
In Silico Analysis of SARS-CoV-2 Spike Proteins of Different Field Variants




Abstract
1. Introduction
2. Methods
2.1. Viral Strain Selection and Retrieval of Protein Sequence
2.2. Mutations Identification in SARS-CoV-2 Genomes
2.3. Protein Secondary Structure
- Secondary structure prediction and solvent accessibility;
- Topology (TMSEG);
- Disered region (Meta-Disorder);
- Protein binding (ProNA);
- Disulfide bond;
- Conversation score;
- Disordered region (Meta-Disorder).
2.4. Tertiary Structure
2.5. Comparison of Pakistani Variants with Reference Strain
3. Results
3.1. Mutations Identified in the Sequenced SARS-CoV-2 Genomes
3.2. Protein Secondary Structure
3.3. Tertiary Structure
3.4. Comparison of Pakistani Variants with Reference Strain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shaikh, S.S.; Jose, A.P.; Nerkar, D.A.; Kv, M.V.; Shaikh, S.K. COVID-19 pandemic crisis—A complete outline of SARS-CoV-2. Future J. Pharm. Sci. 2020, 6, 116. [Google Scholar] [CrossRef]
- Wormser, G.P.; Aitken, C. Clinical Virology, 3rd Edition Edited by D. D. Richman, R. J. Whitley, and F. G. Hayden Washington, DC: ASM Press, 2009. 1408 pp, Illustrated. $259.59 (hardcover). Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 50, 1692. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef]
- Arias-Reyes, C.; Zubieta-DeUrioste, N.; Poma-Machicao, L.; Aliaga-Raduan, F.; Carvajal-Rodriguez, F.; Dutschmann, M.; Schneider-Gasser, E.M.; Zubieta-Calleja, G.; Soliz, J. Does the pathogenesis of SARS-CoV-2 virus decrease at high-altitude? Respir. Physiol. Neurobiol. 2020, 277, 103443. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Zhang, X.; Hasoksuz, M.; Nagesha, H.S.; Haynes, L.M.; Fang, Y.; Lu, S.; Saif, L.J. Two-way antigenic cross-reactivity between severe acute respiratory syndrome coronavirus (SARS-CoV) and group 1 animal CoVs is mediated through an antigenic site in the N-terminal region of the SARS-CoV nucleoprotein. J. Virol. 2007, 81, 13365–13377. [Google Scholar] [CrossRef]
- Graham, R.L.; Sparks, J.S.; Eckerle, L.D.; Sims, A.C.; Denison, M.R. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008, 133, 88–100. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer TA, P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Mihindukulasuriya, K.A.; Wu, G.; St Leger, J.; Nordhausen, R.W.; Wang, D. Identification of a Novel Coronavirus from a Beluga Whale by Using a Panviral Microarray. J. Virol. 2008, 82, 5084–5088. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lai, K.K.Y.; Huang, Y.; Lee, P.; Luk, G.S.M.; Dyrting, K.C.; Chan, K.-H.; Yuen, K.-Y. Comparative Analysis of Complete Genome Sequences of Three Avian Coronaviruses Reveals a Novel Group 3c Coronavirus. J. Virol. 2009, 83, 908–917. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef]
- Masters, P.S. Coronavirus genomic RNA packaging. Virology 2019, 537, 198–207. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel Coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Sanavia, T.; Birolo, G.; Montanucci, L.; Turina, P.; Capriotti, E.; Fariselli, P. Limitations challenges in protein stability prediction upon genome variations: Towards future applications in precision medicine. Comput. Struct. Biotechnol. J. 2020, 18, 1968–1979. [Google Scholar] [CrossRef]
- Babcock, G.J.; Esshaki, D.J.; Thomas, W.D.; Ambrosino, D.M. Amino Acids 270 to 510 of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Are Required for Interaction with Receptor. J. Virol. 2004, 78, 4552–4560. [Google Scholar] [CrossRef]
- Prabakaran, P.; Gan, J.; Feng, Y.; Zhu, Z.; Choudhry, V.; Xiao, X.; Ji, X.; Dimitrov, D.S. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 2006, 281, 15829–15836. [Google Scholar] [CrossRef]
- Souza, P.F.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.; Lima, K.R.; Costa, M.B.; Farias, I.R.; Lima, L.B.; Montenegro, R.C. The human pandemic coronaviruses on the show: The spike glycoprotein as the main actor in the coronaviruses play. Int. J. Biol. Macromol. 2021, 179, 1–19. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R.; Biology, S. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Hu, J.; He, C.L.; Gao, Q.Z.; Zhang, G.J.; Cao, X.X.; Long, Q.X.; Deng, H.J.; Huang, L.Y.; Chen, J.; Wang, K.; et al. D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. BioRxiv 2020. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Iii KH, D.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Rehman, Z.; Haider, A.; Badar, N.; Ammar, M.; Ahad, A.; Suleman, R.; Salman, M. Genomic Diversity of SARS-CoV-2 in Pakistan during Fourth Wave of Pandemic. MedRxiv 2021, 92. Available online: https://www.medrxiv.org/content/10.1101/2021.09.30.21264343v1%0Ahttps://www.medrxiv.org/content/10.1101/2021.09.30.21264343v1.abstract (accessed on 3 October 2021). [CrossRef]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020, 74, 4–7. [Google Scholar] [CrossRef]
- Weissman, D.; Alameh, M.-G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS-CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2021, 29, 23–31.e4. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
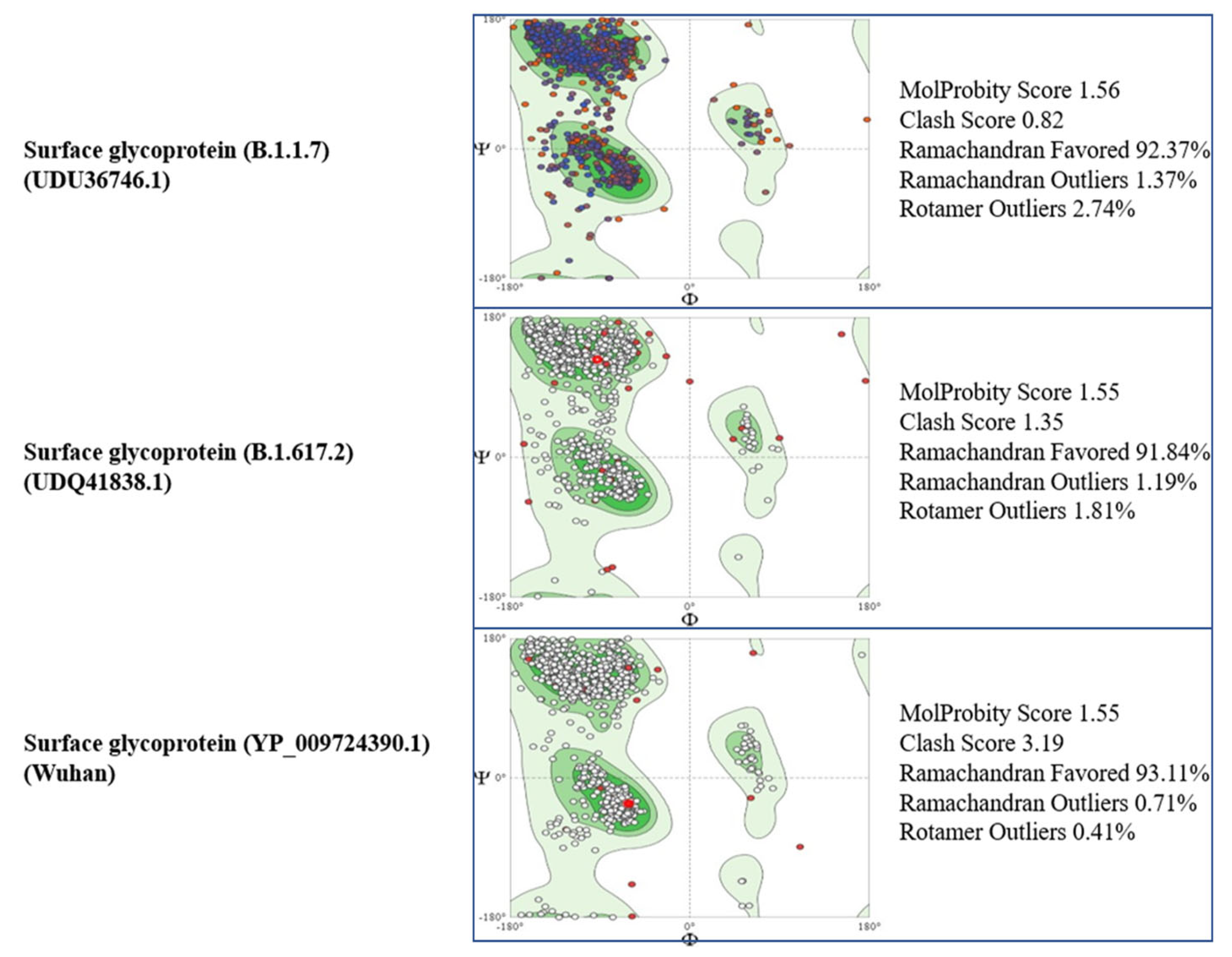{kind=link}
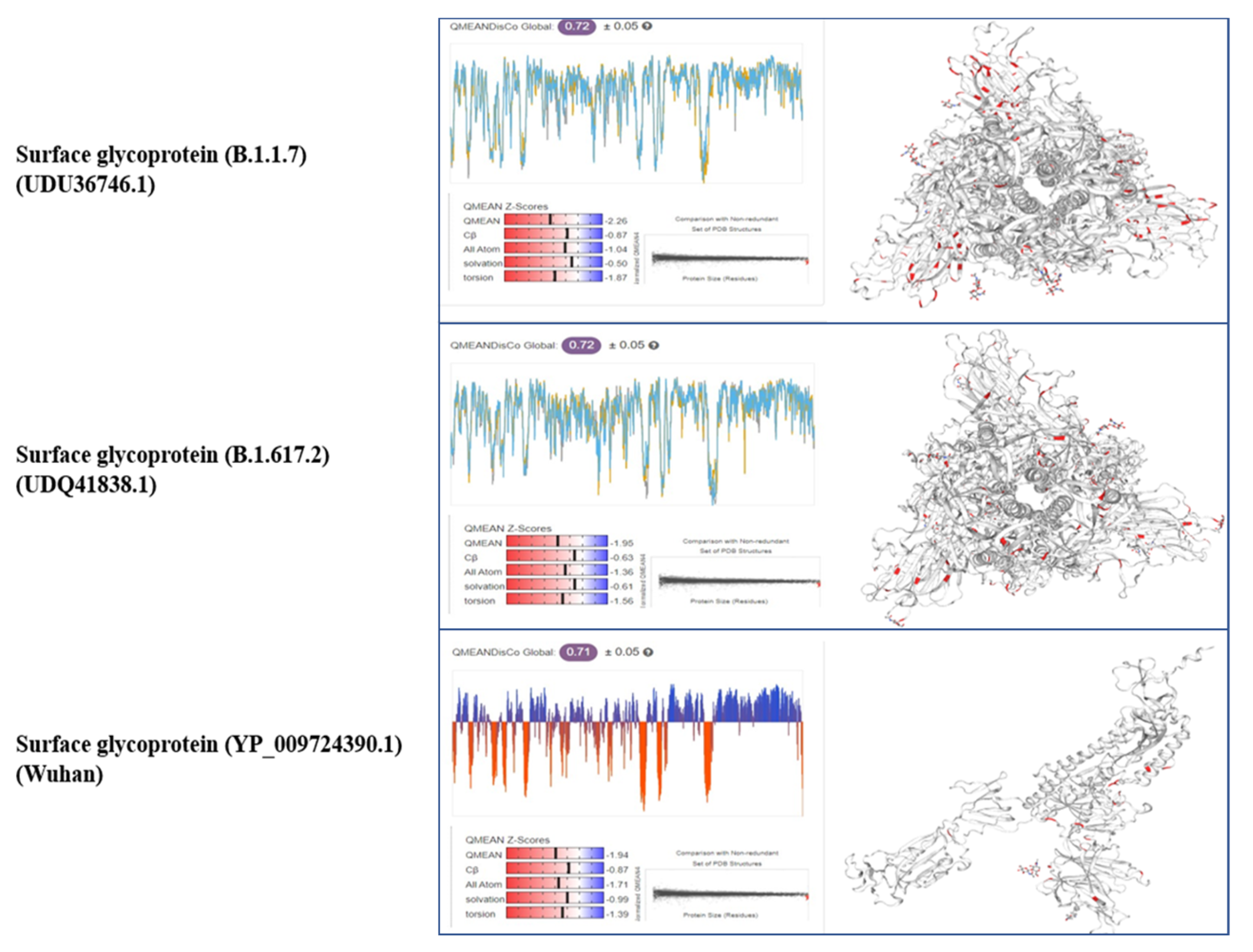{kind=link}
{kind=link}
| Proteins | Variants | Accession ID | Alpha Helix (Hh) | Extended Strand (Ee) | Beta Turn (Tt) | Random Coil (Cc) |
|---|---|---|---|---|---|---|
| Envelope protein | B.1.617.21 | UDU36748.1 | 33 is 44% | 20 is 26.67% | 7 is 9.33% | 15 is 20% |
| B.1.1.7 | UDQ41840.1 | 33 is 44% | 20 is 26.67% | 7 is 9.33% | 15 is 20% | |
| Wuhan | YP_009724392. 1 | 33 is 44% | 20 is 26.67% | 7 is 9.33% | 15 is 20% | |
| Membrane | B.1.617.21 | UDU36749.1 | 81 is 36.49% | 48 is 21.62% | 13 is 5.86% | 80 is 3 6.04% |
| B.1.1.7 | UDQ41841.1 | 77 is 34.68% | 47 is 21.17% | 15 is 6.76% | 83 is 37.39% | |
| Wuhan | YP_009724393. 1 | 77 is 34.68% | 47 is 21.17% | 15 is 6.76% | 83 is 37.39% | |
| Nucleocapsid | B.1.617.21 | UDU36754.1 | 87 is 20.76% | 70 is 16.71% | 32 is 7.64% | 230 is 54.89% |
| B.1.1.7 | UDQ41846.1 | 73 is 17.42% | 75 is 17.9% | 29 is 6.92% | 242 is 57.76% | |
| Wuhan | YP_009724397. 2 | 89 is 21.24% | 70 is 16.71% | 29 is 6.92% | 231 is 55.13% | |
| ORF10 Protein | B.1.617.21 | UDU36755.1 | 11 is 28.95% | 14 is 36.84% | 2 is 5.26% | 11 is 28.95% |
| B.1.1.7 | UDQ41847.1 | 11 is 28.95% | 14 is 36.84% | 2 is 5.26% | 11 is 28.95% | |
| Wuhan | YP_009725255. 1 | 11 is 28.95% | 12 is 31.58% | 2 is 5.26% | 13 is 34.21% | |
| ORF1a | B.1.617.21 | UDU36745.1 | 1773 is 40.25% | 911 is 20.68% | 353 is 8.01% | 1368 is 31.06% |
| B.1.1.7 | UDQ41837.1 | 1773 is 40.25% | 895 is 20.33% | 351 is 7.97% | 1383 is 31.42% | |
| Wuhan | YP_009725295.1 | 1776 is 40.32% | 909 is 20.64% | 351 is7. 97% | 1369 is 31.08% | |
| ORF1ab Polyprotein | B.1.617.21 | UDU36744.1 | 1937 is 38.75% | 1106 is 22.12% | 493 is 9.86% | 1463 is 29.27% |
| B.1.1.7 | UDQ41836.1 | 1944 is 38.91 | 1106 is 22.14% | 497 is 9.95% | 1449 is 29% | |
| Wuhan | YP_009724389.1 | 1908 is 40.39% | 949 is 20.09% | 345 is 7.30% | 1522 is 32.22% | |
| ORF3a Protein | B.1.617.21 | UDU36747.1 | 72 is 26.18% | 80 is 29.09% | 28 is 10.18% | 95 is 34.55% |
| B.1.1.7 | UDQ41839.1 | 72 is 26.18% | 82 is 29.82% | 28 is 10.18% | 93 is 33.82% | |
| Wuhan | YP_009724391.1 | 72 is 26.18% | 82 is 29.82% | 28 is 10.18% | 93 is 33.82% | |
| ORF6 Protein | B.1.617.21 | UDU36750.1 | 43 is 70.49% | 6 is 9.84% | 5 is 8.2% | 7 is 11.48% |
| B.1.1.7 | UDQ41842.1 | 43 is 70.49% | 6 is 9.84% | 5 is 8.2% | 7 is 11.48% | |
| Wuhan | YP_009724394.1 | 43 is 70.49% | 6 is 9.84% | 5 is 8.2% | 7 is 11.48% | |
| ORF7a Protein | B.1.617.21 | UDU36751.1 | 53 is 43.8% | 24 is 19.83% | 11 is 9.09% | 33 is 27.27% |
| B.1.1.7 | UDQ41843.1 | 52 is 42.98% | 23 is 19.01% | 12 is 9.92% | 34 is 28.1% | |
| Wuhan | YP_009724395.1 | 52 is 42.98% | 23 is 19.01% | 12 is 9.92% | 34 is 28.1% | |
| ORF7b | B.1.617.21 | UDU36752.1 | 32 is 74.42% | 1 is 2.33% | 1 is 2.33% | 9 is 20.93% |
| B.1.1.7 | UDQ41844.1 | 32 is 74.42% | 1 is 2.33% | 1 is 2.33% | 9 is 20.93% | |
| Wuhan | YP_009725318.1 | 32 is 74.42% | 1 is 2.33% | 1 is 2.33% | 9 is 20.93% | |
| ORF8 protein | B.1.617.21 | UDU36753.1 | 24 is 19.83% | 43 is 35.54% | 6 is 4.96% | 48 is 39.67% |
| - | - | - | - | - | - | |
| Wuhan | YP_009724396.1 | 24 is 19.83% | 43 is 35.54% | 6 is 4.96% | 48 is 39.67% | |
| Surface Glycoprotein | B.1.617.21 | UDU36746.1 | 381 is 29.98% | 271 is 21.32% | 40 is 3.15% | 579 is 45.55% |
| B.1.1.7 | UDQ41838.1 | 377 is 29.69% | 284 is 22.36% | 45 is 3.54% | 564 is 44.41% | |
| Wuhan | YP_009724390.1 | 364 is 28.59% | 296 is 23.25% | 43 is 3.38% | 570 is 44.78% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haseeb, M.; Amir, A.; Ikram, A. In Silico Analysis of SARS-CoV-2 Spike Proteins of Different Field Variants. Vaccines 2023, 11, 736. https://doi.org/10.3390/vaccines11040736
Haseeb M, Amir A, Ikram A. In Silico Analysis of SARS-CoV-2 Spike Proteins of Different Field Variants. Vaccines. 2023; 11(4):736. https://doi.org/10.3390/vaccines11040736
Chicago/Turabian StyleHaseeb, Muhammad, Afreenish Amir, and Aamer Ikram. 2023. "In Silico Analysis of SARS-CoV-2 Spike Proteins of Different Field Variants" Vaccines 11, no. 4: 736. https://doi.org/10.3390/vaccines11040736
APA StyleHaseeb, M., Amir, A., & Ikram, A. (2023). In Silico Analysis of SARS-CoV-2 Spike Proteins of Different Field Variants. Vaccines, 11(4), 736. https://doi.org/10.3390/vaccines11040736