1. Introduction
The SARS-CoV-2 pandemic highlights an urgent need for vaccine platforms that can efficiently control viral outbreaks [
1,
2]. It is important that vaccines can be rapidly and efficiently made available to large populations and that protective immune responses are achieved after vaccination. Enveloped virus-like particles (eVLPs) show promise as a potential vaccine platform to combat infectious diseases because they structurally and antigenically mimic the authentic virus but lack the viral genome [
3,
4,
5,
6,
7,
8]. eVLPs are different from inactivated virus vaccines, which contain the viral genome and are usually made non-infectious by exposure to chemical or physical inactivating agents. Such chemical or physical treatment, such as the most commonly used inactivating agent, formalin, can induce irreversible changes in viral antigens, resulting in poor immunogenicity and weak cell-mediated and mucosal immune responses [
9]. Multiple studies have shown that repetitive display of antigenic epitopes on the surface of eVLPs that range in size from ~20 to ~200 nm can facilitate optimal migration to lymph nodes and uptake by antigen-presenting cells, particularly dendritic cells (DCs). Enhanced uptake and processing by DCs enable eVLPs to induce powerful immune responses [
3,
4,
10,
11,
12]. Despite this, the production of eVLPs has remained challenging. For example, the current eVLP purification process is intricate and unscalable [
3,
4,
10,
13,
14,
15]. The genetic delivery of eVLP through immunization with a DNA construct (DNA vaccine platform) or with a synthetic mRNA (mRNA vaccine platform) can bypass the eVLP purification process. DNA and mRNA vaccine platforms have proven to be rapidly scalable and available for the vaccination of large populations [
16,
17].
To circumvent the challenges of in vitro eVLP preparation, we immunized mice with eVLPs displaying SARS-CoV-2 spikes through genetic delivery with a DNA vaccine platform [
18] and evaluated if genetic delivery of eVLPs could elicit strong humoral immune responses in mice as previously reported from immunization with in vitro purified eVLPs [
6]. We compared humoral immune responses induced by a single-gene-transcript eVLP vaccine platform, by a multiple-gene-transcript eVLP vaccine platform, and by a protein subunit vaccine platform, the latter of which had the same sequence as that utilized in a SARS-CoV-2 mRNA vaccine [
17,
19], expressed as a membrane-bound form, which has shown success in combating COVID-19. Our study demonstrates that the genetic delivery of eVLPs could provide a rapid and efficient means to deliver eVLPs and that a different vaccine platform may have a different mechanism of inducing neutralizing antibodies, which could potentially inspire new vaccines that elicit strong and durable neutralizing immunity.
2. Materials and Methods
2.1. Animal
BALB/c mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA), and all animals were housed and cared for in accordance with American Association for Accreditation of Laboratory Animal Care standards in accredited facilities.
2.2. Cell Line
HEK293T cells (#CRL3216, ATCC, VA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM)-formulated optimal cell growth medium containing 12% inactivated fetal bovine serum (FBS) and 100 U/mL streptomycin–penicillin (ABI Scientific, Sterling, VA, USA) at 37 °C and 5% CO2.
2.3. Preparation of DNA Constructs
DNA construct of S-2P-TM, a full-length of WA-1 SARS-CoV-2 prefusion-stabilized spike gene with two proline substitutions at residues 986–987 including spike ectodomain (S-2P, amino acid residues 1–1208) and transmembrane cytoplasm tail domain (TM, amino acid residues 1209–1273), encoded a cell-membrane-anchored S-2P (GenBank: MN908947). The DNA construct of S-2Pᵦ-TM was the Beta variant of spike (B.1.351) [
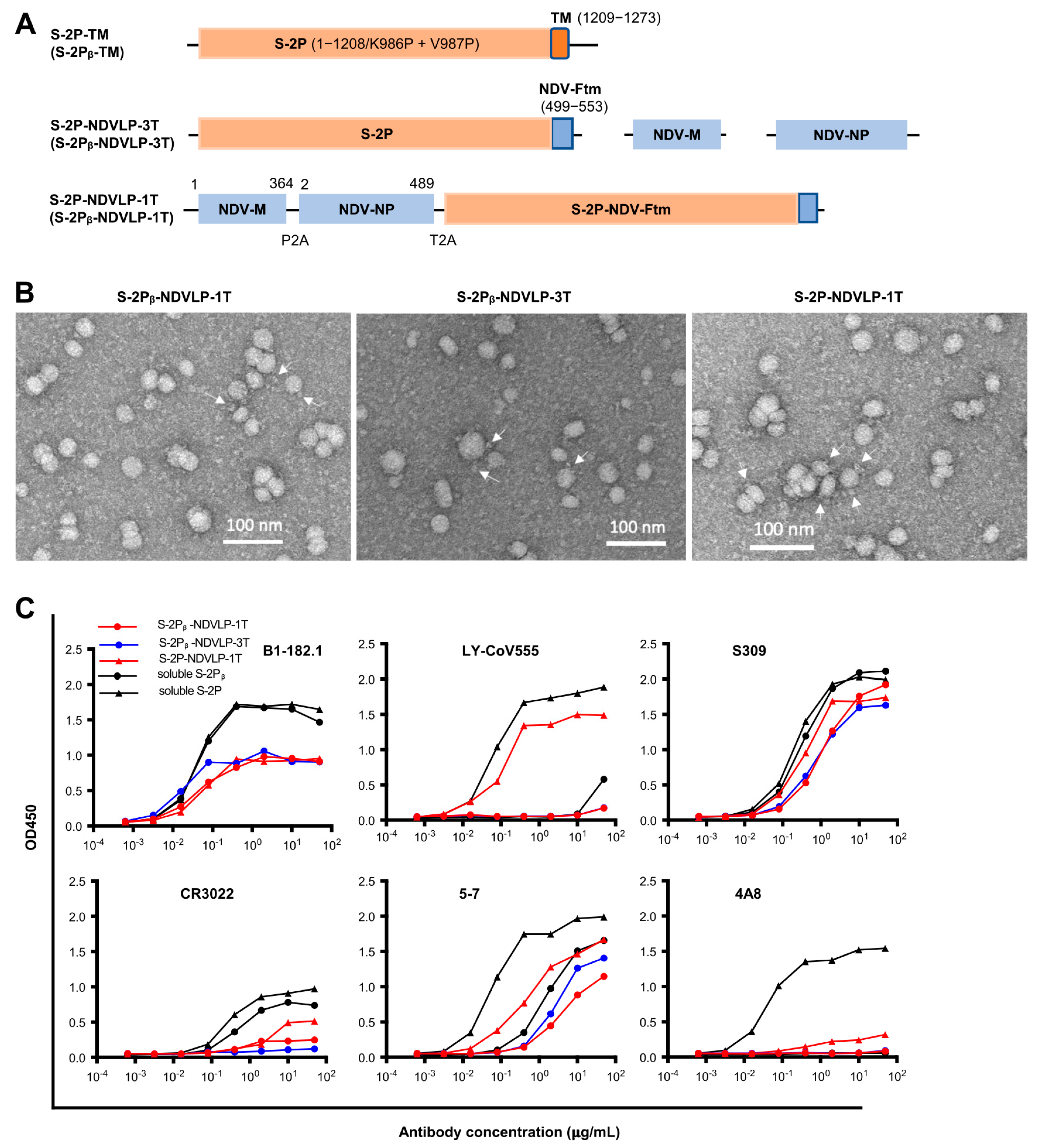
20]. The DNA construct of S-2P-NDV-Ftm or S-2Pᵦ-NDV-Ftm, a chimeric gene including a SARS-CoV-2 prefusion-stabilized spike ectodomain gene (S-2P or S-2Pᵦ, amino acid residues 1–1208) fused with an NDV fusion protein (NDV-F, avian avulavirus 1, GenBank: AMQ09764) transmembrane-cytoplasmic domain gene (Ftm, amino acid residues 499–553), encoded a cell-membrane-anchored S-2P or S-2Pᵦ [
6]. To make a DNA construct of a single-gene-transcript S-2P-NDVLP-1T or S-2Pᵦ-NDVLP-1T, three genes of the NDV matrix (NDV-M, amino acid residues 1–364, and avian avulavirus 1; GenBank: AMQ09763), nucleoprotein (NDV-NP, amino acid residues 2–489, and avian avulavirus 1; GenBank: AMQ09761), and S-2P-NDV-Ftm or S-2Pᵦ-NDV-Ftm were linked by two self-cleavage peptide sequences (P2A and T2A) as a monocistronic DNA molecule, which encoded a single polypeptide that was cleaved into three individual proteins of NDV-M, NDV-NP, and cell-membrane-anchored S-2P or S-2Pᵦ during post-translation processing in the cell, with exclusive self-assembly into secreted S-2P-NDVLP or S-2Pᵦ-NDVLP. Multiple-gene-transcript DNA constructs of S-2P-NDVLP-3T or S-2Pᵦ-NDVLP-3T were composed of three individual genes of NDV-M, NDV-NP, and S-2P-NDV-Ftm or S-2Pᵦ-NDV-Ftm, which directly encoded three individual proteins of NDV-M, NDV-NP, and cell-membrane-anchored S-2P or S-2Pᵦ, respectively.
To make a DNA construct of single-gene-transcript S-2P-CoV2VLP-1T, the envelope gene (E), matrix gene (M), and full-length prefusion-stabilized spike gene (S-2P-TM) from a WA-1 SARS-CoV2 isolate were linked by two self-cleavage peptide sequences (P2A and T2A) as a monocistronic DNA molecule, which encoded a single polypeptide that was cleaved into three individual proteins of E, M, and cell-membrane-anchored S-2P during post-translation processing in the cell, with exclusive self-assembly into secreted S-2P-CoV2VLP. Multiple-gene-transcript DNA constructs of S-2P-CoV2VLP-3T were composed of WA-1 SARS-CoV2 and three individual genes of E, M, and S-2P-TM, directly encoding three individual proteins of E, M, and cell-membrane-anchored S-2P, respectively. Expression control for the DNA construct of S-2P or S-2Pᵦ was achieved with the gene of the prefusion-stabilized spike ectodomain (amino acid residues 1–1208) from the SARS-CoV2 WA-1 or Beta isolate. Each gene was codon-optimized, synthesized, and cloned into a VRC8400 vector (CMV/R expression vector developed and used in house) as a plasmid DNA construct (GeneImmune Biotechnology, New York, NY, USA).
2.4. Cell Culture Microplate-Formatted Expression
First, 2.5 × 104 log-phase HEK 293T cells in 100 μL of RealFect Expression medium (ABI Scientific, Sterling, VA, USA) per well were inoculated in a 96-well cell culture microplate (Corning Scientific, Corning, NY, USA) and allowed to grow for 24 h at 37 °C under 5% CO2. Immediately before transfection, 40 μL of spent medium per well was removed, and 250 ng of plasmid DNA encoding either eVLP or soluble protein in 10 μL of Opti-MEM Reduced Serum medium (Thermo Fisher Scientific, Rockville, MD, USA) was mixed with 0.75 μL of TrueFect Max transfection reagent (United Biosystems, Bethesda, MD, USA) in 10 μL of Opti-MEM Reduced Serum medium at room temperature (RT) for 15 min, and then mixed with growing cells in each well in the 96-well cell culture microplate. Transfected cells were incubated at 37 °C under 5% CO2 for 6–12 h, then fed with 25 μL of CelBooster medium (Cell Growth Enhancer for adherent cells, ABI Scientific) per well, with an additional 10% FBS and 3 × 100 U/mL streptomycin–penicillin. Starting from day 3 post transfection, the supernatants in the wells of a 96-well cell culture microplate were sampled, and the yields of eVLPs were measured by an eVLP ELISA assessment with a SARS-CoV-2 neutralizing antibody (S309).
2.5. Expression and Purification of eVLP
First 7 × 10
6 log-phase growing HEK293T cells were seeded in a T75-cm
2 flask (Corning Scientific) and cultured at 37 °C under 5% CO
2 for 24 h, achieving 85% cell confluency. Prior to transfection, the spent culture medium in the flask was replaced with 8 mL of fresh RealFect expression medium, and 25 μg of plasmid DNA in 1.1 mL of serum-free Opti-MEM medium was mixed with 75 μL of TrueFect transfection reagent in 1.1 mL of serum-free Opti-MEM for 15 min at RT, then mixed with growing cells in a T75-cm
2 flask. Transfected cells were incubated at 37 °C under 5% CO
2 for 6–12 h, then fed with 5 mL of CelBooster medium, with an additional 10% FBS and 3 × 100 U/mL streptomycin–penicillin. Three days after transfection, the culture supernatant in transfected cell flasks was harvested, and eVLPs were purified by polyethylene glycol (PEG)-mediated precipitation and sucrose gradient ultracentrifugation method as follows. Harvested supernatant was clarified by centrifugation at 1500×
g at 4 °C for 30 min, followed by filtration through a 0.8 μm filter, slow mixing with ¼ volume of Universal PEG Virus Precipitation Solution (ABI Scientific), and gentle shaking at 4 °C for 2 h. Then, the supernatant was incubated at 4 °C overnight. After centrifugation of the supernatant–PEG mixture at 2000×
g for 30 min at 4 °C, the top phase solution was removed, and precipitated eVLPs in pellet form were resuspended in MES buffer (150 mL NaCl, 20 mM MES, pH 6.0) and transferred onto the top of a discontinuous sucrose gradient in a 12 mL ultracentrifuge tube (Beckman Coulter, Brea, CA, USA) with 65% sucrose on the bottom and 10% sucrose on the top and a total of 6 layers (65%, 50%, 40%, 30%, 20%, and 10%; 1.5 mL/layer), followed by ultracentrifugation at 36,000 rpm at 4 °C for 12 h (Beckman SW28.1 rotor) [
6,
21]. eVLPs were sedimented in 10–20% and 40% sucrose layers, as determined by eVLP ELISA assessment with a SARS-CoV-2 neutralizing antibody (S309), and stored at −80 °C for analyses. The concentration of eVLPs was measured with a Micro BCA Protein Assay kit (Themo Fisher Scientific).
2.6. Negative Staining Electron Microscopy
The ultracentrifugation-purified eVLP samples were applied to carbon-coated copper grids that were subjected to glow discharging immediately before the experiment. The drop volume was 4.8 µL. After a brief incubation, the drop was removed with filter paper. Three washes with buffer containing 10 mM HEPES (pH 7) and 150 mM NaCl were performed in the same manner. After the final wash, adsorbed material was negatively stained by consecutively applying three drops of 0.7% uranyl formate. Images were recorded at a nominal magnification of 57,000× using a Thermo Scientific Talos F200C electron microscope equipped with a Ceta camera and operated at 200 kV. The pixel size was 0.25 nm.
2.7. Preparation of Prefusion-Stabilized SARS-CoV-2 Spike-Soluble Trimeric Glycoprotein (S-2P or S-2Pᵦ)
The expression plasmid design and protein production of S-2P or S-2Pᵦ trimer protein were performed as described previously [
22]. Briefly, each protein sequence was fused with an 8-mer His-tagger sequence at the carboxyl terminus, and the DNA-encoding code was optimized and synthesized by GeneImmune Biotechnology. Each His-tagged protein was expressed in 293 Freestyle cells by transient transfection for 6 days at 37 °C. On days 1 and 3 post transfection, an enriched feed medium, Cell Growth Enhancer for suspension cells (ABI Scientific, Sterling, VA, USA), was added into the culture at 5% culture volume. Following expression, the cell supernatant was collected and filtered and applied to nickel affinity resin. The protein-bound resin was washed extensively with an increasing imidazole concentration and eluted with 300 mM imidazole buffer. His-tagged HRV3c protease was added to the nickel elution and incubated overnight at 4 °C. The cleaved protein was then concentrated using spin filters and applied to a Superdex S-200 gel filtration column and equilibrated in PBS. The S-2P or S-2Pᵦ protein was eluted primarily in a single peak corresponding to a trimeric size. The peak fraction was collected, concentrated to 1 mg/mL, and flash-frozen in liquid nitrogen prior to storage at −80 °C.
2.8. Preparation of SARS-CoV-2 Neutralizing Antibodies
Antibody heavy-chain and light-chain sequences were codon-optimized, synthesized, and cloned into a VRC8400-based IgG1 vector and co-expressed by transient transfection within Expi293 cells (Thermo Fisher Scientific) according to the manufacturer’s recommendation. Briefly, 50 μg plasmid-encoding heavy-chain genes and 50 μg plasmid-encoding light-chain genes were mixed with 300 μL of Turbo293 transfection reagent (SPEED BioSystems, Gaithersburg, MD, USA) for 15 min, added to 100 mL of cells at a concentration of 2.5 × 106/mL, and incubated in a shaker incubator at 120 rpm and 37 °C under 9% CO2. On days 1 and 3 post transfection, an enriched feed medium, AbBooster Antibody Expression Enhancer for suspension cells (ABI Scientific), was added into the culture at a 10% culture volume. After 5 days post transfection, cell culture supernatant was harvested and purified with a Protein A (GE Healthcare, Chicago, IL, USA) column. The antibody was eluted using IgG Elution Buffer (Thermo Fisher Scientific) and brought to neutral pH (7.0) with 1 M Tris-HCl (pH 8.0). Eluted antibodies were dialyzed against PBS overnight before use.
2.9. eVLP ELISA Assessment
First, 96-well ELISA plates (Nunc Maxisorp, Thermo Fisher Scientific) were coated with 100 μL/well of lectin (Galanthus Nivalis, SIGMA, St. Louis, MO, USA) at a concentration of 5 μg/mL in PBS overnight at 4 °C, followed by blocking with a standard blocking buffer, 1% BSA, and 0.05% Tween in PBS. Following washing, 33 μL of eVLP expression supernatant was mixed with 67 μL of PBS per well and incubated in precoated lectin plates for 1.5 h at RT. Following washing, 100 μL/well of SARS-CoV-2 neutralizing antibodies (S309 or others) at a concentration of 10 μg/mL was incubated in the plates for 50 min at RT. Then, bound antibodies were detected by incubation with a goat anti-human IgG Fc fragment–horseradish peroxidase (HRP) conjugate (Jackson Immunoresearch, West Grove, PA, USA) at 1 × 104 dilution in blocking buffer, as per the manufacture’s instructions, for 30 min at RT. Following washing, the bound anti-human IgG Fc fragment–HRP conjugate was detected with HRP substrate 3,5,3′5′-tetramethylbenzidine (TMB) (BioFX TMB, SurModics, Eden Prairie, MN, USA) for 10 min at RT. The reaction was stopped with 0.5 N sulfuric acid. The optical density at 450 nm (OD450) was read with a spectrophotometer (SpectroPlus, Molecular Devices, San Jose, CA, USA).
2.10. eVLP Antigenic Analysis
First, 96-well ELISA plates (Nunc Maxisorp, Thermo Fisher Scientific) were coated with 100 μL per well of lectin (Galanthus Nivalis, SIGMA) at a concentration of 5 μg/mL in PBS overnight at 4 °C, followed by blocking with a standard blocking buffer, 1% BSA, and 0.05% Tween in PBS. Following washing, 100 μL per well of purified eVLP at a concentration of 25 μg/mL or S-2P or S-2Pᵦ trimer protein at a concentration of 1 μg/mL was incubated in precoated lectin plates for 1.5 h at RT. Following washing, 100 μL per well of serially diluted SARS-CoV-2 neutralizing antibodies at a concentrations ranging from 0.00064 μg/mL to 50 μg/mL was incubated in the plates for 50 min at RT. Then, bound antibodies were detected by incubation with a goat anti-human IgG Fc fragment–HRP conjugate (Jackson Immunoresearch, PA) at 1 × 104 dilution in blocking buffer for 30 min at RT. Following washes, the bound anti-human IgG Fc fragment–HRP conjugate was detected with the HRP substrate of BioFX TMB for 10 min at RT. The reaction was stopped with 0.5 N sulfuric acid. The optical density (OD450) was read at 450 nm with a spectrophotometer (SpectroPlus, Molecular Devices). OD450 values versus serial dilution of SARS-CoV-2 neutralizing antibodies was illustrated using GraphPad Prism v8 software (GraphPad Software, San Diego, CA, USA).
2.11. Mouse Immunization
Female BALB/c mice, aged 8–12 weeks (Jackson Laboratories, Bar Harbor, ME, USA), were immunized intramuscularly by injection and electroporation (BTX AgilePulse, Holliston, MA, USA) with indicated doses of plasmid DNA at week(s) 0, 4 (n = 10 per group), or 20 (n = 5 per group), respectively. The mice were injected with 100 μL of plasmid DNA into the medial large muscle (m. vastus medialis) of each hind limb, followed by electroporation. Two doses comprising different amounts of plasmid DNA were used: a high dose of 50 μg per immunization per mouse and a low dose of 10 μg per immunization per mouse. For immunization of S-2Pᵦ-NDVLP-3T, three individual plasmid DNAs were mixed in the same plasmid molar ratio. Mice were bled at week(s) 0, 2, 6, 10, 14, 18, and 22, and sera were collected accordingly for serological analysis.
2.12. SARS-CoV-2 Spike-Specific Serum IgG Assay
WA-1 or Beta SARS-CoV-2 spike-specific IgG titers were assessed with an antibody titration ELISA as described previously [
6]. Briefly, ELISA plates (Thermo Fisher Scientific, 442404) were coated with 1 µg/mL of SARS-CoV-2 WA-1 S-2P or S-2Pᵦ in PBS (pH 7.4) at 4 °C for 16 h. The plates were washed with PBST three times, then blocked with 5% skim milk in 1 × PBST at RT for 2 h. The sera were diluted by 100-fold in 5% skim milk in PBST. A further serial 4-fold dilution for week-0 and week-2 sera or 6-fold dilution for week-6 sera was applied to the 100-fold dilution preparations. The plates were incubated with diluted sera at RT for 1 h. The HRP-conjugated anti-mouse secondary antibody (Thermo Fisher Scientific, 1/2000 dilution) was used to detect the antibody responses. The endpoint titers were calculated as the dilution that yielded an optical density equivalent to 4× background (secondary antibody alone).
2.13. Pseudovirus Neutralization Titer Assay
WA-1 or Beta SARS-CoV-2 neutralization titers were measured using a lentivirus-based SARS-CoV-2 pseudovirus assay. Pseudoviruses were generated by co-transfection of transducing plasmid pHR’ CMV-Luc encoding a luciferase reporter, lentivirus packaging plasmid pCMVd8.2, and a TMPRSS2 plasmid and S plasmids from SARS-CoV-2 WA-1 isolate (GenBank: MN908947.3), and B.1.351 isolate (Beta) into HEK293T/17 cells (ATCC CRL-11268) using Lipofectamine 3000 transfection reagent (L3000-001, ThermoFisher Scientific) [
17,
20,
23]. Heat-inactivated serum was mixed with the titrated pseudoviruses, incubated, then added to preplated 293T-ACE2 cells (provided by Dr. Michael Farzan) in triplicate. Following a 2 h incubation, wells were replenished with 150 µL of fresh medium. Then, 72 h later, the cells were lysed, and the luciferase activity was recorded in relative light units (RLUs). Percent neutralization and neutralization ID
50s were calculated using GraphPad Prism 9.0.2.
2.14. Intracellular Cytokine Staining and Flow Cytometry Assay
Mouse splenocytes were obtained by using a gentleMACS tissue dissociator (Miltenyi Biotec, Gaithersburg, MD, USA), followed by 70 µm filtration and density gradient centrifugation using Fico/Lite-LM medium (Atlanta Biologicals, Flowery Branch, GA, USA). Cells from each mouse spleen were resuspended in R10 media (RPMI 1640 supplemented with pen–strep antibiotic, 10% HI-FBS, Glutamax, and HEPES) and incubated for 6 h at 37 °C with protein transport inhibitor cocktail (eBioscience, San Diego, CA, USA) under three conditions: no peptide (DMSO only) stimulation or stimulated with one of two spike peptide pools (S1 and S2 peptide pools, 85% pure, JPT Peptide Technologies, Berlin, Germany). Peptide pools were used at a final concentration of 2 µg/mL for each peptide. After stimulation, cells were washed with PBS prior to staining with LIVE/DEAD Fixable Blue Dead Cell Stain (Invitrogen) for 20 min at RT, then washed in FC buffer (PBS supplemented with 2% HI-FBS and 0.05% NaN3). Cells were then resuspended in BD Fc Block (clone 2.4G2) for 5 min at RT prior to staining with a surface stain cocktail containing the following antibodies purchased from BD and Biolegend: I-A/I-E (M5/114.15.2) PE, CD8a (53-6.7) BUV805, CD44 (IM7) BUV395, CD62L (MEL-14) BV605, and CD4 (RM4-5) BV480 in brilliant stain buffer (BD). After a 15 min incubation at RT, cells were washed with FC buffer, then fixed and permeabilized using a BD Cytofix/Cytoperm fixation/permeabilization solution kit according to manufacturer’s instructions. Cells were washed using perm/wash solution and stained with Fc Block (5 min at RT), followed by intracellular staining (30 min at 4 °C) using a cocktail of the following antibodies purchased from BD, Biolegend, or eBioscience: CD3e (17A2) BUV737, IFN-γ (XMG1.2) BV650, TNF-α (MP6-XT22) BV711, IL-2 (JES6-5H4) BV421, IL-4 (11B11) Alexa Fluor 488, IL-5 (TRFK5) APC, and IL-13 (eBio13A) PE-Cy7 in 1× perm/wash diluted with brilliant stain buffer. Finally, cells were washed in perm/wash solution and resuspended in 0.5% PFA-FC stain buffer prior to running on a Symphony A5 flow cytometer (BD Biosciences). Analysis was performed using FlowJo software, version 10.6.2, according to the gating strategy. Background cytokine expression in the no-peptide condition was subtracted from that measured in the S1 and S2 peptide pools for each individual mouse.
2.15. Statistical Analysis
All experimental data were subjected to statistical analyses using GraphPad Prism v9 software. IgG endpoint titers or neutralization ID50 values were calculated using non-linear dose–response regression analysis. Multiple comparisons between groups were calculated using a two-way ANOVA test in GraphPad Prism v9. A t test was used to test differences between the groups. For T-cell assay results, Kruskal–Wallis and post hoc Mann–Whitney U tests with Bonferroni correction and two-way repeated-measures ANOVA tests with multiple post hoc comparisons and Dunnett’s correction were used to detect significant differences between the groups.
4. Discussion
The current study demonstrates that the profiles of humoral immune responses in animals achieved with the genetic delivery of a single-gene-transcript eVLP platform were comparable to those attained with the immunization of in vitro purified eVLPs in a previously reported study [
6]. Comparison of genetic delivery of eVLP with the previously reported immunization of in vitro purified eVLP is summarized in
Table 2. GMT comparison analysis shows the neutralizing titer (GMT of 4552) induced with the immunization of 50 μg of in vitro purified S-2P-NDVLP to be superior in terms of neutralization relative to the neutralizing titer (GMT of 2949) achieved with genetic delivery of the analogous S-2P-NDVLP-1T, although the ratio of in vitro purified S-2P-NDVLP-derived GMT (4552) to S-2P-derived GMT (1178) was comparable to the ratio of S-2P-NDVLP-1T-derived GMT (2949) to S-2P-TM-derived GMT (476). It is plausible that the GMT variability between these two eVLP delivery methods is due to differences in the amount of genetically delivered eVLPs and in vitro purified eVLPs. Analysis of in vitro eVLP expression revealed that the production of 50 μg of in vitro purified S-2P-NDVLP required approximately 200 μg of either S-2P-NDVLP-1T plasmid or S-2P-NDVLP-3T plasmid. Overall, after a single dose of immunization in mice, both eVLP delivery platforms elicited modest pseudovirus neutralizing titers, while their counterparts, protein subunits of S-2P-TM, S-2Pᵦ-TM, or S-2P, did not. After a second immunization in mice, both eVLP delivery platforms elicited higher pseudovirus neutralizing titers than their protein subunit counterparts. The genetic delivery of eVLPs resulted in a high peak of neutralizing titers but of shorter duration (
Figure 3B). Moreover, genetic delivery of S-2Pᵦ-NDVLP-3T elicited a combination of stronger and longer-lasting pseudovirus neutralizing titers than S-2Pᵦ-TM or S-2Pᵦ-NDVLP-1T. The neutralizing titers induced by cell-membrane-anchored S-2P or S-2Pᵦ were modest and appeared later but lasted longer, as shown in
Figure 3B. The observation that the Beta vaccine showed greater neutralization of the Beta pseudovirus than did the WT vaccine (50 μg;
Figure 2) suggests a vaccine-to-viral strain correlation, as well as the antigenic specificity of these vaccines.
Genetic delivery bypasses the challenges of purification and instability of eVLP-based vaccines in vitro [
3,
4,
10,
18,
29], enabling the use of eVLP-based vaccines for the vaccination of large populations, although the results obtained in mice need to be confirmed in non-human primates (and humans) [
30]. However, it remains to be determined whether the eVLP-based vaccines described here can offer sterilizing immunity against SARS-CoV-2 infection.
In addition, the genetic delivery of a DNA construct of S-2P-TM or S-2P-NDVLP-1T generated high spike-specific T-cell responses. Recent studies have shown that SARS-CoV-2-specific T-cell responses are essential for viral clearance, provide robust memory, mediate recognition of viral variants, and may effectively prevent initial viral infection in concert with antibody responses [
31]. In the current study, the frequency of spike-specific T cells after DNA immunization was higher than that previously reported in mice immunized with Moderna mRNA-1273 [
17,
32]. This finding is consistent with the clinical observation that robust T-cell responses were induced after dual mRNA vaccination [
33,
34]. This observation may provide additional insight into the role of T-cell responses after genetic delivery in protection against SARS-CoV-2 infection.
The current study also intended to examine the special features inducing neutralizing immunity through various vaccine platforms, specifically the humoral immune responses achieved by the genetic delivery of a single-gene-transcript eVLP platform of S-2P-NDVLP-1T/S-2Pᵦ-NDVLP-1T, of a multiple-gene-transcript eVLP platform of S-2Pᵦ-NDVLP-3T, and of a protein subunit platform of S-2P-TM or S-2Pᵦ-TM in mice. Based on the presented results and prior knowledge [
4,
35,
36,
37,
38,
39,
40], we discussed the putative mechanistic models and special features of each vaccine platform in inducing neutralizing immunity after the first genetic delivery (
Figure 5).
Cell-membrane-anchored S-2P or S-2Pᵦ functions as an immobile particle antigen, while S-2P-NDVLP or S-2Pᵦ-NDVLP functions as a mobile particle antigen. The genetic delivery of S-2P-TM or S-2Pᵦ-TM was unable to elicit a pseudovirus neutralizing titer after the first genetic delivery at week 2 (
Figure 2C) but able to induce higher levels of spike-specific CD4+ T cells in lymphoid tissues, such as in the spleen (
Figure 4C). Thus, the levels of spike-specific CD4+ T cells reflect the levels of spike-specific DCs (DC+), and membrane-anchored S-2P or S-2Pᵦ on the muscle cell surfaces localizes a considerable amount of DCs in muscle tissue, where tissue-localized DCs interact with membrane-anchored S-2P or S-2Pᵦ and are activated as spike-specific DCs (DC+). The spike-specific DCs migrate into lymphoid tissues and activate CD4+ T cells as spike-specific CD4+ T cells, which then trigger CD4+ T-cell-dependent humoral immune response (
Figure 5A). The mechanism resulting in spike-specific DCs/CD4+ T-cell response being unable to elicit an observed neutralizing titer after the first genetic delivery remains to be determined. In contrast, eVLPs, as mobile particles, directly migrated into lymph nodes, activated B cells, and led to modest pseudovirus neutralizing titers after the first genetic delivery at week 2, suggesting a T-cell-independent humoral immune response (
Figure 5B). The genetic delivery of S-2Pᵦ-NDVLP-3T dominantly produced both mobile particles of S-2Pᵦ-NDVLP and immobile particles of cell-membrane-anchored S-2Pᵦ in muscle tissue. The observation of relatively low pseudovirus neutralizing titers after the first genetic delivery of S-2Pᵦ-NDVLP-3T at week 2 (
Figure 2C) inspired us to speculate that cell-membrane-anchored S-2Pᵦ results in a higher amount of tissue-localized DCs, which restrict further S-2Pᵦ-NDVLPs in the muscle tissue. In turn, a lower amount of free S-2Pᵦ-NDVLPs migrates into lymph nodes, resulting in a low pseudovirus neutralizing titer (
Figure 5C). It is also possible that more potent neutralizing titers were achieved due to the spike S-2P antigen being expressed in a “natural viral membrane” fully conformational state (like “authentic” form) on the eVLPs, while the membrane-anchored S-2P may not have been in a fully native form on the host cell surfaces. All these possibilities remain to be determined in further studies.
The second genetic delivery of S-2Pᵦ-NDVLP-3T robustly boosted both cell-membrane-anchored, S-2Pᵦ-mediated, and S-2Pᵦ-NDVLP-mediated humoral immune responses, which correlate with strong and long-lasting pseudovirus neutralization titers (
Figure 2F and
Figure 3B). This observation may imply that combined immunization with two different platform vaccines can synergically achieve the best humoral immune responses. In the current study, S-2Pᵦ-NDVLP-3T produced two platform vaccines in vivo—cell-membrane-anchored S-2Pᵦ and S-2Pᵦ-NDVLP—which may be functionally similar to combined immunization with S-2Pᵦ-TM and S-2Pᵦ-NDVLP-1T. Such observations have also been reported with heterologous vaccination with both ChAdOx1 and mRNA1273, which achieved expanded and enhanced humoral immune responses [
41]. The results achieved in the current study not only help us to better understand the genetic delivery of different vaccine platforms, resulting in different immunogenicity outcomes, but also provide a practical strategy for eVLP-based vaccine development that should be simple, rapid, scalable, and efficient for the vaccination of large populations.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}