
Self-Amplifying RNA: A Second Revolution of mRNA Vaccines against COVID-19


Abstract
1. Introduction
1.1. COVID-19 and SARS-CoV-2
1.2. Approved COVID-19 Vaccines

{kind=link}
{kind=link}
| Vaccine Name | Company | Country of Origin | Platform a | Antigen b | First Approval | Reference |
|---|---|---|---|---|---|---|
| mRNA-1273 | Moderna | USA | mRNA LNPs | Pre-fusion S | 2020 | [12] |
| Moderna 2023–2024 Formula | Moderna | USA | mRNA LNPs | Pre-fusion S (Omicron XBB.1.5) | 2023 | [15] |
| BNT162b2 | Pfizer/ BioNTech | Germany/USA | mRNA LNPs | Pre-fusion S | 2020 | [13] |
| Pfizer 2023–2024 Formula | Pfizer/ BioNTech | Germany/USA | mRNA LNPs | Pre-fusion S (Omicron XBB.1.5) | 2023 | [15] |
| AZD1222 | AstraZeneca/Oxford | UK | ChAdOX1 | Native S | 2021 | [6] |
| Ad26COVS1 | Janssen | Holland | Ad26 | Pre-fusion S | 2021 | [7] |
| Gam-COVID-Vax Sputnik V | Gamaleya Research Institute | Russia | Ad26/Ad5 | Native S | 2020 | [16] |
| NVX-CoV2373 | Novavax | USA | Protein-based | Pre-fusion S | 2021 | [17] |
| CoronaVac | SinovacBiotech | China | Inactivated virus | Whole inactivated virus | 2021 | [18] |
| Convidecia™ Ad5-nCoV | CanSino | China | Ad5 | Native S | 2021 | [19] |
| ZyCov-D | Zydus Cadila/ India’s Depart. Biotechnology | India | Plasmid DNA | Native S | 2021 | [20] |
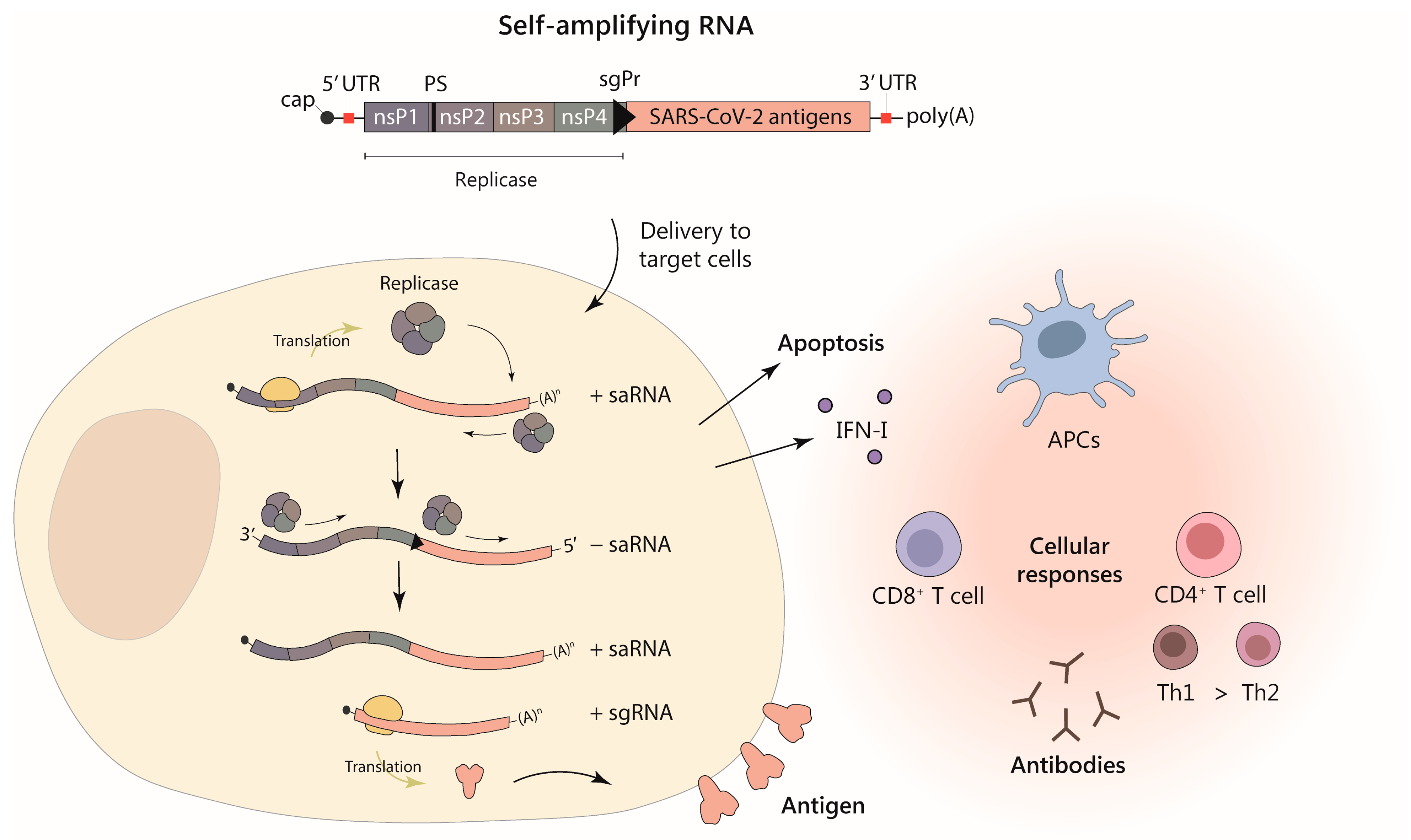
1.3. Self-Amplifying RNA
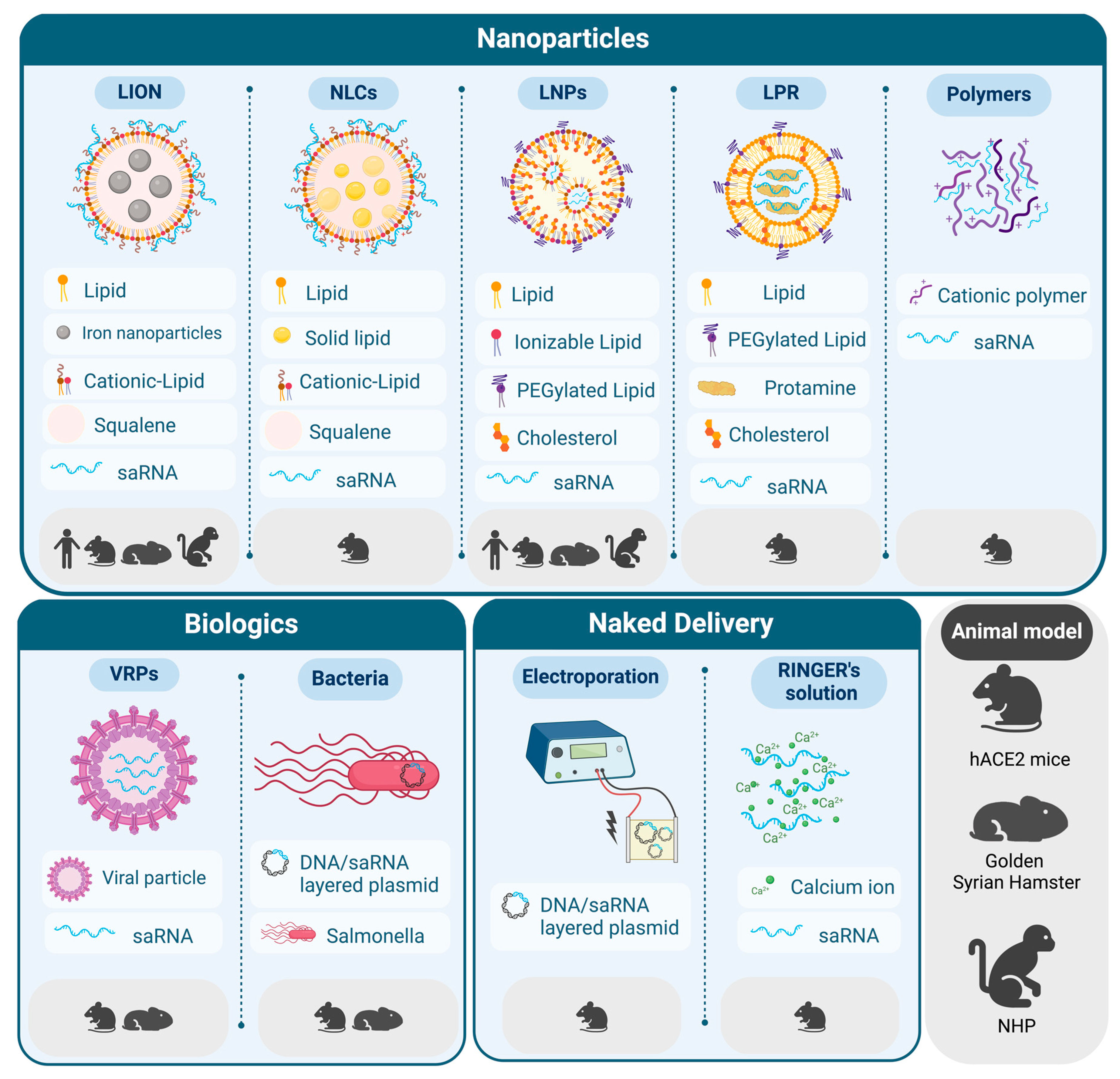
2. Delivery Strategies for saRNA Vaccines against SARS-CoV-2
2.1. LNPs for the Delivery of saRNA
2.2. New Trends in Lipid-Based Formulations for saRNA Delivery
2.3. Beyond Lipids: Other Delivery Strategies for saRNA Vaccines
2.4. Toxicology Studies
| Vector |
Vaccine Formulation | Encoded Antigen |
Vaccination Schedule |
Animal Model | Main Results | Ref. |
|---|---|---|---|---|---|---|
| VEEV | LNP | Prefusion stabilized S protein | IM, 0.015–1.5 μg, prime/boost (3-week gap) | Mouse | nAbs, T cell response (Th1) | [38] |
| IM, 0.03–3 μg, prime/boost (3-week gap) | Hamster | Partial protection against infection | [38] | |||
| IM, 0.01–10 μg, prime/boost (4-week gap) | Mouse | nAbs, T cell response (Th1) | [40] | |||
| IM, 5 μg, prime/boost (4-week gap) | Hamster | Protection from severe disease | [41] | |||
| Unmodified protein | IM, 0.2–10 μg, only prime | Mouse | nAbs, T cell response (Th1) | [42] | ||
| hACE2 Mouse | Protection against infection | |||||
| RBD-TM (Wuhan- Hu-1) | IM, 1 μg, prime/boost (4-week gap) | Mouse | Cross-reactive Abs, specific CD4+ and CD8+ T-cell responses | [85] | ||
| IM, 50 μg, prime/boost (4-week gap) | Cynomolgus monkey | Protection against infection | ||||
| RBD-TM (gamma) | IM, 10 μg, prime/boost (4-week gap) | Hamster | Cross-nAbs, antigen-specific B and T cells, prevent weight loss | |||
| NLC | Prefusion stabilized protein | IM, 1–30 μg, only prime or prime/boost (3-week gap) | Mouse | Cross-nAbs, Th1 response | [46] | |
| LION | Unmodified protein | IM, 0.1–10 μg, prime/boost (4-week gap) | nAbs, T cell responses (Th1) | [47] | ||
| IM, 250 μg only prime or 50 μg prime/boost (4-week gap) | Pigtail macaque | nAbs, modest T cell responses | ||||
| IM, 5 to 50 μg, prime/boost (4 to 20-week gap) | nAbs, partial protection from infection, protection from disease | [48] | ||||
| IM, 5 or 25 μg, only prime | SIV-infected pigtail macaque | nAbs, modest T cell responses | [49] | |||
| S protein from different VoCs | IM, 1 μg, prime/boost (4-week gap) | Mouse | Cross-nAbs | [53] | ||
| IM, 20 μg, prime/boost (4-week gap) | Hamster | Cross-nAbs, partial protection from infection and disease | ||||
| LPR | RBD | SC, 2 μg, prime/boost (4- week gap) | Mouse | nAbs, extended antigen expression in dLNs | [57] | |
| LNP + Ad | Full-length S protein | IM, 108 i.u. Ad prime, and 1μg RNA boost (4-week gap) | Mouse | nAbs, cytotoxic T cells and Th1 | [86] | |
| Codon-optimized S protein | SC, 10 μg, prime/boost (8-week gap) | Mouse | nAbs, specific T-cell responses | [87] | ||
| IM bilateral, 3–30 μg, prime/boost (4-week gap) | Rhesus macaque | |||||
| LNP + OX40 agonist | Prefusion stabili- zed trimeric S protein | IM, 1 μg, prime/boost (4-week gap) | Mouse | Specific CD4+ and CD8+ T-cell responses | [88] | |
| VRP | Prefusion stabilized S protein (Omicron) | IP, IM or IN, 1 × 106 VRPs, prime/two boosts (2-week gap) | nAbs, Th1 (IP route), protection against infection | [77] | ||
| Hamster | nAbs, Th1, protection against disease | |||||
| Optimized S protein | IM, 107 VRPs, prime/two boosts (3-week gap) | Guinea pig | nAbs | [79] | ||
| SC, 5 × 107 VRPs, prime/boost (3-week gap) | Cat | nAbs, protection against infection and transmission | ||||
| DNA/SFV replicon | Naked, Ep | Unmodified or prefusion stabilized S protein | ID, 10 μg, prime/boost (4-week gap) | Mouse | nAbs, T cell response (Th1) | [63] |
| Salmonella | RBD, HR, M, nsp13 | IM, 1 × 10⁷ CFU, only prime, or oral, 1 × 10⁸ CFU, prime/boost (2-week gap) | nAbs, specific CD4+ and CD8+ T-cell responses | [71] | ||
| IM, 2 × 10⁷ CFU, only prime, or oral, 2 × 10⁸ CFU, prime/boost (2-week gap) | Hamster | Cross-nAbs, IgA, protection against infection and disease |
3. Strategies to Increase Efficacy of saRNA Vaccines
3.1. Multi-Antigenic Vaccines
3.2. Optimization of Antigen Selection
3.3. Optimization of saRNA Molecules
3.4. Heterologous Vaccination Regimens
3.5. Combination with Immunostimulatory Molecules
4. saRNA-Based COVID-19 Vaccines in Clinical Trials
4.1. Vaccines Based on S Protein
4.2. Vaccines Based on the RBD
4.3. Multi-Antigenic Vaccines
4.4. Heterologous Vaccination
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- WHO COVID-19 Dashboard. Available online: https://data.who.int/dashboards/covid19/deaths?n=c (accessed on 25 February 2024).
- Tang, D.; Comish, P.; Kang, R. The Hallmarks of COVID-19 Disease. PLoS Pathog. 2020, 16, e1008536. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.-D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron Variant of SARS-CoV-2: Genomics, Transmissibility, and Responses to Current COVID-19 Vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Li, D.; Sempowski, G.D.; Saunders, K.O.; Acharya, P.; Haynes, B.F. SARS-CoV-2 Neutralizing Antibodies for COVID-19 Prevention and Treatment. Annu. Rev. Med. 2022, 73, 1–16. [Google Scholar] [CrossRef]
- Sheridan, C. First COVID-19 DNA Vaccine Approved, Others in Hot Pursuit. Nat. Biotechnol. 2021, 39, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and Immunogenicity of the ChAdOx1 NCoV-19 Vaccine against SARS-CoV-2: A Preliminary Report of a Phase 1/2, Single-Blind, Randomised Controlled Trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Daian e Silva, D.S.d.O.; da Fonseca, F.G. The Rise of Vectored Vaccines: A Legacy of the COVID-19 Global Crisis. Vaccines 2021, 9, 1101. [Google Scholar] [CrossRef]
- Offord, C.; Cohen, J. Award Honors Pair for MRNA Work Key to COVID-19 Vaccines. Science 2023, 382, 22. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gong, L.; Wang, P.; Zhao, X.; Zhao, F.; Zhang, Z.; Li, Y.; Huang, W. Recent Advances in Lipid Nanoparticles for Delivery of MRNA. Pharmaceutics 2022, 14, 2682. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.E.; Frenck, R.W.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Fiolet, T.; Kherabi, Y.; MacDonald, C.-J.; Ghosn, J.; Peiffer-Smadja, N. Comparing COVID-19 Vaccines for Their Characteristics, Efficacy and Effectiveness against SARS-CoV-2 and Variants of Concern: A Narrative Review. Clin. Microbiol. Infect. 2022, 28, 202–221. [Google Scholar] [CrossRef]
- Regan, J.J.; Moulia, D.L.; Link-Gelles, R.; Godfrey, M.; Mak, J.; Najdowski, M.; Rosenblum, H.G.; Shah, M.M.; Twentyman, E.; Meyer, S.; et al. Use of Updated COVID-19 Vaccines 2023-2024 Formula for Persons Aged ≥ 6 Months: Recommendations of the Advisory Committee on Immunization Practices—United States, September 2023. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 1140–1146. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Zubkova, O.V.; Tukhvatullin, A.I.; Shcheblyakov, D.V.; Dzharullaeva, A.S.; Grousova, D.M.; Erokhova, A.S.; Kovyrshina, A.V.; Botikov, A.G.; et al. Safety and Immunogenicity of an RAd26 and RAd5 Vector-Based Heterologous Prime-Boost COVID-19 Vaccine in Two Formulations: Two Open, Non-Randomised Phase 1/2 Studies from Russia. Lancet 2020, 396, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.T.; Galiza, E.P.; Baxter, D.N.; Boffito, M.; Browne, D.; Burns, F.; Chadwick, D.R.; Clark, R.; Cosgrove, C.; Galloway, J.; et al. Safety and Efficacy of NVX-CoV2373 Covid-19 Vaccine. N. Engl. J. Med. 2021, 385, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Tanriover, M.D.; Doğanay, H.L.; Akova, M.; Güner, H.R.; Azap, A.; Akhan, S.; Köse, Ş.; Erdinç, F.Ş.; Akalın, E.H.; Tabak, Ö.F.; et al. Efficacy and Safety of an Inactivated Whole-Virion SARS-CoV-2 Vaccine (CoronaVac): Interim Results of a Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial in Turkey. Lancet 2021, 398, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, J.; Zhang, Z.; Wu, J.; Zhang, J.; Hu, H.; Zhu, T.; Zhang, J.; Luo, L.; Fan, P.; et al. Safety, Tolerability, and Immunogenicity of an Aerosolised Adenovirus Type-5 Vector-Based COVID-19 Vaccine (Ad5-NCoV) in Adults: Preliminary Report of an Open-Label and Randomised Phase 1 Clinical Trial. Lancet Infect. Dis. 2021, 21, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, A.; Bhate, S.; Ramaiah, V.; Deshpande, S.; Giri, K.; Phophle, H.; Supe, P.; Godara, I.; Revanna, R.; Nagarkar, R.; et al. Efficacy, Safety, and Immunogenicity of the DNA SARS-CoV-2 Vaccine (ZyCoV-D): The Interim Efficacy Results of a Phase 3, Randomised, Double-Blind, Placebo-Controlled Study in India. Lancet 2022, 399, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Self-Copying RNA Vaccine Wins First Full Approval: What’s Next? Nature 2023, 624, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Quetglas, J.I.; Ruiz-Guillen, M.; Aranda, A.; Casales, E.; Bezunartea, J.; Smerdou, C. Alphavirus Vectors for Cancer Therapy. Virus Res. 2010, 153, 179–196. [Google Scholar] [CrossRef]
- Smerdou, C.; Liljeström, P. Non-Viral Amplification Systems for Gene Transfer: Vectors Based on Alphaviruses. Curr. Opin. Mol. Ther. 1999, 1, 244–251. [Google Scholar]
- Liljeström, P.; Garoff, H. A New Generation of Animal Cell Expression Vectors Based on the Semliki Forest Virus Replicon. Bio/Technology 1991, 9, 1356–1361. [Google Scholar] [CrossRef]
- Urban, C.; Rhême, C.; Maerz, S.; Berg, B.; Pick, R.; Nitschke, R.; Borner, C. Apoptosis Induced by Semliki Forest Virus Is RNA Replication Dependent and Mediated via Bak. Cell Death Differ. 2008, 15, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Quetglas, J.I.; Reboredo, M.; Dubrot, J.; Rodriguez-Madoz, J.R.; Mancheño, U.; Casales, E.; Riezu-Boj, J.I.; Ruiz-Guillen, M.; Ochoa, M.C.; et al. Strict Requirement for Vector-Induced Type I Interferon in Efficacious Antitumor Responses to Virally Encoded IL12. Cancer Res. 2015, 75, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Näslund, T.I.; Kostic, L.; Nordström, E.K.; Chen, M.; Liljeström, P. Role of Innate Signalling Pathways in the Immunogenicity of Alphaviral Replicon-Based Vaccines. Virol. J. 2011, 8, 36. [Google Scholar] [CrossRef]
- Lundstrom, K. Self-Replicating RNA Viruses for Vaccine Development against Infectious Diseases and Cancer. Vaccines 2021, 9, 187. [Google Scholar] [CrossRef]
- Lundstrom, K. Alphaviruses in Cancer Immunotherapy. Int. Rev. Cell Mol. Biol. 2023, 379, 143–168. [Google Scholar] [CrossRef]
- Lundstrom, K. Applications of Self-Replicating RNA. Int. Rev. Cell Mol. Biol. 2022, 372, 97–157. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Wu, Y.; Rui, X.; Yang, Y.; Ling, C.; Liu, S.; Liu, S.; Wang, Y. Animal Models for COVID-19: Advances, Gaps and Perspectives. Signal Transduct. Target. Ther. 2022, 7, 220. [Google Scholar] [CrossRef]
- Garber, K. Alnylam Launches Era of RNAi Drugs. Nat. Biotechnol. 2018, 36, 777–778. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral Delivery of Self-Amplifying RNA Vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Anderluzzi, G.; Lou, G.; Woods, S.; Schmidt, S.T.; Gallorini, S.; Brazzoli, M.; Johnson, R.; Roberts, C.W.; O’Hagan, D.T.; Baudner, B.C.; et al. The Role of Nanoparticle Format and Route of Administration on Self-Amplifying MRNA Vaccine Potency. J. Control. Release 2022, 342, 388–399. [Google Scholar] [CrossRef]
- Kairuz, D.; Samudh, N.; Ely, A.; Arbuthnot, P.; Bloom, K. Production, Characterization, and Assessment of Permanently Cationic and Ionizable Lipid Nanoparticles for Use in the Delivery of Self-Amplifying RNA Vaccines. Pharmaceutics 2023, 15, 1173. [Google Scholar] [CrossRef]
- Kim, B.; Hosn, R.R.; Remba, T.; Yun, D.; Li, N.; Abraham, W.; Melo, M.B.; Cortes, M.; Li, B.; Zhang, Y.; et al. Optimization of Storage Conditions for Lipid Nanoparticle-Formulated Self-Replicating RNA Vaccines. J. Control. Release 2023, 353, 241–253. [Google Scholar] [CrossRef]
- Tregoning, J.S.; Stirling, D.C.; Wang, Z.; Flight, K.E.; Brown, J.C.; Blakney, A.K.; McKay, P.F.; Cunliffe, R.F.; Murugaiah, V.; Fox, C.B.; et al. Formulation, Inflammation, and RNA Sensing Impact the Immunogenicity of Self-Amplifying RNA Vaccines. Mol. Ther.-Nucleic Acids 2023, 31, 29–42. [Google Scholar] [CrossRef]
- Maruggi, G.; Mallett, C.P.; Westerbeck, J.W.; Chen, T.; Lofano, G.; Friedrich, K.; Qu, L.; Sun, J.T.; McAuliffe, J.; Kanitkar, A.; et al. A Self-Amplifying MRNA SARS-CoV-2 Vaccine Candidate Induces Safe and Robust Protective Immunity in Preclinical Models. Mol. Ther. 2022, 30, 1897–1912. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron Extensively but Incompletely Escapes Pfizer BNT162b2 Neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef] [PubMed]
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-Amplifying RNA SARS-CoV-2 Lipid Nanoparticle Vaccine Candidate Induces High Neutralizing Antibody Titers in Mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef] [PubMed]
- Frise, R.; Baillon, L.; Zhou, J.; Kugathasan, R.; Peacock, T.P.; Brown, J.C.; Samnuan, K.; McKay, P.F.; Shattock, R.J.; Barclay, W.S. A Self-Amplifying RNA Vaccine Protects against SARS-CoV-2 (D614G) and Alpha Variant of Concern (B.1.1.7) in a Transmission-Challenge Hamster Model. Vaccine 2022, 40, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- de Alwis, R.; Gan, E.S.; Chen, S.; Leong, Y.S.; Tan, H.C.; Zhang, S.L.; Yau, C.; Low, J.G.H.; Kalimuddin, S.; Matsuda, D.; et al. A Single Dose of Self-Transcribing and Replicating RNA-Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity in Mice. Mol. Ther. 2021, 29, 1970–1983. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of Lipid Nanoparticle Formulations for Exterior Complexation and in Vivo Delivery of SaRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef]
- Gerhardt, A.; Voigt, E.; Archer, M.; Reed, S.; Larson, E.; Van Hoeven, N.; Kramer, R.; Fox, C.; Casper, C. A Flexible, Thermostable Nanostructured Lipid Carrier Platform for RNA Vaccine Delivery. Mol. Ther.-Methods Clin. Dev. 2022, 25, 205–214. [Google Scholar] [CrossRef]
- Voigt, E.A.; Gerhardt, A.; Hanson, D.; Jennewein, M.F.; Battisti, P.; Reed, S.; Singh, J.; Mohamath, R.; Bakken, J.; Beaver, S.; et al. A Self-Amplifying RNA Vaccine against COVID-19 with Long-Term Room-Temperature Stability. NPJ Vaccines 2022, 7, 136. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.B.; et al. An Alphavirus-Derived Replicon RNA Vaccine Induces SARS-CoV-2 Neutralizing Antibody and T Cell Responses in Mice and Nonhuman Primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef]
- O’Connor, M.A.; Hawman, D.W.; Meade-White, K.; Leventhal, S.; Song, W.; Randall, S.; Archer, J.; Lewis, T.B.; Brown, B.; Fredericks, M.N.; et al. A Replicon RNA Vaccine Can Induce Durable Protective Immunity from SARS-CoV-2 in Nonhuman Primates after Neutralizing Antibodies Have Waned. PLOS Pathog. 2023, 19, e1011298. [Google Scholar] [CrossRef]
- O’Connor, M.A.; Erasmus, J.H.; Randall, S.; Archer, J.; Lewis, T.B.; Brown, B.; Fredericks, M.; Groenier, S.; Iwayama, N.; Ahrens, C.; et al. A Single Dose SARS-CoV-2 Replicon RNA Vaccine Induces Cellular and Humoral Immune Responses in Simian Immunodeficiency Virus Infected and Uninfected Pigtail Macaques. Front. Immunol. 2021, 12, 800723. [Google Scholar] [CrossRef]
- Britton, A.; Embi, P.J.; Levy, M.E.; Gaglani, M.; DeSilva, M.B.; Dixon, B.E.; Dascomb, K.; Patel, P.; Schrader, K.E.; Klein, N.P.; et al. Effectiveness of COVID-19 MRNA Vaccines Against COVID-19–Associated Hospitalizations Among Immunocompromised Adults During SARS-CoV-2 Omicron Predominance—VISION Network, 10 States, December 2021—August 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1335–1342. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Vokó, Z.; Surján, O.; Rákóczi, É.; Szamosi, S.; Szűcs, G.; Szekanecz, É.; Müller, C.; Kiss, Z. Effectiveness and Waning of Protection with the BNT162b2 Vaccine against the SARS-CoV-2 Delta Variant in Immunocompromised Individuals. Front. Immunol. 2023, 14, 1247129. [Google Scholar] [CrossRef]
- Nikoloudis, A.; Neumann, I.J.; Buxhofer-Ausch, V.; Machherndl-Spandl, S.; Binder, M.; Kaynak, E.; Milanov, R.; Nocker, S.; Stiefel, O.; Strassl, I.; et al. Successful SARS-CoV-2 MRNA Vaccination Program in Allogeneic Hematopoietic Stem Cell Transplant Recipients—A Retrospective Single-Center Analysis. Vaccines 2023, 11, 1534. [Google Scholar] [CrossRef]
- Hawman, D.W.; Meade-White, K.; Archer, J.; Leventhal, S.S.; Wilson, D.; Shaia, C.; Randall, S.; Khandhar, A.P.; Krieger, K.; Hsiang, T.-Y.; et al. SARS-CoV2 Variant-Specific Replicating RNA Vaccines Protect from Disease Following Challenge with Heterologous Variants of Concern. eLife 2022, 11, e75537. [Google Scholar] [CrossRef]
- Low, J.G.; de Alwis, R.; Chen, S.; Kalimuddin, S.; Leong, Y.S.; Mah, T.K.L.; Yuen, N.; Tan, H.C.; Zhang, S.L.; Sim, J.X.Y.; et al. A Phase I/II Randomized, Double-Blinded, Placebo-Controlled Trial of a Self-Amplifying Covid-19 MRNA Vaccine. NPJ Vaccines 2022, 7, 161. [Google Scholar] [CrossRef]
- Pollock, K.M.; Cheeseman, H.M.; Szubert, A.J.; Libri, V.; Boffito, M.; Owen, D.; Bern, H.; McFarlane, L.R.; O’Hara, J.; Lemm, N.-M.; et al. Safety and Immunogenicity of a Self-Amplifying RNA Vaccine against COVID-19: COVAC1, a Phase I, Dose-Ranging Trial. eClinicalMedicine 2022, 44, 101262. [Google Scholar] [CrossRef]
- Kimura, T.; Leal, J.M.; Simpson, A.; Warner, N.L.; Berube, B.J.; Archer, J.F.; Park, S.; Kurtz, R.; Hinkley, T.; Nicholes, K.; et al. A Localizing Nanocarrier Formulation Enables Multi-Target Immune Responses to Multivalent Replicating RNA with Limited Systemic Inflammation. Mol. Ther. 2023, 31, 2360–2375. [Google Scholar] [CrossRef]
- Lin, G.; Yan, H.; Sun, J.; Zhao, J.; Zhang, Y. Self-Replicating RNA Nanoparticle Vaccine Elicits Protective Immune Responses against SARS-CoV-2. Mol. Ther.-Nucleic Acids 2023, 32, 650–666. [Google Scholar] [CrossRef]
- Johansson, D.X.; Ljungberg, K.; Kakoulidou, M.; Liljeström, P. Intradermal Electroporation of Naked Replicon RNA Elicits Strong Immune Responses. PLoS ONE 2012, 7, e29732. [Google Scholar] [CrossRef]
- Cu, Y.; Broderick, K.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.; Ulmer, J.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying MRNA Vaccines by Electroporation in Situ. Vaccines 2013, 1, 367–383. [Google Scholar] [CrossRef]
- Silva-Pilipich, N.; Lasarte-Cía, A.; Lozano, T.; Martín-Otal, C.; Lasarte, J.J.; Smerdou, C. Intratumoral Electroporation of a Self-Amplifying RNA Expressing IL-12 Induces Antitumor Effects in Mouse Models of Cancer. Mol. Ther. Nucleic Acids 2022, 29, 387–399. [Google Scholar] [CrossRef]
- Kohno, A.; Emi, N.; Kasai, M.; Tanimoto, M.; Saito, H. Semliki Forest Virus-Based DNA Expression Vector: Transient Protein Production Followed by Cell Death. Gene Ther. 1998, 5, 415–418. [Google Scholar] [CrossRef][Green Version]
- Berglund, P.; Smerdou, C.; Fleeton, M.N.; Tubulekas, L.; Liljeström, P. Enhancing Immune Responses Using Suicidal DNA Vaccines. Nat. Biotechnol. 1998, 16, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Szurgot, I.; Hanke, L.; Sheward, D.J.; Vidakovics, L.P.; Murrell, B.; McInerney, G.M.; Liljeström, P. DNA-Launched RNA Replicon Vaccines Induce Potent Anti-SARS-CoV-2 Immune Responses in Mice. Sci. Rep. 2021, 11, 3125. [Google Scholar] [CrossRef] [PubMed]
- Nordström, E.K.L.; Forsell, M.N.E.; Barnfield, C.; Bonin, E.; Hanke, T.; Sundström, M.; Karlsson, G.B.; Liljeström, P. Enhanced Immunogenicity Using an Alphavirus Replicon DNA Vaccine against Human Immunodeficiency Virus Type 1. J. Gen. Virol. 2005, 86, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wei, Q.; Cai, L.; Yang, X.; Xing, C.; Tan, F.; Leavenworth, J.W.; Liang, S.; Liu, W. Alphavirus Replicon DNA Vectors Expressing Ebola GP and VP40 Antigens Induce Humoral and Cellular Immune Responses in Mice. Front. Microbiol. 2018, 8, 2662. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, P.; García-Arriaza, J.; Zusinaite, E.; Szurgot, I.; Männik, A.; Kraus, A.; Ustav, M.; Merits, A.; Esteban, M.; Liljeström, P.; et al. DNA-Launched RNA Replicon Vaccines Induce Potent Anti-Ebolavirus Immune Responses That Can Be Further Improved by a Recombinant MVA Boost. Sci. Rep. 2018, 8, 12459. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.Q.; Pérez, P.; Ljungberg, K.; Sorzano, C.Ó.S.; Gómez, C.E.; Liljeström, P.; Esteban, M.; García-Arriaza, J. Potent Anti-Hepatitis C Virus (HCV) T Cell Immune Responses Induced in Mice Vaccinated with DNA-Launched RNA Replicons and Modified Vaccinia Virus Ankara-HCV. J. Virol. 2019, 93, e00055. [Google Scholar] [CrossRef] [PubMed]
- van de Wall, S.; Ljungberg, K.; Ip, P.P.; Boerma, A.; Knudsen, M.L.; Nijman, H.W.; Liljeström, P.; Daemen, T. Potent Therapeutic Efficacy of an Alphavirus Replicon DNA Vaccine Expressing Human Papilloma Virus E6 and E7 Antigens. Oncoimmunology 2018, 7, e1487913. [Google Scholar] [CrossRef]
- Amano, T.; Yu, H.; Amano, M.; Leyder, E.; Badiola, M.; Ray, P.; Kim, J.; Ko, A.C.; Achour, A.; Weng, N.; et al. Controllable Self-Replicating RNA Vaccine Delivered Intradermally Elicits Predominantly Cellular Immunity. iScience 2023, 26, 106335. [Google Scholar] [CrossRef]
- Jawalagatti, V.; Kirthika, P.; Lee, J.H. Oral MRNA Vaccines Against Infectious Diseases- A Bacterial Perspective [Invited]. Front. Immunol. 2022, 13, 884862. [Google Scholar] [CrossRef]
- Jawalagatti, V.; Kirthika, P.; Park, J.-Y.; Hewawaduge, C.; Lee, J.H. Highly Feasible Immunoprotective Multicistronic SARS-CoV-2 Vaccine Candidate Blending Novel Eukaryotic Expression and Salmonella Bactofection. J. Adv. Res. 2022, 36, 211–222. [Google Scholar] [CrossRef]
- Jawalagatti, V.; Kirthika, P.; Hewawaduge, C.; Yang, M.; Park, J.-Y.; Oh, B.; Lee, J.H. Bacteria-Enabled Oral Delivery of a Replicon-Based MRNA Vaccine Candidate Protects against Ancestral and Delta Variant SARS-CoV-2. Mol. Ther. 2022, 30, 1926–1940. [Google Scholar] [CrossRef]
- Jawalagatti, V.; Kirthika, P.; Hewawaduge, C.; Park, J.-Y.; Yang, M.-S.; Oh, B.; So, M.Y.; Kim, B.; Lee, J.H. A Simplified SARS-CoV-2 Mouse Model Demonstrates Protection by an Oral Replicon-Based MRNA Vaccine. Front. Immunol. 2022, 13, 811802. [Google Scholar] [CrossRef]
- Ashraf, M.U.; Kim, Y.; Kumar, S.; Seo, D.; Ashraf, M.; Bae, Y.-S. COVID-19 Vaccines (Revisited) and Oral-Mucosal Vector System as a Potential Vaccine Platform. Vaccines 2021, 9, 171. [Google Scholar] [CrossRef]
- Blakney, A.K.; Zhu, Y.; McKay, P.F.; Bouton, C.R.; Yeow, J.; Tang, J.; Hu, K.; Samnuan, K.; Grigsby, C.L.; Shattock, R.J.; et al. Big Is Beautiful: Enhanced SaRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano 2020, 14, 5711–5727. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Hu, K.; Samnuan, K.; Jain, N.; Brown, A.; Thomas, A.; Rogers, P.; Polra, K.; Sallah, H.; et al. Polymeric and Lipid Nanoparticles for Delivery of Self-Amplifying RNA Vaccines. J. Control. Release 2021, 338, 201–210. [Google Scholar] [CrossRef]
- Zhang, H.-Q.; Zhang, Y.-N.; Zhang, Z.-R.; Chen, X.-L.; Hu, Y.-Y.; Shi, Y.-J.; Wang, J.; Deng, C.; Zhang, B.; Li, X.-D.; et al. An Alphavirus Replicon Particle Delivering Prefusion-Stabilized Spike Protein Provides Potent Immunoprotection against SARS-CoV-2 Omicron Variant. Signal Transduct. Target. Ther. 2022, 7, 390. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Zhang, Z.-R.; Zhang, H.-Q.; Li, N.; Zhang, Q.-Y.; Li, X.-D.; Deng, C.-L.; Deng, F.; Shen, S.; Zhu, B.; et al. Different Pathogenesis of SARS-CoV-2 Omicron Variant in Wild-Type Laboratory Mice and Hamsters. Signal Transduct. Target. Ther. 2022, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Langereis, M.A.; Albulescu, I.C.; Stammen-Vogelzangs, J.; Lambregts, M.; Stachura, K.; Miller, S.; Bosco-Lauth, A.M.; Hartwig, A.E.; Porter, S.M.; Allen, M.; et al. An Alphavirus Replicon-Based Vaccine Expressing a Stabilized Spike Antigen Induces Protective Immunity and Prevents Transmission of SARS-CoV-2 between Cats. NPJ Vaccines 2021, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Q.; Zhang, Z.-R.; Zhang, H.-Q.; Zhang, Y.-N.; Zeng, X.-Y.; Zhang, Q.-Y.; Deng, C.-L.; Li, X.-D.; Zhang, B.; Ye, H.-Q. Intranasal Delivery of Replicating MRNA Encoding Neutralizing Antibody against SARS-CoV-2 Infection in Mice. Signal Transduct. Target. Ther. 2021, 6, 369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Zhang, H.-Q.; Wang, G.-F.; Zhang, Z.-R.; Li, J.-Q.; Chen, X.-L.; Hu, Y.-Y.; Zeng, X.-Y.; Shi, Y.-J.; Wang, J.; et al. Intranasal Delivery of Replicating MRNA Encoding HACE2-Targeting Antibody against SARS-CoV-2 Omicron Infection in the Hamster. Antivir. Res. 2023, 209, 105507. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.; Laxton, C.; Horscroft, N.; Richard, V.; Thomas, A.; Parkinson, T. Comparison of Rat and Human Responses to Toll-like Receptor 7 Activation. J. Interferon Cytokine Res. 2009, 29, 113–126. [Google Scholar] [CrossRef]
- Donahue, D.A.; Ballesteros, C.; Maruggi, G.; Glover, C.; Ringenberg, M.A.; Marquis, M.; Ben Abdeljelil, N.; Ashraf, A.; Rodriguez, L.-A.; Stokes, A.H. Nonclinical Safety Assessment of Lipid Nanoparticle-and Emulsion-Based Self-Amplifying MRNA Vaccines in Rats. Int. J. Toxicol. 2023, 42, 37–49. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-Generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef]
- Komori, M.; Nogimori, T.; Morey, A.L.; Sekida, T.; Ishimoto, K.; Hassett, M.R.; Masuta, Y.; Ode, H.; Tamura, T.; Suzuki, R.; et al. SaRNA Vaccine Expressing Membrane-Anchored RBD Elicits Broad and Durable Immunity against SARS-CoV-2 Variants of Concern. Nat. Commun. 2023, 14, 2810. [Google Scholar] [CrossRef]
- Spencer, A.J.; McKay, P.F.; Belij-Rammerstorfer, S.; Ulaszewska, M.; Bissett, C.D.; Hu, K.; Samnuan, K.; Blakney, A.K.; Wright, D.; Sharpe, H.R.; et al. Heterologous Vaccination Regimens with Self-Amplifying RNA and Adenoviral COVID Vaccines Induce Robust Immune Responses in Mice. Nat. Commun. 2021, 12, 2893. [Google Scholar] [CrossRef]
- Rappaport, A.R.; Hong, S.-J.; Scallan, C.D.; Gitlin, L.; Akoopie, A.; Boucher, G.R.; Egorova, M.; Espinosa, J.A.; Fidanza, M.; Kachura, M.A.; et al. Low-Dose Self-Amplifying MRNA COVID-19 Vaccine Drives Strong Protective Immunity in Non-Human Primates against SARS-CoV-2 Infection. Nat. Commun. 2022, 13, 3289. [Google Scholar] [CrossRef]
- Duhen, R.; Beymer, M.; Jensen, S.M.; Abbina, S.; Abraham, S.; Jain, N.; Thomas, A.; Geall, A.J.; Hu, H.M.; Fox, B.A.; et al. OX40 Agonist Stimulation Increases and Sustains Humoral and Cell-Mediated Responses to SARS-CoV-2 Protein and SaRNA Vaccines. Front. Immunol. 2022, 13, 896310. [Google Scholar] [CrossRef]
- McCafferty, S.; Haque, A.K.M.A.; Vandierendonck, A.; Weidensee, B.; Plovyt, M.; Stuchlíková, M.; François, N.; Valembois, S.; Heyndrickx, L.; Michiels, J.; et al. A Dual-Antigen Self-Amplifying RNA SARS-CoV-2 Vaccine Induces Potent Humoral and Cellular Immune Responses and Protects against SARS-CoV-2 Variants through T Cell-Mediated Immunity. Mol. Ther. 2022, 30, 2968–2983. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef]
- Pourseif, M.M.; Parvizpour, S.; Jafari, B.; Dehghani, J.; Naghili, B.; Omidi, Y. A Domain-Based Vaccine Construct against SARS-CoV-2, the Causative Agent of COVID-19 Pandemic: Development of Self-Amplifying MRNA and Peptide Vaccines. BioImpacts 2020, 11, 65–84. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Ghosh, P.; Patra, P.; Patra, B.C.; Lee, S.-S.; Chakraborty, C. Bioengineering of Novel Non-Replicating MRNA (NRM) and Self-Amplifying MRNA (SAM) Vaccine Candidates Against SARS-CoV-2 Using Immunoinformatics Approach. Mol. Biotechnol. 2022, 64, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Komori, M.; Morey, A.L.; Quiñones-Molina, A.A.; Fofana, J.; Romero, L.; Peters, E.; Matsuda, K.; Gummuluru, S.; Smith, J.F.; Akahata, W.; et al. Incorporation of 5 Methylcytidine Alleviates Innate Immune Response to Self-Amplifying RNA Vaccine. bioRxiv Prepr. Serv. Biol. 2023. [Google Scholar] [CrossRef]
- McGee, J.E.; Kirsch, J.R.; Kenney, D.; Chavez, E.; Shih, T.-Y.; Douam, F.; Wong, W.W.; Grinstaff, M.W. Complete Substitution with Modified Nucleotides Suppresses the Early Interferon Response and Increases the Potency of Self-Amplifying RNA. bioRxiv Prepr. Serv. Biol. 2023. [Google Scholar] [CrossRef]
- Aboshi, M.; Matsuda, K.; Kawakami, D.; Kono, K.; Kazami, Y.; Sekida, T.; Komori, M.; Morey, A.L.; Suga, S.; Smith, J.F.; et al. Safety and Immunogenicity of VLPCOV-02, a SARS-CoV-2 Self-Amplifying RNA Vaccine with a Modified Base, 5-Methylcytosine. iScience 2024, 27, 108964. [Google Scholar] [CrossRef] [PubMed]
- Deming, M.E.; Lyke, K.E. A ‘Mix and Match’ Approach to SARS-CoV-2 Vaccination. Nat. Med. 2021, 27, 1510–1511. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.; Verma, M.; Voigt, E.; Battisti, P.; Beaver, S.; Reed, S.; Dinkins, K.; Mody, S.; Zakin, L.; Tanaka, S.; et al. Heterologous SaRNA Prime, DNA Dual-Antigen Boost SARS-CoV-2 Vaccination Elicits Robust Cellular Immunogenicity and Cross-Variant Neutralizing Antibodies. Front. Immunol. 2022, 13, 910136. [Google Scholar] [CrossRef] [PubMed]
- Szubert, A.J.; Pollock, K.M.; Cheeseman, H.M.; Alagaratnam, J.; Bern, H.; Bird, O.; Boffito, M.; Byrne, R.; Cole, T.; Cosgrove, C.A.; et al. COVAC1 Phase 2a Expanded Safety and Immunogenicity Study of a Self-Amplifying RNA Vaccine against SARS-CoV-2. eClinicalMedicine 2023, 56, 101823. [Google Scholar] [CrossRef]
- Kitonsa, J.; Kamacooko, O.; Ruzagira, E.; Nambaziira, F.; Abaasa, A.; Serwanga, J.; Gombe, B.; Lunkuse, J.; Naluyinda, H.; Tukamwesiga, N.; et al. A Phase I COVID-19 Vaccine Trial among SARS-CoV-2 Seronegative and Seropositive Individuals in Uganda Utilizing a Self-Amplifying RNA Vaccine Platform: Screening and Enrollment Experiences. Hum. Vaccines Immunother. 2023, 19, 2240690. [Google Scholar] [CrossRef]
- Pulendran, B. Systems Vaccinology: Probing Humanity’s Diverse Immune Systems with Vaccines. Proc. Natl. Acad. Sci. USA 2014, 111, 12300–12306. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Scott, M.K.D.; Hagan, T.; Li, C.; Feng, Y.; Wimmers, F.; Grigoryan, L.; Trisal, M.; Edara, V.V.; Lai, L.; et al. Systems Vaccinology of the BNT162b2 MRNA Vaccine in Humans. Nature 2021, 596, 410–416. [Google Scholar] [CrossRef]
- Pepini, T.; Pulichino, A.-M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef]
- Huysmans, H.; Zhong, Z.; De Temmerman, J.; Mui, B.L.; Tam, Y.K.; Mc Cafferty, S.; Gitsels, A.; Vanrompay, D.; Sanders, N.N. Expression Kinetics and Innate Immune Response after Electroporation and LNP-Mediated Delivery of a Self-Amplifying MRNA in the Skin. Mol. Ther. Nucleic Acids 2019, 17, 867–878. [Google Scholar] [CrossRef]
- Ong, E.Z.; Yee, J.X.; Ooi, J.S.G.; Syenina, A.; de Alwis, R.; Chen, S.; Sim, J.X.Y.; Kalimuddin, S.; Leong, Y.S.; Chan, Y.F.Z.; et al. Immune Gene Expression Analysis Indicates the Potential of a Self-Amplifying Covid-19 MRNA Vaccine. NPJ Vaccines 2022, 7, 154. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-Amplifying RNA Vaccines for Infectious Diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Oda, Y.; Kumagai, Y.; Kanai, M.; Iwama, Y.; Okura, I.; Minamida, T.; Yagi, Y.; Kurosawa, T.; Greener, B.; Zhang, Y.; et al. Immunogenicity and Safety of a Booster Dose of a Self-Amplifying RNA COVID-19 Vaccine (ARCT-154) versus BNT162b2 MRNA COVID-19 Vaccine: A Double-Blind, Multicentre, Randomised, Controlled, Phase 3, Non-Inferiority Trial. Lancet Infect. Dis. 2023, S1473-3099(23)00650-3. [Google Scholar] [CrossRef]
- Tregoning, J.S. LION: Taming RNA Vaccine Inflammation. Mol. Ther. 2023, 31, 2557. [Google Scholar] [CrossRef]
- Akahata, W.; Sekida, T.; Nogimori, T.; Ode, H.; Tamura, T.; Kono, K.; Kazami, Y.; Washizaki, A.; Masuta, Y.; Suzuki, R.; et al. Safety and Immunogenicity of SARS-CoV-2 Self-Amplifying RNA Vaccine Expressing an Anchored RBD: A Randomized, Observer-Blind Phase 1 Study. Cell Rep. Med. 2023, 4, 101134. [Google Scholar] [CrossRef]
- Palmer, C.D.; Scallan, C.D.; Kraemer Tardif, L.D.; Kachura, M.A.; Rappaport, A.R.; Koralek, D.O.; Uriel, A.; Gitlin, L.; Klein, J.; Davis, M.J.; et al. GRT-R910: A Self-Amplifying MRNA SARS-CoV-2 Vaccine Boosts Immunity for ≥6 Months in Previously-Vaccinated Older Adults. Nat. Commun. 2023, 14, 3274. [Google Scholar] [CrossRef]
- Elliott, T.; Cheeseman, H.M.; Evans, A.B.; Day, S.; McFarlane, L.R.; O’Hara, J.; Kalyan, M.; Amini, F.; Cole, T.; Winston, A.; et al. Enhanced Immune Responses Following Heterologous Vaccination with Self-Amplifying RNA and MRNA COVID-19 Vaccines. PLoS Pathog. 2022, 18, e1010885. [Google Scholar] [CrossRef]
- First Self-Amplifying MRNA Vaccine Approved. Nat. Biotechnol. 2024, 42, 4. [CrossRef]
- Li, Y.; Teague, B.; Zhang, Y.; Su, Z.; Porter, E.; Dobosh, B.; Wagner, T.; Irvine, D.J.; Weiss, R. In Vitro Evolution of Enhanced RNA Replicons for Immunotherapy. Sci. Rep. 2019, 9, 6932. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; McKay, P.F.; Bouton, C.R.; Hu, K.; Samnuan, K.; Shattock, R.J. Innate Inhibiting Proteins Enhance Expression and Immunogenicity of Self-Amplifying RNA. Mol. Ther. 2021, 29, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Ricke, D.O. Two Different Antibody-Dependent Enhancement (ADE) Risks for SARS-CoV-2 Antibodies. Front. Immunol. 2021, 12, 640093. [Google Scholar] [CrossRef] [PubMed]
- Okuya, K.; Hattori, T.; Saito, T.; Takadate, Y.; Sasaki, M.; Furuyama, W.; Marzi, A.; Ohiro, Y.; Konno, S.; Hattori, T.; et al. Multiple Routes of Antibody-Dependent Enhancement of SARS-CoV-2 Infection. Microbiol. Spectr. 2022, 10, e01553-21. [Google Scholar] [CrossRef]


| Vaccine Name | Clinical Trial Type | Vaccine Formulation | Encoded Antigen | Administration Schedule | Main Results | Ref. |
|---|---|---|---|---|---|---|
| LNP-nCoV saRNA (COVAC1) | Phase I | LNP | Prefusion stabilized S protein | IM, 0.1–10 μg, prime and boost, 4-week gap | 39–61% seroconversion rates. Reactogenicity with 10 μg dose | [55] |
| Phase II | IM, 1 μg (prime) & 10 μg (boost), 14-week gap | 80% seroconversion rates | [98] | |||
| Phase I | IM, 1 μg (prime) & 10 μg (boost), 14-week gap followed by two doses of licenced vaccine (BNT162b2 or AZD1222) | Higher nAb titers among saRNA recipients with a history of COVID-19 compared to licenced vaccines | [110] | |||
| LNP-nCoV saRNA-02 | IM, 5 μg, prime and boost, 4-week gap | Ongoing trial | [99] | |||
| ARCT-021 (LUNAR-COV-19) | LUNAR LNP | Codon-optimized S protein | IM, 1–10 μg, one dose | 80–100% seroconversion rate. Robust Th1 type responses | [54] | |
| Phase II | IM, 3 and 5 μg, prime and boost, 4-week gap | After second dose, no rise in nAbs | [54] | |||
| ARCT-154 | Phase III | S protein (D614G variant) | IM, fourth-dose boost after licenced vaccine (BNT162b2 or mRNA-1273) | Immune responses comparable or superior to BNT162b2 against Omicron BA.4/5 variant. Authorized in Japan in 2023 | [106] | |
| GRT-R910 | Phase I | LNP | Prefusion S (Wuhan Hu-1) and N, M & ORF3 T cell epitopes | IM, 10–30 μg, one dose boost after licenced vaccine (AZD1222, one or two doses) | Binding Abs and nAbs against the original strain and VoCs. Boosted AZD1222-induced T cell responses | [109] |
| VLPCOV-01 | RBD-TM | IM, 0.3 or 1 μg, one dose boost after licenced vaccine (BNT162b2) | Sustained immune responses comparable to BNT162b2. CD4+ CD8+ T-cell responses. nAbs against VoCs | [108] | ||
| VLPCOV-02 | LNP (5-mC modified saRNA) | RBD-TM (gamma VoC) | IM, 1–15 μg, one dose boost after licenced vaccine (BNT162b2) | Immune responses comparable to BNT162b2. CD4+ and CD8+ T-cell responses. and nAbs against VoCs | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Pilipich, N.; Beloki, U.; Salaberry, L.; Smerdou, C. Self-Amplifying RNA: A Second Revolution of mRNA Vaccines against COVID-19. Vaccines 2024, 12, 318. https://doi.org/10.3390/vaccines12030318
Silva-Pilipich N, Beloki U, Salaberry L, Smerdou C. Self-Amplifying RNA: A Second Revolution of mRNA Vaccines against COVID-19. Vaccines. 2024; 12(3):318. https://doi.org/10.3390/vaccines12030318
Chicago/Turabian StyleSilva-Pilipich, Noelia, Uxue Beloki, Laura Salaberry, and Cristian Smerdou. 2024. "Self-Amplifying RNA: A Second Revolution of mRNA Vaccines against COVID-19" Vaccines 12, no. 3: 318. https://doi.org/10.3390/vaccines12030318
APA StyleSilva-Pilipich, N., Beloki, U., Salaberry, L., & Smerdou, C. (2024). Self-Amplifying RNA: A Second Revolution of mRNA Vaccines against COVID-19. Vaccines, 12(3), 318. https://doi.org/10.3390/vaccines12030318