "Immunoinformatics Study: Multi-Epitope Based Vaccine Design from SARS-CoV-2 Spike Glycoprotein" Vaccines 11, no. 2: 399. https://doi.org/10.3390/vaccines11020399