
Cohort-Specific Peptide Reagents Broaden Depth and Breadth Estimates of the CD8 T Cell Response to HIV-1 Gag Potential T Cell Epitopes

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
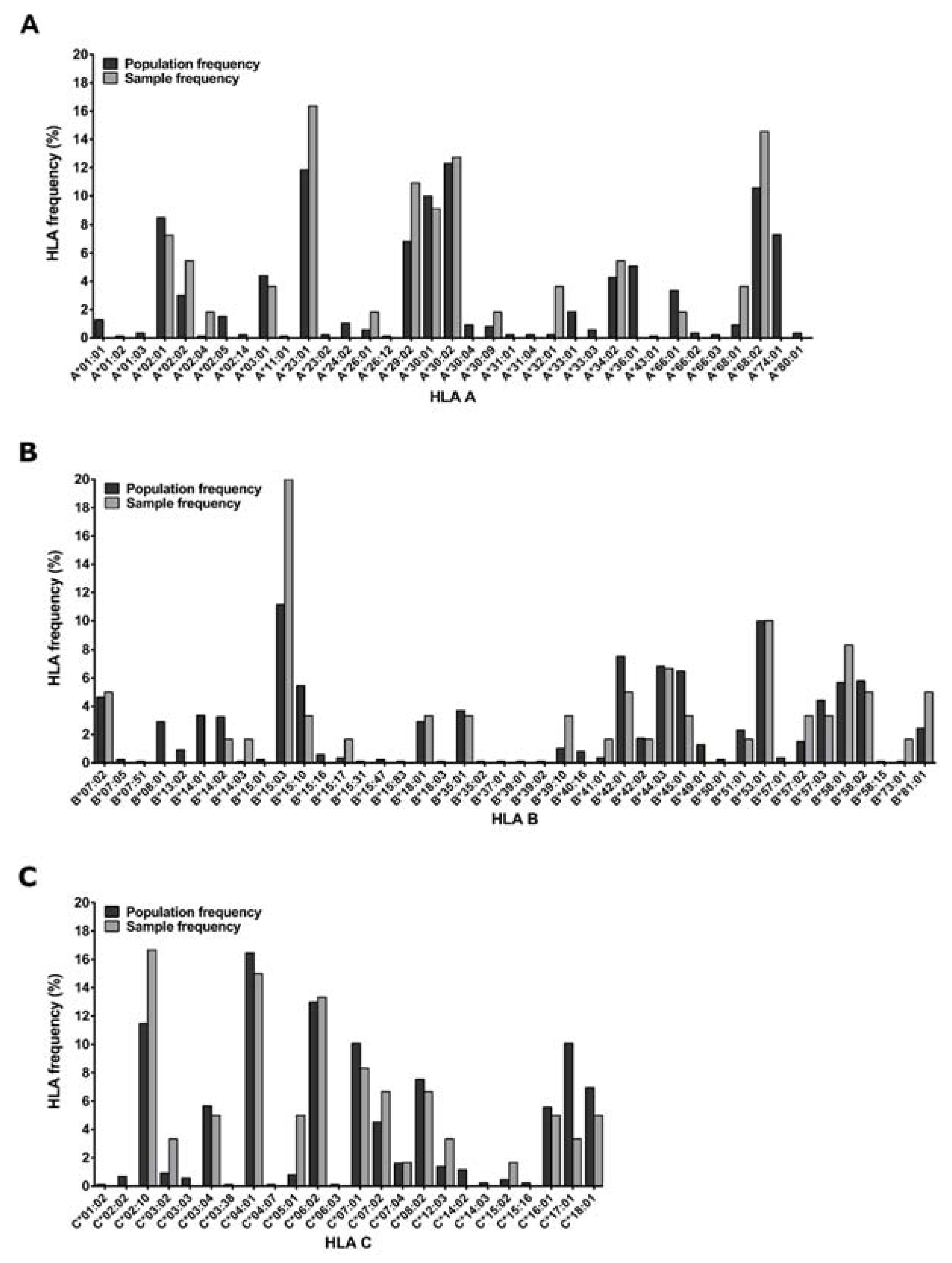
2.1. Study Participants
2.2. PBMC Processing
2.3. CD8 T Cell Expansion
2.4. HIV-1 Gag Peptide Sets and Pools
2.5. ELISpot Assays and Mapping Strategy
2.6. Statistical Analysis
3. Results
3.1. Epitope Mapping of CD8 T Cell Responses
3.2. Epitope Identification and Analysis of Breadth
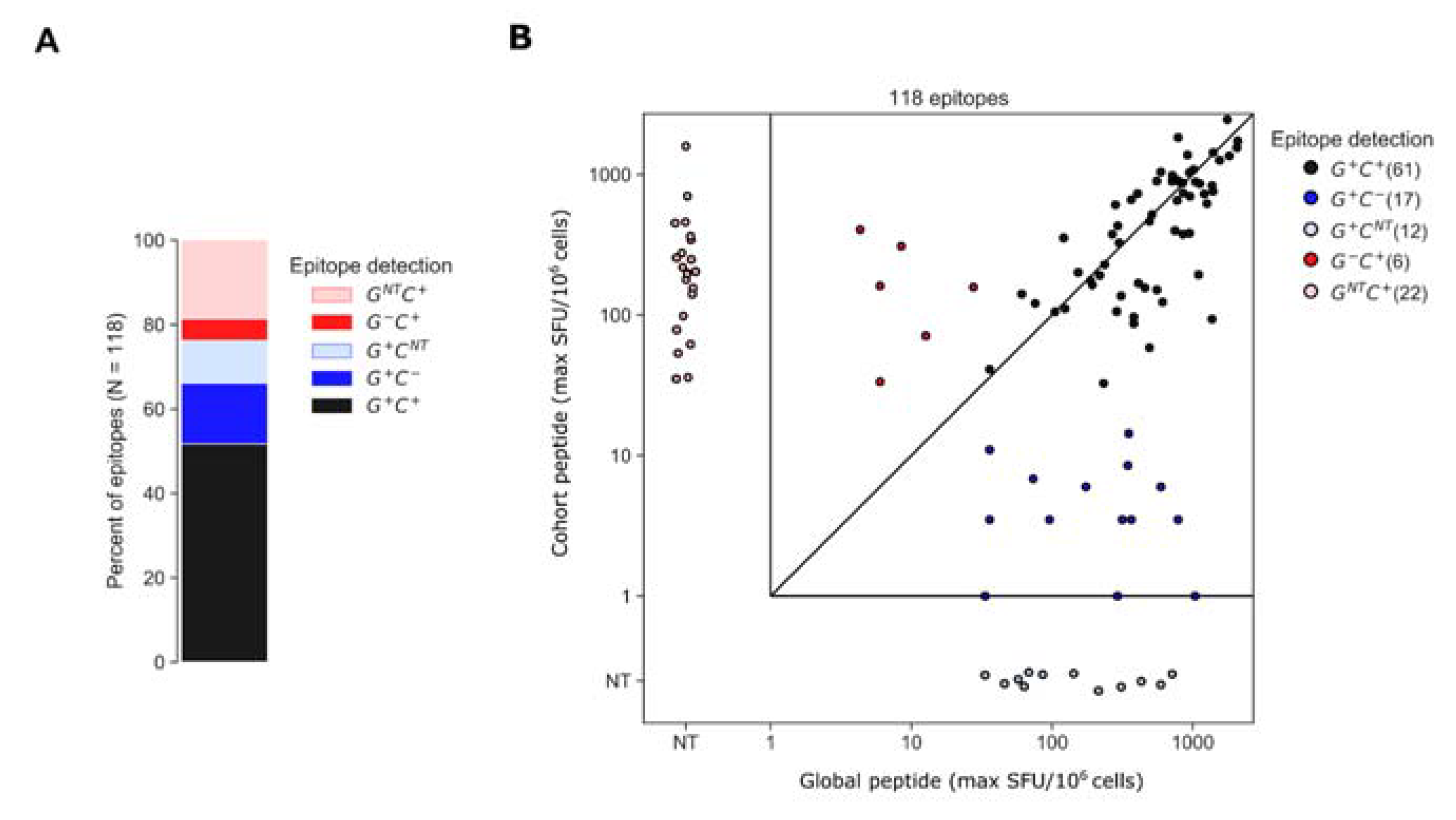
3.3. Assessment of Epitopes Detected by Global or Cohort Peptides or Both
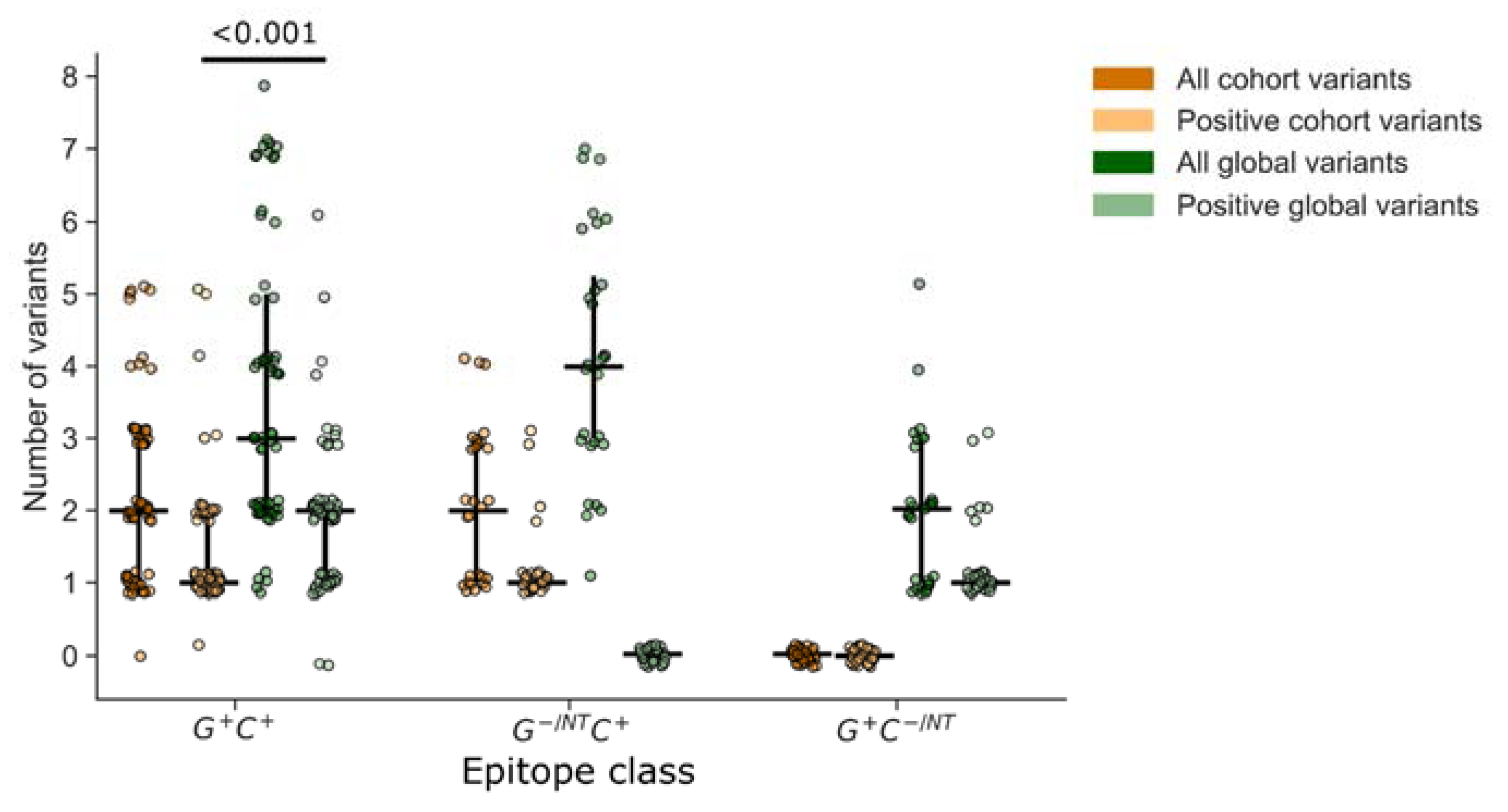
3.4. Analysis of Epitope Response Depth
3.5. Importance of Non-Consensus Peptides in Epitope Mapping
3.6. Accounting for Missed Responses
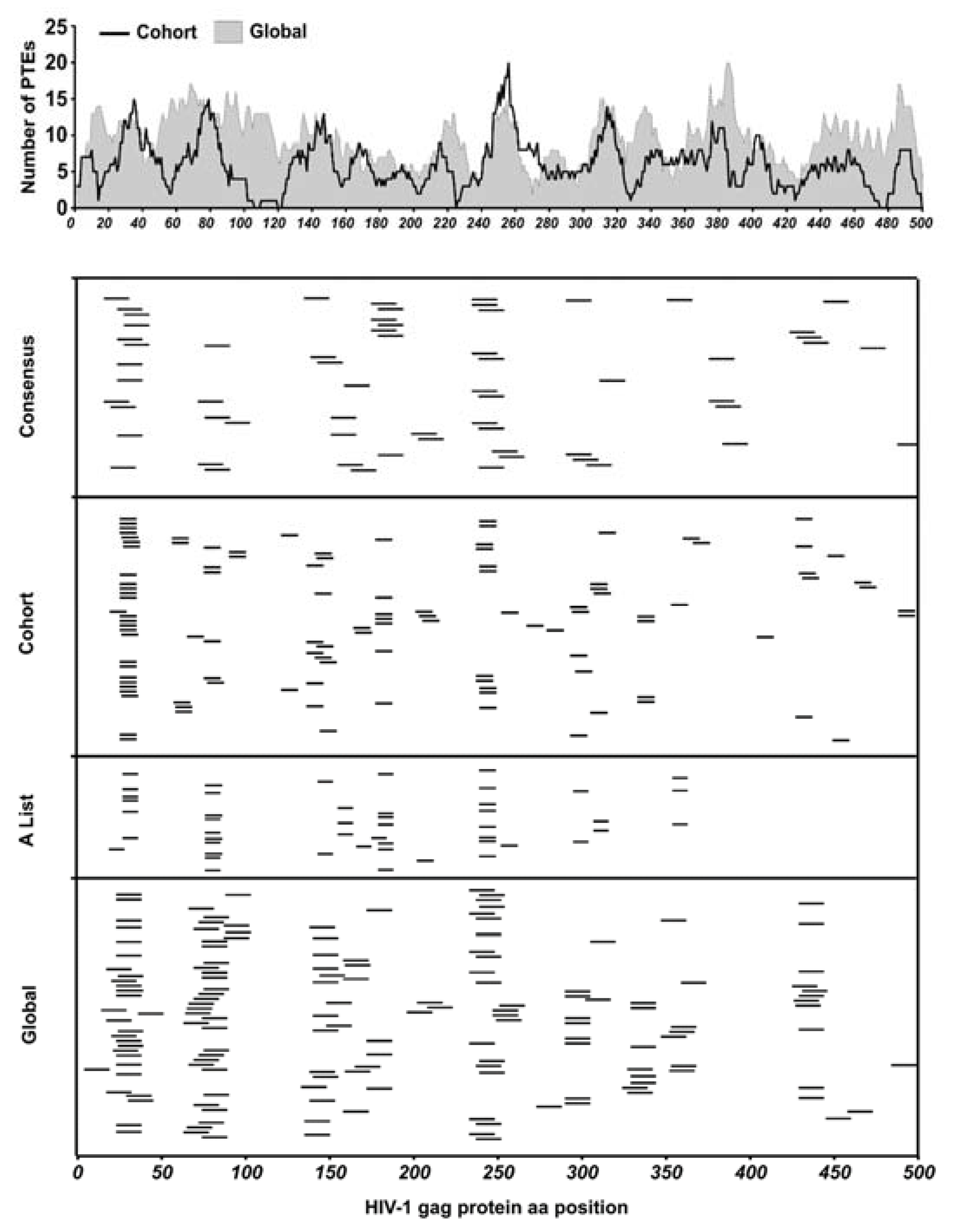
3.7. Epitope Location within Global 15mer Peptides
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martins, M.A.; Shin, Y.C.; Gonzalez-Nieto, L.; Domingues, A.; Gutman, M.J.; Maxwell, H.S.; Castro, I.; Magnani, D.M.; Ricciardi, M.; Pedreño-Lopez, N.; et al. Vaccine-induced immune responses against both Gag and Env improve control of simian immunodeficiency virus replication in rectally challenged rhesus macaques. PLoS Pathog. 2017, 13, e1006529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, A.-M.C.; Holst, P.J. Increased T cell breadth and antibody response elicited in prime-boost regimen by viral vector encoded homologous SIV Gag/Env in outbred CD1 mice. J. Transl. Med. 2016, 14, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.E.; Kuroda, M.J.; Santra, S.; Sasseville, V.G.; Simon, M.A.; Lifton, M.A.; Racz, P.; Tenner-Racz, K.; Dalesandro, M.; Scallon, B.J.; et al. Control of Viremia in Simian Immunodeficiency Virus Infection by CD8 + Lymphocytes. Science 1979, 283, 857–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Veenhuis, R.T.; May, M.; Luna, K.A.; Kirkpatrick, A.R.; Laeyendecker, O.; Cox, A.L.; Carrington, M.; Bailey, J.R.; et al. Factors Associated with the Control of Viral Replication and Virologic Breakthrough in a Recently Infected HIV-1 Controller. eBioMedicine 2017, 16, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.S.; Altfeld, M.; Poon, S.H.; Phillips, M.N.; Wilkes, B.M.; Eldridge, R.L.; Robbins, G.K.; D’Aquila, R.T.; Goulder, P.J.R.; Walker, B.D. Immune control of HIV-1 after early treatment of acute infection. Nature 2000, 407, 523–526. [Google Scholar] [CrossRef]
- Koofhethile, C.K.; Ndhlovu, Z.M.; Thobakgale-Tshabalala, C.; Prado, J.G.; Ismail, N.; Mncube, Z.; Mkhize, L.; van der Stok, M.; Yende, N.; Walker, B.D.; et al. CD8 + T Cell Breadth and Ex Vivo Virus Inhibition Capacity Distinguish between Viremic Controllers with and without Protective HLA Class I Alleles. J. Virol. 2016, 90, 6818–6831. [Google Scholar] [CrossRef] [Green Version]
- Betts, M.R.; Nason, M.C.; West, S.M.; De Rosa, S.C.; Migueles, S.A.; Abraham, J.; Lederman, M.M.; Benito, J.M.; Goepfert, P.A.; Connors, M.; et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 2006, 107, 4781–4789. [Google Scholar] [CrossRef] [Green Version]
- Borrow, P.; Lewicki, H.; Hahn, B.H.; Shaw, G.M.; Oldstone, M.B. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994, 68, 6103–6110. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, T.C.; Valentine, L.E.; Yant, L.J.; Rakasz, E.G.; Piaskowski, S.M.; Furlott, J.R.; Weisgrau, K.L.; Burwitz, B.; May, G.E.; León, E.J.; et al. Subdominant CD8 + T-Cell Responses Are Involved in Durable Control of AIDS Virus Replication. J. Virol. 2007, 81, 3465–3476. [Google Scholar] [CrossRef] [Green Version]
- Migueles, S.A.; Laborico, A.C.; Shupert, W.L.; Sabbaghian, M.S.; Rabin, R.; Hallahan, C.W.; Van Baarle, D.; Kostense, S.; Miedema, F.; McLaughlin, M.; et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 2002, 3, 1061–1068. [Google Scholar] [CrossRef]
- Carrington, M.; Bontrop, R.E. Effects of MHC Class I on HIV/SIV Disease in Primates. AIDS 2002, 16, S105–S114. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Zhang, Y.; Xu, K.Y.; Yan, H.; James, I.; Peng, Y.; Blais, M.-E.; Gaudieri, S.; Chen, X.; Lun, W.; et al. Extensive HLA-driven viral diversity following a narrow-source HIV-1 outbreak in rural China. Blood 2011, 118, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wood, N.; Bhattacharya, T.; Keele, B.F.; Giorgi, E.; Liu, M.; Gaschen, B.; Daniels, M.; Ferrari, G.; Haynes, B.F.; McMichael, A.; et al. HIV Evolution in Early Infection: Selection Pressures, Patterns of Insertion and Deletion, and the Impact of APOBEC. PLoS Pathog. 2009, 5, e1000414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, D.; Friedrich, T.; Hughes, A.; Allen, T.M.; Watkins, D. Understanding cytotoxic T-lymphocyte escape during simian immunodeficiency virus infection. Immunol. Rev. 2001, 183, 115–126. [Google Scholar] [CrossRef]
- Kwong, P.D.; Mascola, J.R.; Nabel, G.J. The changing face of HIV vaccine research. J. Int. AIDS Soc. 2012, 15, 17407. [Google Scholar] [CrossRef]
- Nabel, G.J.; Kwong, P.D.; Mascola, J.R. Progress in the rational design of an AIDS vaccine. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2759–2765. [Google Scholar] [CrossRef]
- Rolland, M.; Nickle, D.C.; Mullins, J.I. HIV-1 Group M Conserved Elements Vaccine. PLoS Pathog. 2007, 3, e157. [Google Scholar] [CrossRef] [Green Version]
- Gaschen, B.; Taylor, J.; Yusim, K.; Foley, B.; Gao, F.; Lang, D.; Novitsky, V.; Haynes, B.; Hahn, B.H.; Bhattacharya, T.; et al. Diversity Considerations in HIV-1 Vaccine Selection. Science 2002, 296, 2354–2360. [Google Scholar] [CrossRef]
- Novitsky, V.; Smith, U.R.; Gilbert, P.; McLane, M.F.; Chigwedere, P.; Williamson, C.; Ndung’U, T.; Klein, I.; Chang, S.-Y.; Peter, T.; et al. Human Immunodeficiency Virus Type 1 Subtype C Molecular Phylogeny: Consensus Sequence for an AIDS Vaccine Design? J. Virol. 2002, 76, 5435–5451. [Google Scholar] [CrossRef] [Green Version]
- Yusim, K.; Kesmir, C.; Gaschen, B.; Addo, M.M.; Altfeld, M.; Brunak, S.; Chigaev, A.; Detours, V.; Korber, B. Clustering Patterns of Cytotoxic T-Lymphocyte Epitopes in Human Immunodeficiency Virus Type 1 (HIV-1) Proteins Reveal Imprints of Immune Evasion on HIV-1 Global Variation. J. Virol. 2002, 76, 8757–8768. [Google Scholar] [CrossRef] [Green Version]
- Santra, S.; Liao, H.X.; Zhang, R.; Muldoon, M.; Watson, S.; Fischer, W.; Theiler, J.; Szinger, J.; Balachandran, H.; Buzby, A.; et al. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat. Med. 2010, 16, 324–328. [Google Scholar] [CrossRef]
- Bakari, M.; Aboud, S.; Nilsson, C.; Francis, J.; Buma, D.; Moshiro, C.; Aris, E.A.; Lyamuya, E.F.; Janabi, M.; Godoy-Ramirez, K.; et al. Broad and potent immune responses to a low dose intradermal HIV-1 DNA boosted with HIV-1 recombinant MVA among healthy adults in Tanzania. Vaccine 2011, 29, 8417–8428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frahm, N.; Kiepiela, P.; Adams, S.; Linde, C.H.; Hewitt, H.S.; Sango, K.; Feeney, M.E.; Addo, M.M.; Lichterfeld, M.; Lahaie, M.P.; et al. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nat. Immunol. 2006, 7, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Currier, J.R.; Herrmann, E.; Haule, A.; Kuta, E.; McCutchan, F.; Njovu, L.; Geis, S.; Hoffmann, O.; Maboko, L.; et al. CD8 T-Cell Recognition of Multiple Epitopes within Specific Gag Regions Is Associated with Maintenance of a Low Steady-State Viremia in Human Immunodeficiency Virus Type 1-Seropositive Patients. J. Virol. 2007, 81, 2440–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altfeld, M.; Addo, M.M.; Shankarappa, R.; Lee, P.K.; Allen, T.M.; Yu, X.G.; Rathod, A.; Harlow, J.; O’Sullivan, K.; Johnston, M.N.; et al. Enhanced Detection of Human Immunodeficiency Virus Type 1-Specific T-Cell Responses to Highly Variable Regions by Using Peptides Based on Autologous Virus Sequences. J. Virol. 2003, 77, 7330–7340. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Malhotra, U.; Gilbert, P.B.; Hawkins, N.R.; Duerr, A.C.; McElrath, J.M.; Corey, L.; Self, S.G. Peptide selection for human immunodeficiency virus type 1 CTL-based vaccine evaluation. Vaccine 2006, 24, 6893–6904. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, M.; Cong, H. Advances in the study of HLA-restricted epitope vaccines. Hum. Vaccines Immunother. 2013, 9, 2566–2577. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, U.; Li, F.; Nolin, J.; Allison, M.; Zhao, H.; Mullins, J.I.; Self, S.; McElrath, M.J. Enhanced Detection of Human Immunodeficiency Virus Type 1 (HIV-1) Nef-Specific T Cells Recognizing Multiple Variants in Early HIV-1 Infection. J. Virol. 2007, 81, 5225–5237. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Røder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a Method for Quantitative Predictions of Peptide Binding to Any HLA-A and -B Locus Protein of Known Sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef] [Green Version]
- Landais, E.; Huang, X.; Havenar-Daughton, C.; Murrell, B.; Price, M.A.; Wickramasinghe, L.; Ramos, A.; Bian, C.B.; Simek, M.; Allen, S.; et al. Broadly Neutralizing Antibody Responses in a Large Longitudinal Sub-Saharan HIV Primary Infection Cohort. PLoS Pathog. 2016, 12, e1005369. [Google Scholar] [CrossRef] [Green Version]
- Amornkul, P.N.; Karita, E.; Kamali, A.; Rida, W.N.; Sanders, E.J.; Lakhi, S.; Price, M.; Kilembe, W.; Cormier, E.; Anzala, O.; et al. Disease progression by infecting HIV-1 subtype in a seroconverter cohort in sub-Saharan Africa. AIDS 2013, 27, 2775–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamali, A.; Price, M.A.; Lakhi, S.; Karita, E.; Inambao, M.; Sanders, E.J.; Anzala, O.; Latka, M.H.; Bekker, L.-G.; Kaleebu, P.; et al. Creating an African HIV Clinical Research and Prevention Trials Network: HIV Prevalence, Incidence and Transmission. PLoS ONE 2015, 10, e0116100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Tang, S.; Lobashevsky, E.; Myracle, A.D.; Fideli, U.; Aldrovandi, G.; Allen, S.; Musonda, R.; Kaslow, R.A.; Zambia-UAB HIV Research Project, 2002. Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1. J. Virol. 2002, 76, 8276–8284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelo, C.M.; Dalel, J.A.; Hayes, P.; Fernandez, N.; Fiore-Gartland, A.; Kilembe, W.; Tang, J.; Streatfield, C.; Gilmour, J.; Hunter, E. Comprehensive epitope mapping using polyclonally expanded human CD8 T cells and a two-step ELISpot assay for testing large peptide libraries. J. Immunol. Methods 2020, 491, 112970. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.; Fernandez, N.; Ochsenbauer, C.; Dalel, J.; Hare, J.; King, D.; Black, L.; Streatfield, C.; Kakarla, V.; Macharia, G.; et al. Breadth of CD8 T-cell mediated inhibition of replication of diverse HIV-1 transmitted-founder isolates correlates with the breadth of recognition within a comprehensive HIV-1 Gag, Nef, Env and Pol potential T-cell epitope (PTE) peptide set. PLoS ONE 2021, 16, e0260118. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.; Colvin, R.B. Selective reduction and proliferation of the CD4+ and CD8+ T cell subsets with bispecific monoclonal antibodies: Evidence for inter-T cell-mediated cytolysis. Clin. Immunol. Immunopathol. 1991, 58, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Spentzou, A.; Bergin, P.; Gill, D.; Cheeseman, H.; Ashraf, A.; Kaltsidis, H.; Cashin-Cox, M.; Anjarwalla, I.; Steel, A.; Higgs, C.; et al. Viral Inhibition Assay: A CD8 T Cell Neutralization Assay for Use in Clinical Trials of HIV-1 Vaccine Candidates. J. Infect. Dis. 2010, 201, 720–729. [Google Scholar] [CrossRef]
- Carlson, J.M.; Schaefer, M.; Monaco, D.C.; Batorsky, R.; Claiborne, D.T.; Prince, J.; Deymier, M.J.; Ende, Z.S.; Klatt, N.R.; DeZiel, C.E.; et al. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 2014, 345, 1254031. [Google Scholar] [CrossRef] [Green Version]
- Llano, A.; Williams, A.; Olvera, A.; Silva-Arrieta, S.; Brander, C. Best-Characterized HIV-1 CTL Epitopes: The 2013 Update. HIV Mol. Immunol. 2013, 2013, 3–25. [Google Scholar]
- Fiore-Gartland, A.; Manso, B.A.; Friedrich, D.P.; Gabriel, E.E.; Finak, G.; Moodie, Z.; Hertz, T.; De Rosa, S.C.; Frahm, N.; Gilbert, P.B.; et al. Pooled-Peptide Epitope Mapping Strategies Are Efficient and Highly Sensitive: An Evaluation of Methods for Identifying Human T Cell Epitope Specificities in Large-Scale HIV Vaccine Efficacy Trials. PLoS ONE 2016, 11, e0147812. [Google Scholar] [CrossRef]
- Gill, D.K.; Huang, Y.; Levine, G.L.; Sambor, A.; Carter, D.K.; Sato, A.; Kopycinski, J.; Hayes, P.; Hahn, B.; Birungi, J.; et al. Equivalence of ELISpot Assays Demonstrated between Major HIV Network Laboratories. PLoS ONE 2010, 5, e14330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boaz, M.J.; Hayes, P.; Tarragona, T.; Seamons, L.; Cooper, A.; Birungi, J.; Kitandwe, P.; Semaganda, A.; Kaleebu, P.; Stevens, G.; et al. Concordant Proficiency in Measurement of T-Cell Immunity in Human Immunodeficiency Virus Vaccine Clinical Trials by Peripheral Blood Mononuclear Cell and Enzyme-Linked Immunospot Assays in Laboratories from Three Continents. Clin. Vaccine Immunol. 2009, 16, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, T.; Ahmed, H.; Friedrich, D.P.; Casimiro, D.R.; Self, S.G.; Corey, L.; McElrath, M.J.; Buchbinder, S.; Horton, H.; Frahm, N.; et al. HIV-1 Vaccine-Induced T-Cell Reponses Cluster in Epitope Hotspots that Differ from Those Induced in Natural Infection with HIV-1. PLoS Pathog. 2013, 9, e1003404. [Google Scholar] [CrossRef]
- Currier, J.R.; Robb, M.L.; Michael, N.L.; Marovich, M.A. Defining epitope coverage requirements for T cell-based HIV vaccines: Theoretical considerations and practical applications. J. Transl. Med. 2011, 9, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draenert, R.; Brander, C.; Xu, G.Y.; Altfeld, M.; Verrill, C.L.; Feeney, M.E.; Walker, B.D.; Goulder, P.J. Impact of intrapeptide epitope location on CD8 T cell recognition: Implications for design of overlapping peptide panels. AIDS 2004, 18, 871–876. [Google Scholar] [CrossRef]
- Hemelaar, J. Implications of HIV diversity for the HIV-1 pandemic. J. Infect. 2013, 66, 391–400. [Google Scholar] [CrossRef]
- Paraskevis, D.; Hatzakis, A. Global molecular epidemiology of HIV-1: The chameleon challenge. Lancet Infect. Dis. 2019, 19, 114–115. [Google Scholar] [CrossRef]
- Stephenson, K.; Barouch, D.H. A global approach to HIV-1 vaccine development. Immunol. Rev. 2013, 254, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Hulot, S.L.; Korber, B.; Giorgi, E.E.; Vandergrift, N.; Saunders, K.O.; Balachandran, H.; Mach, L.V.; Lifton, M.A.; Pantaleo, G.; Tartaglia, J.; et al. Comparison of Immunogenicity in Rhesus Macaques of Transmitted-Founder, HIV-1 Group M Consensus, and Trivalent Mosaic Envelope Vaccines Formulated as a DNA Prime, NYVAC, and Envelope Protein Boost. J. Virol. 2015, 89, 6462–6480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, P.J.; Cox, J.H.; Coleman, A.R.; Fernandez, N.; Bergin, P.J.; Kopycinski, J.T.; Nitayaphan, S.; Pitisuttihum, P.; de Souza, M.; Duerr, A.; et al. Adenovirus-based HIV-1 vaccine candidates tested in efficacy trials elicit CD8+ T cells with limited breadth of HIV-1 inhibition. AIDS 2016, 30, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Pira, G.L.; Ivaldi, F.; Moretti, P.; Manca, F. High Throughput T Epitope Mapping and Vaccine Development. J. Biomed. Biotechnol. 2010, 2010, 325720. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Female | 18 * |
| Male | 12 * | |
| Age (years) | Mean (SD) | 34 ± 9 |
| Day post EDI | Mean ± SD | 348 ± 17 |
| CD4 Count (cells/uL) | Mean ± SD | 566 ± 198 |
| Plasma Viral load (copies/mL) | Mean ± SD | 8301 ± 6046 |
| Mode of infection | Heterosexual discordant couples | |
| HIV-1 subtype | Subtype C | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelo, C.M.; Fiore-Gartland, A.; Dalel, J.A.; Hayes, P.; Tang, J.; McGowan, E.; Kilembe, W.; Fernandez, N.; Gilmour, J.; Hunter, E. Cohort-Specific Peptide Reagents Broaden Depth and Breadth Estimates of the CD8 T Cell Response to HIV-1 Gag Potential T Cell Epitopes. Vaccines 2023, 11, 472. https://doi.org/10.3390/vaccines11020472
Michelo CM, Fiore-Gartland A, Dalel JA, Hayes P, Tang J, McGowan E, Kilembe W, Fernandez N, Gilmour J, Hunter E. Cohort-Specific Peptide Reagents Broaden Depth and Breadth Estimates of the CD8 T Cell Response to HIV-1 Gag Potential T Cell Epitopes. Vaccines. 2023; 11(2):472. https://doi.org/10.3390/vaccines11020472
Chicago/Turabian StyleMichelo, Clive M., Andrew Fiore-Gartland, Jama A. Dalel, Peter Hayes, Jianming Tang, Edward McGowan, William Kilembe, Natalia Fernandez, Jill Gilmour, and Eric Hunter. 2023. "Cohort-Specific Peptide Reagents Broaden Depth and Breadth Estimates of the CD8 T Cell Response to HIV-1 Gag Potential T Cell Epitopes" Vaccines 11, no. 2: 472. https://doi.org/10.3390/vaccines11020472