
Immunoinformatics Strategy to Develop a Novel Universal Multiple Epitope-Based COVID-19 Vaccine

 , and
, and
Abstract
:1. Introduction
2. Materials and Procedures
2.1. Sequence Retrieval, Structural Analysis, and Sequence Alignment
2.2. B-Cell Epitopes Prediction
2.3. T-Cell Epitopes Prediction
2.4. Allergenicity and Antigenicity Profiling of Selected T and B-Cell Epitopes
2.5. Analysis of Conservation
2.6. Multi-Epitope Vaccine Construction
2.7. Analysis of Solubility and Physiochemical Properties
2.8. Extrapolation of Secondary and Tertiary Structures
2.9. Validation and Tertiary Structure Improvement
2.10. Docking Evaluation
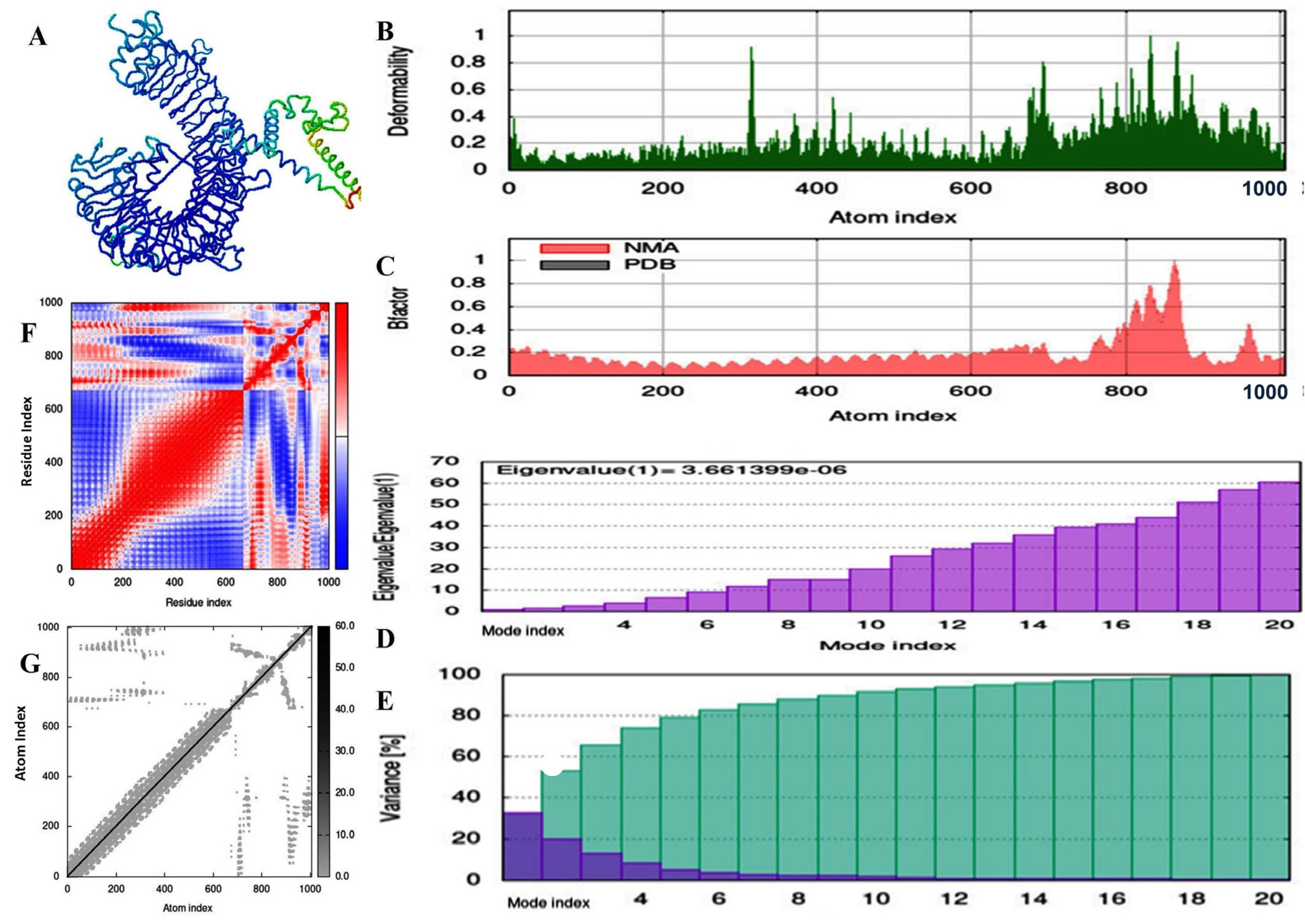
2.11. Molecular Dynamic and Simulation Analysis of Predicted Vaccine Construct
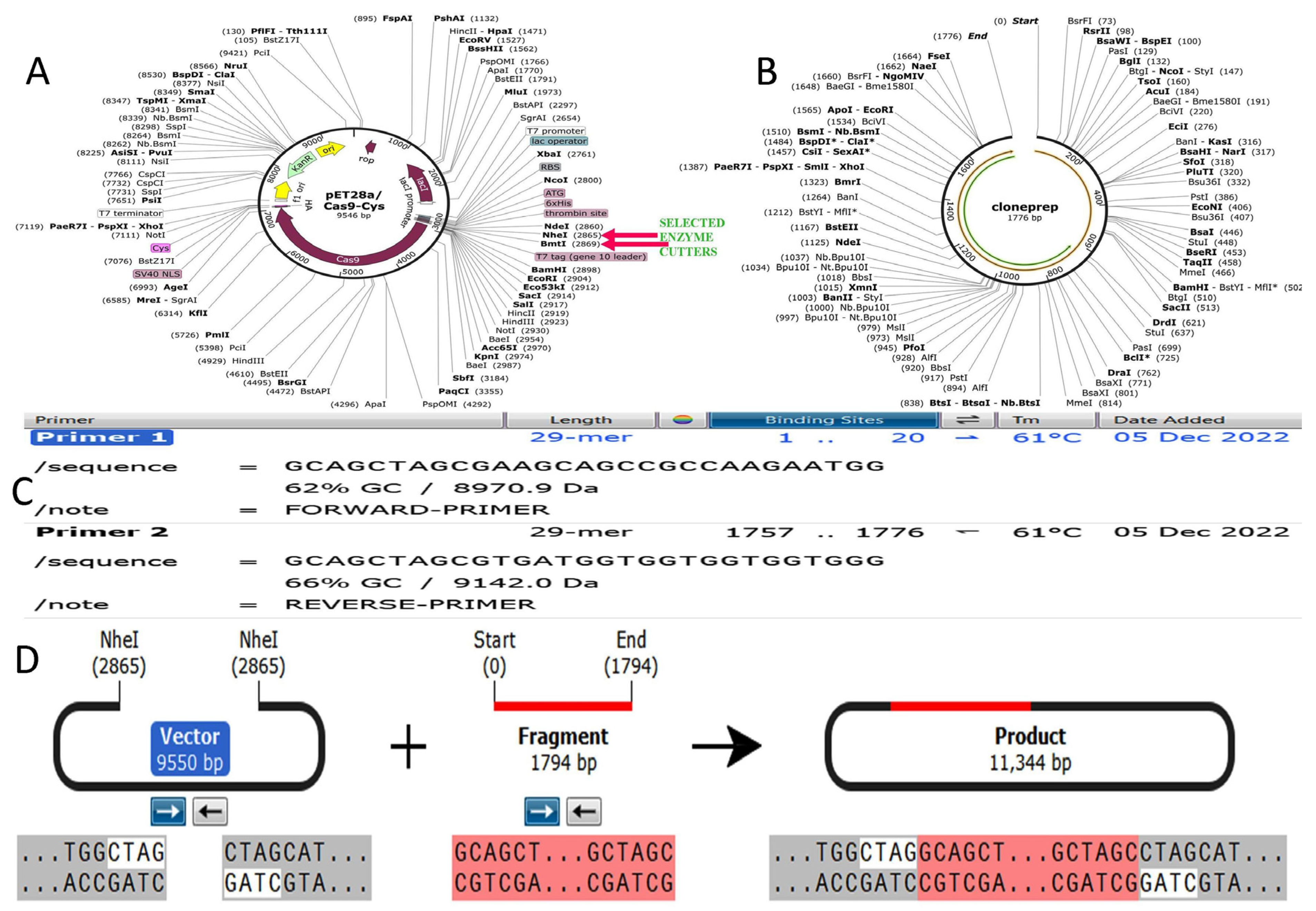
2.12. Codon Optimization of Designed Vaccine Peptide for Expression Analysis
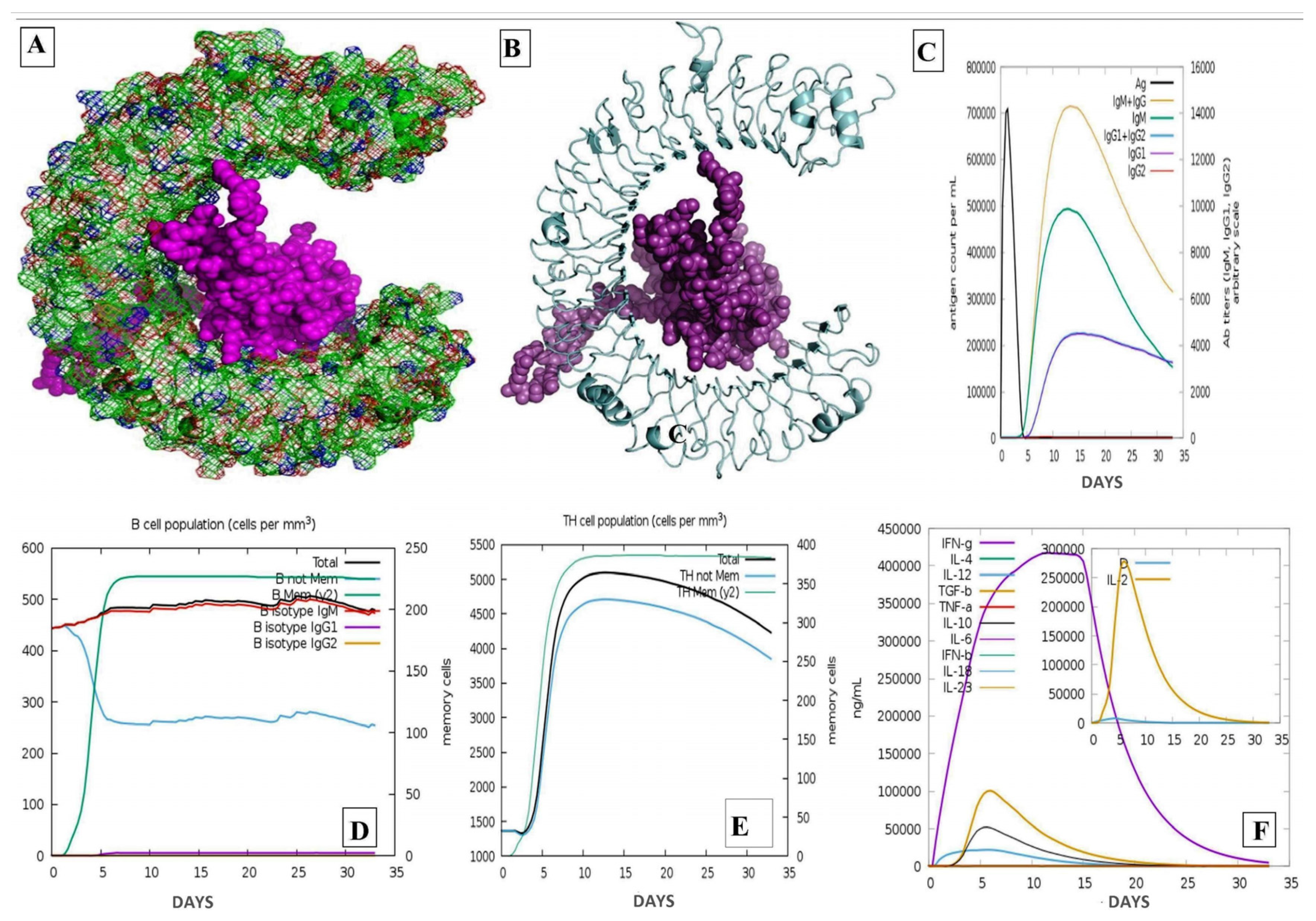
2.13. Analysis of Immune Simulation
3. Results
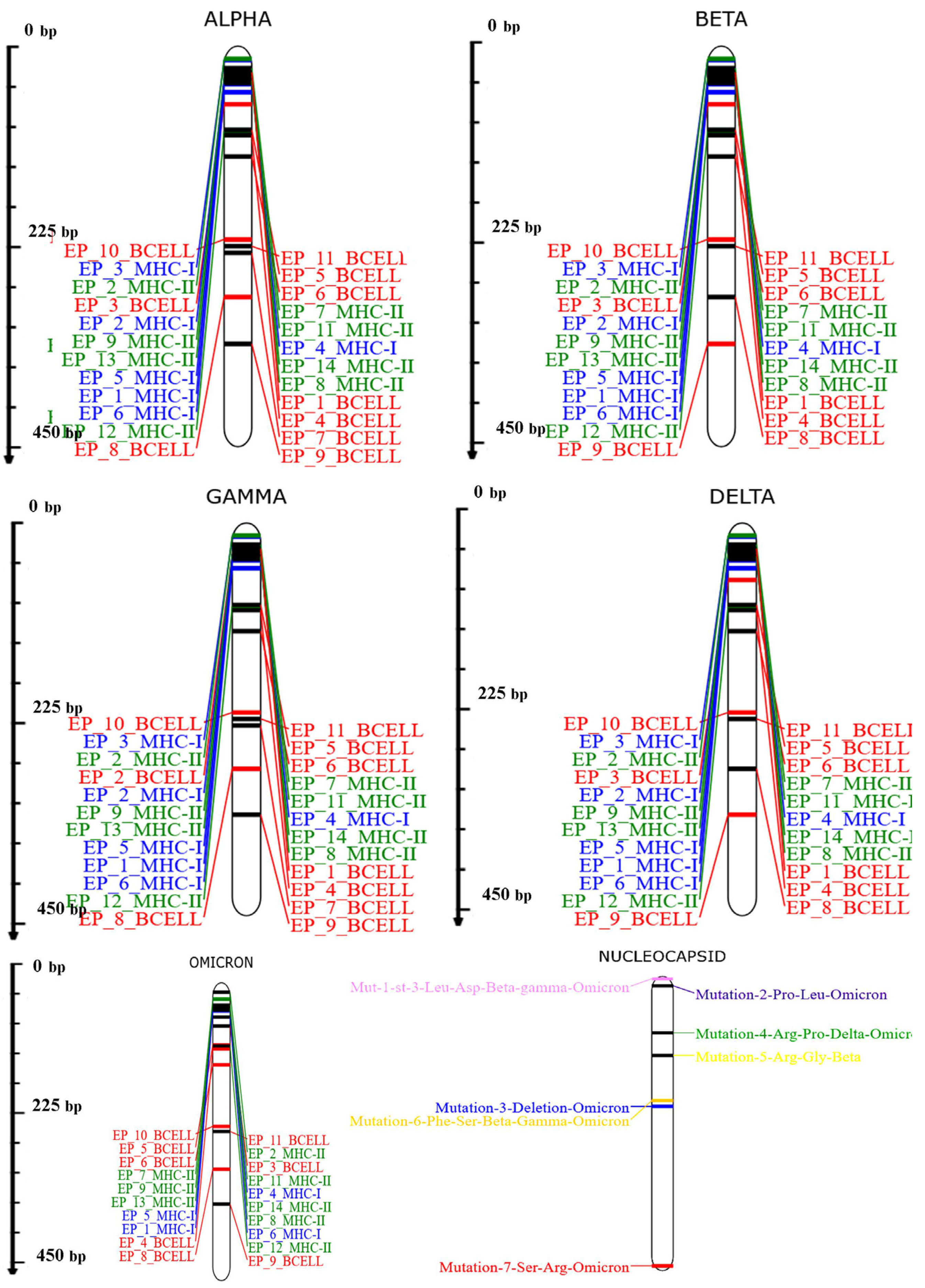
3.1. MSA Analysis and Selection of Conserved Segment for Consideration of Epitopes
3.2. Sequence and Structure Analysis
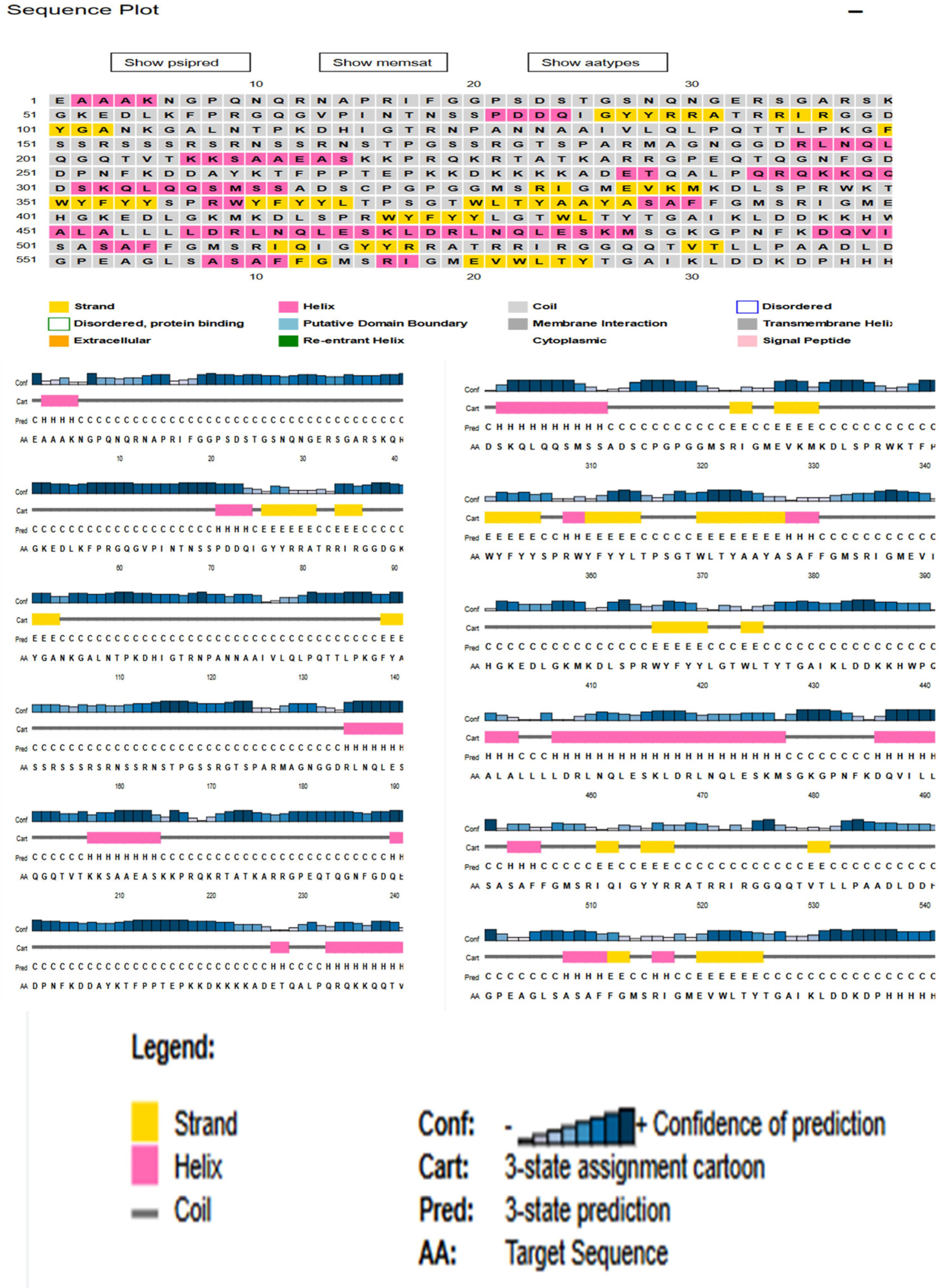
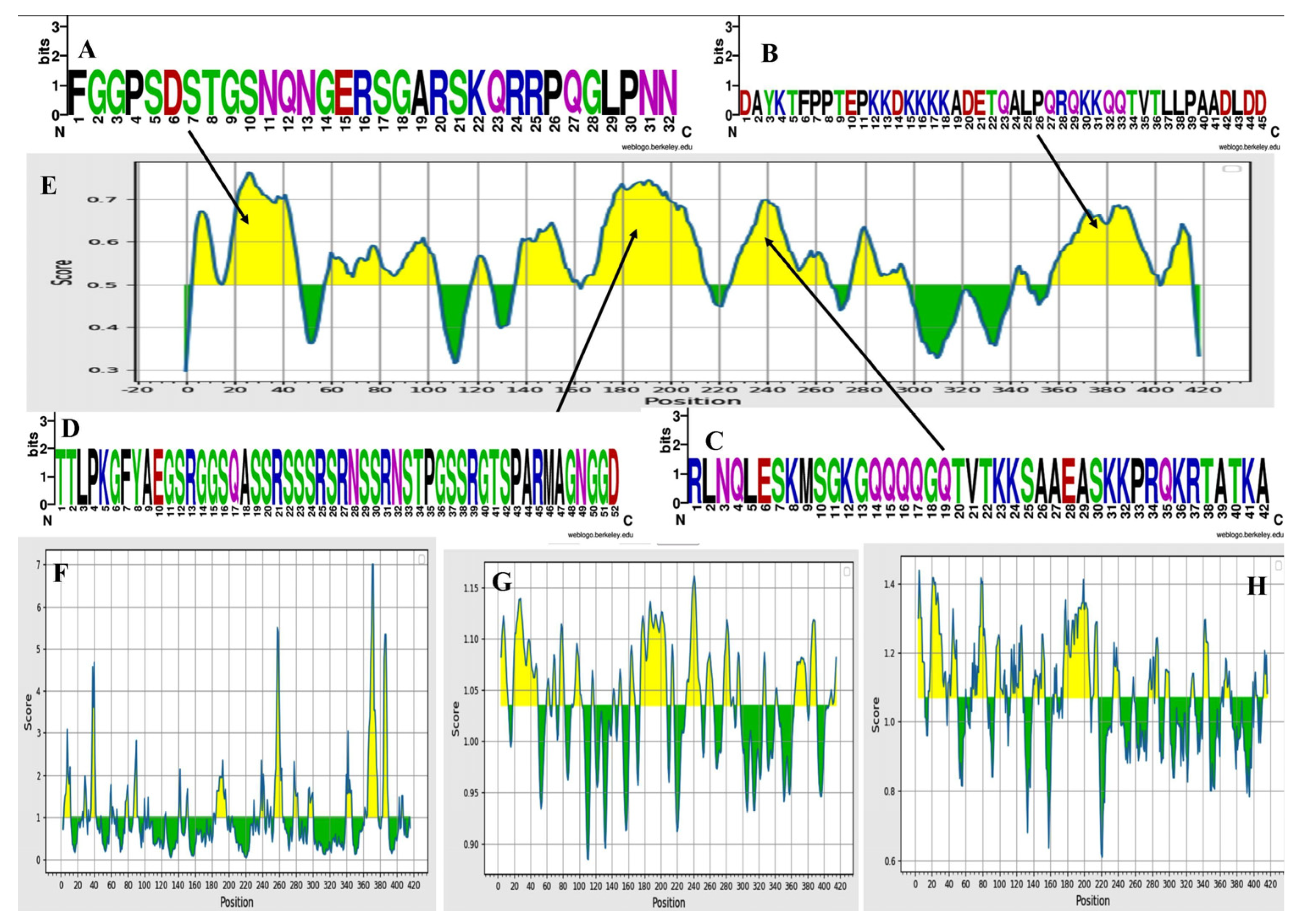
3.3. Physiochemical Analysis, Secondary Structure and Transmembrane Topology Prediction of N Protein
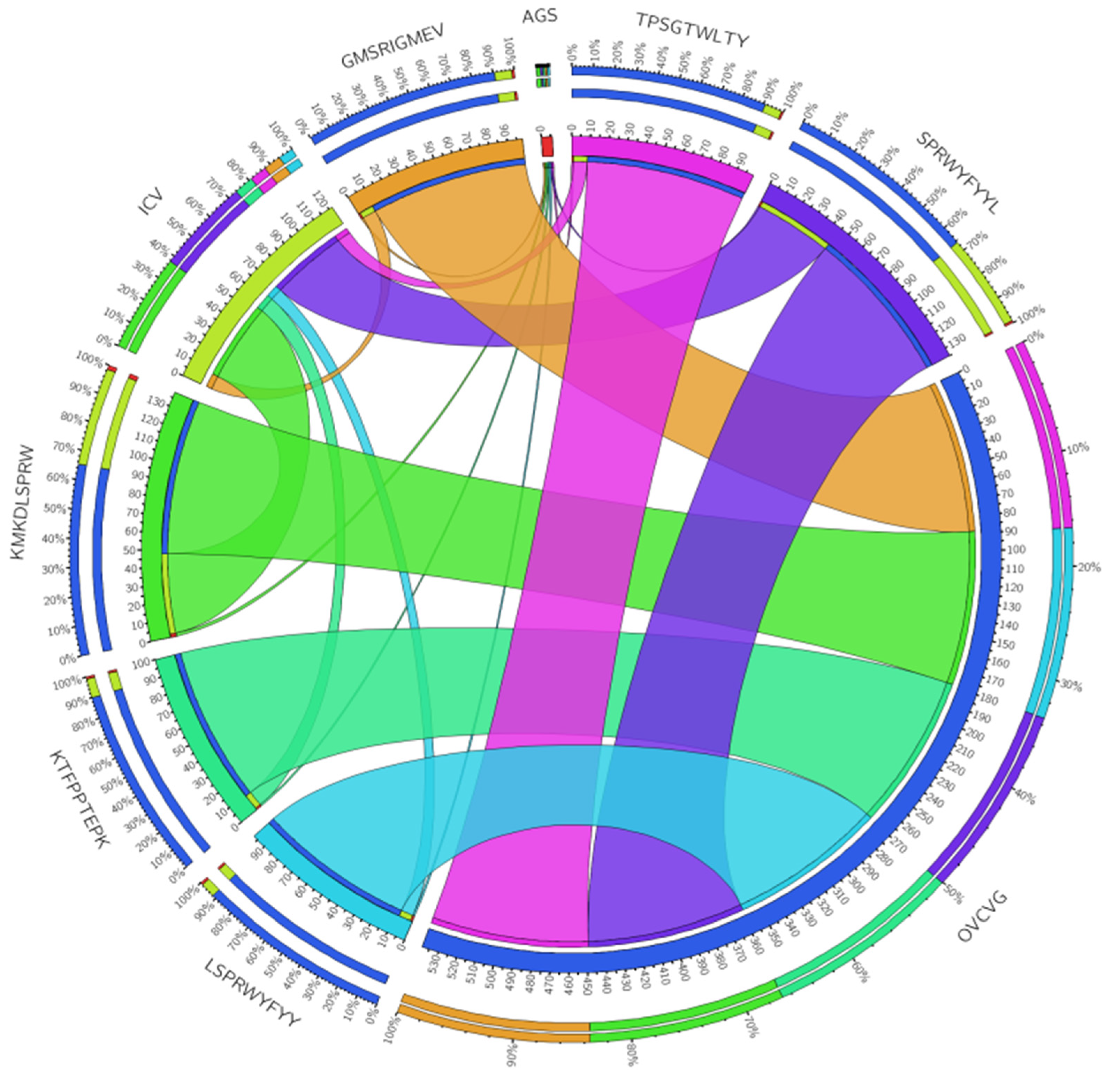
3.4. Linear B Cell Epitope Prediction
3.5. T-Cell Epitope Identification
3.5.1. Prediction of MHC Class-I Binding Profile for Conserved Epitopes
3.5.2. MHC Class II Binding Profile Prediction for Conserved Epitopes
3.6. Assembly of Vaccine Construct
3.7. Investigation of the Population Coverage and Epitope Conservation
3.8. Analysis of Solubility and Physiochemical Properties of Multi-Epitope Subunit
3.9. Antigenicity and Allergenicity Evaluation of the Vaccine Protein
3.10. Secondary Structure Extrapolation
3.11. Protein’s Tertiary Structure Evaluation
3.12. Tertiary Structure Prediction and Validation of Vaccine Construct
3.13. Molecular Docking with Ligand Binding Domain of TLR3
3.14. Codon Optimization and Cloning Expression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, W.; Zong, W.; Wang, F.; Ju, S. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): A review. Mol. Cancer 2020, 19, 100. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Tian, M.; Wu, N.; Yang, X.; Qi, J.; Ren, W.; Li, F.; Bian, H. SARS-CoV-2 and Emerging Variants: Unmasking Structure, Function, Infection, and Immune Escape Mechanisms. Front. Cell. Infect. Microbiol. 2022, 12, 869832. [Google Scholar] [CrossRef] [PubMed]
- Palatnik-de-Sousa, I.; Wallace, Z.S.; Cavalcante, S.C.; Ribeiro, M.P.F.; Silva, J.A.B.M.; Cavalcante, R.C.; Scheuermann, R.H.; Palatnik-de-Sousa, C.B. A novel vaccine based on SARS-CoV-2 CD4+ and CD8+ T cell conserved epitopes from variants Alpha to Omicron. Sci. Rep. 2022, 12, 16731. [Google Scholar] [CrossRef] [PubMed]
- Tarke, A.; Grifoni, A.; Sette, A. Bioinformatic and Experimental Analysis of T Cell Immune Reactivity to SARS-CoV-2 and its Variants. Front. Bioinform. 2022, 2, 876380. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Mentzer, A.J.; O’Connor, D.; Bibi, S.; Chelysheva, I.; Clutterbuck, E.A.; Demissie, T.; Dinesh, T.; Edwards, N.J.; Felle, S.; Feng, S. Human leukocyte antigen alleles associate with COVID-19 vaccine immunogenicity and risk of breakthrough infection. Nat. Med. 2022, 29, 147–157. [Google Scholar] [CrossRef]
- Mullard, A. COVID antibody drugs have saved lives-so why aren’t they more popular? Nature 2022, 606, 854–856. [Google Scholar] [CrossRef]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The SARS coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Pan, Y.; Cai, W.; Cheng, A.; Wang, M.; Yin, Z.; Jia, R. Flaviviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Front. Immunol. 2022, 13, 829433. [Google Scholar] [CrossRef]
- Lien, E.; Ingalls, R.R. Toll-like receptors. Crit. Care Med. 2002, 30, S1–S11. [Google Scholar] [CrossRef]
- Link, A. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigan-Womas, I.; Spadoni, J.L.; Poiret, T.; Taieb, F.; Randrianarisaona, F.; Faye, R.; Mbow, A.A.; Gaye, A.; Dia, N.; Loucoubar, C.; et al. Linear epitope mapping of the humoral response against SARS-CoV-2 in two independent African cohorts. Sci. Rep. 2023, 13, 782. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Ovchinnikov, S.; Park, H.; Kim, D.E.; DiMaio, F.; Baker, D. Protein structure prediction using Rosetta in CASP12. Proteins 2018, 86 (Suppl. S1), 113–121. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Seliger, B.; Ferrone, S. HLA Class I Antigen Processing Machinery Defects in Cancer Cells-Frequency, Functional Significance, and Clinical Relevance with Special Emphasis on Their Role in T Cell-Based Immunotherapy of Malignant Disease. Methods Mol. Biol. 2020, 2055, 325–350. [Google Scholar] [CrossRef]
- Schein, C.H.; Ivanciuc, O.; Braun, W. Bioinformatics approaches to classifying allergens and predicting cross-reactivity. Immunol. Allergy Clin. N. Am. 2007, 27, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef] [PubMed]
- Khade, P.M.; Scaramozzino, D.; Kumar, A.; Lacidogna, G.; Carpinteri, A.; Jernigan, R.L. hdANM: A new comprehensive dynamics model for protein hinges. Biophys. J. 2021, 120, 4955–4965. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Start | End | Peptide | Length |
|---|---|---|---|
| 4 | 15 | NGPQNQRNAPRI | 12 |
| 17 | 48 | FGGPSDSTGSNQNGERSGARSKQRRPQGLPNN | 32 |
| 59 | 105 | HGKEDLKFPRGQGVPINTNSSPDDQIGYYRRATRRIRGGDGKMKDLS | 47 |
| 119 | 127 | AGLPYGANK | 9 |
| 137 | 163 | GALNTPKDHIGTRNPANNAAIVLQLPQ | 27 |
| 165 | 216 | TTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGD | 52 |
| 226 | 267 | RLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKA | 42 |
| 276 | 299 | RRGPEQTQGNFGDQELIRQGTDYK | 24 |
| 343 | 348 | DPNFKD | 6 |
| 358 | 402 | DAYKTFPPTEPKKDKKKKADETQALPQRQKKQQTVTLLPAADLDD | 45 |
| 404 | 416 | SKQLQQSMSSADS | 13 |
| Start | End | Peptide | Length |
|---|---|---|---|
| 52 | 59 | WFTALTQH | 8 |
| 69 | 75 | GQGVPIN | 7 |
| 83 | 89 | QIGYYRR | 7 |
| 106 | 115 | PRWYFYYLGT | 10 |
| 119 | 124 | AGLPYG | 6 |
| 130 | 136 | IIWVATE | 7 |
| 154 | 166 | NAAIVLQLPQGTT | 13 |
| 217 | 227 | AALALLLLDRL | 11 |
| 243 | 249 | GQTVTKK | 7 |
| 267 | 273 | AYNVTQA | 7 |
| 299 | 315 | KHWPQIAQFAPSASAFF | 17 |
| 333 | 339 | YTGAIKL | 7 |
| 347 | 363 | KDQVILLNKHIDAYKTF | 17 |
| 379 | 385 | TQALPQR | 7 |
| 389 | 401 | QQTVTLLPAADLD | 13 |
| 403 | 411 | FSKQLQQSM | 9 |
| Peptide Start | Peptide End | IC50 | Epitopes | Allergenicity | Antigenic Score | Antigenicity |
|---|---|---|---|---|---|---|
| 36 | 44 | 7.43 | GMSRIGMEV | HLA-A*02:03 | 0.6287 | NA |
| 30 | 38 | 46.84 | KMKDLSPRW | HLA-A*32:01 | 1.7462 | NA |
| 11 | 19 | 8.51 | KTFPPTEPK | HLA-A*11:01 | 0.7571 | NA |
| 34 | 42 | 44.28 | LSPRWYFYY | HLA-A*30:02 | 1.2832 | NA |
| 35 | 43 | 8.33 | SPRWYFYYL | HLA-B*07:02 | 0.734 | NA |
| 45 | 53 | 7.45 | TPSGTWLTY | HLA-B*35:01 | 0.145 | NA |
| Peptide Start | Peptide End | IC50 | Epitopes | Allergenicity | Antigenic Score |
|---|---|---|---|---|---|
| 82 | 96 | 44.72 | KHWPQIAQFAPSASA | non allergen | 0.4293 |
| 127 | 141 | 45 | LALLLLDRLNQLESK | non allergen | 0.4293 |
| 311 | 325 | 70.51 | QIGYYRRATRRIRGG | non allergen | 0.4614 |
| 348 | 362 | 71.83 | QQTVTLLPAADLDDF | non allergen | 0.4614 |
| 311 | 325 | 70.51 | QFAPSASAFFGMSRI | non allergen | 0.4658 |
| 311 | 325 | 89.66 | SASAFFGMSRIGMEV | non allergen | 0.6584 |
| 219 | 233 | 54.34 | LDRLNQLESKMSGKG | non allergen | 0.7029 |
| 354 | 368 | 81.97 | RWYFYYLGTGPEAGL | non allergen | 0.7505 |
| 325 | 339 | 8.27 | ASAFFGMSRIGMEVT | non allergen | 0.862 |
| 82 | 96 | 69.21 | PNFKDQVILLNKHID | non allergen | 0.988 |
| 311 | 325 | 39.08 | GTWLTYTGAIKLDDK | non allergen | 0.9934 |
| 82 | 96 | 17.28 | ASWFTALTQHGKEDL | non allergen | 0.4116 |
| 348 | 362 | 38.67 | GKMKDLSPRWYFYYL | non allergen | 1.1625 |
| 349 | 363 | 95.13 | WLTYTGAIKLDDKDP | non allergen | 1.2787 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamjan, N.A.; Lohani, M.; Khan, M.F.; Khan, S.; Algaissi, A. Immunoinformatics Strategy to Develop a Novel Universal Multiple Epitope-Based COVID-19 Vaccine. Vaccines 2023, 11, 1090. https://doi.org/10.3390/vaccines11061090
Khamjan NA, Lohani M, Khan MF, Khan S, Algaissi A. Immunoinformatics Strategy to Develop a Novel Universal Multiple Epitope-Based COVID-19 Vaccine. Vaccines. 2023; 11(6):1090. https://doi.org/10.3390/vaccines11061090
Chicago/Turabian StyleKhamjan, Nizar A., Mohtashim Lohani, Mohammad Faheem Khan, Saif Khan, and Abdullah Algaissi. 2023. "Immunoinformatics Strategy to Develop a Novel Universal Multiple Epitope-Based COVID-19 Vaccine" Vaccines 11, no. 6: 1090. https://doi.org/10.3390/vaccines11061090