
Local Enrichment with Convergence of Enriched T-Cell Clones Are Hallmarks of Effective Peptide Vaccination against B16 Melanoma
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice Vaccination and Tumor Model
2.2. Tissue Processing and In Vitro Restimulation
2.3. Flow Cytometric Analysis
2.4. TCR Library Preparation and Repertoire Sequencing
2.5. TCR Repertoire Analysis
3. Results
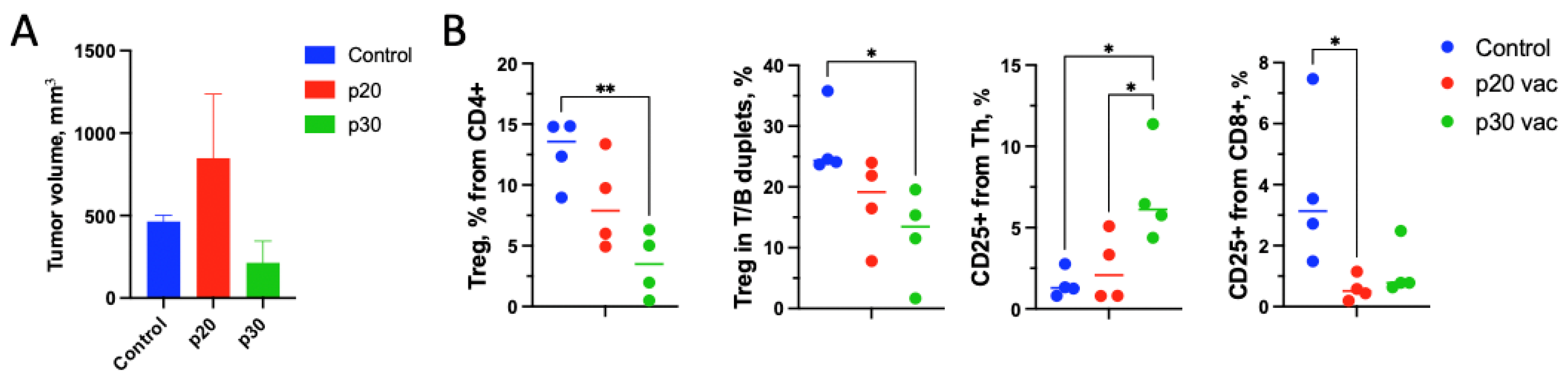
3.1. Pre-Vaccination with the p20 Peptide Stimulates Tumor Growth
3.2. T-Cells from p20-Vaccinated Mice Are More Responsive to In Vitro Restimulation Compared to p30
3.3. p30 Vaccine Provides Better Th Tumor Response and Treg Suppression In Vivo
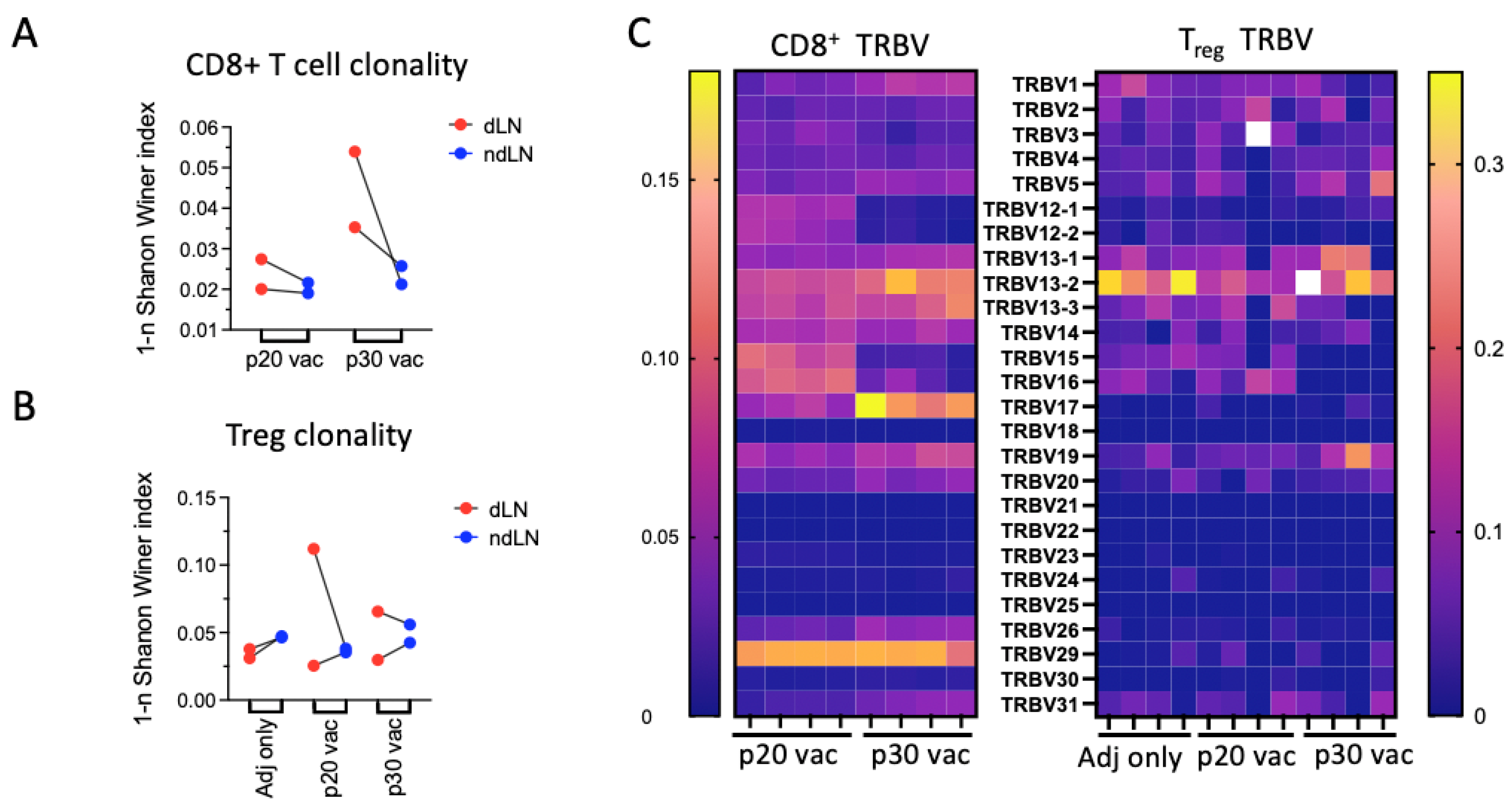
3.4. p30 but Not p20: Vaccine Elicits Clonal Site-Specific Tumor Response
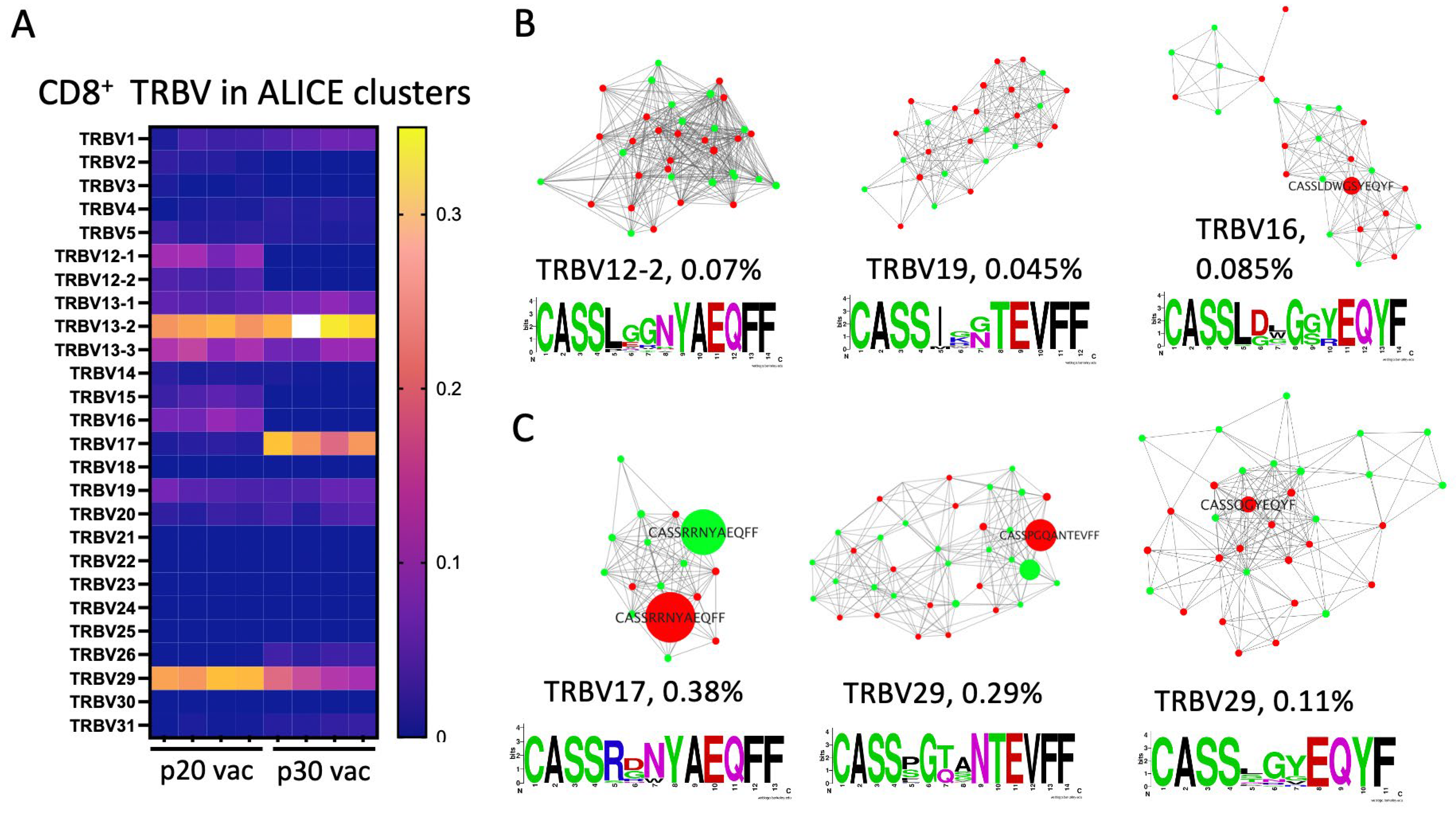
3.5. Major dLN Expanded Clones in p30 but Not in p20 Have Clusters of Convergent Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- van Poelgeest, M.I.E.; Welters, M.J.P.; van Esch, E.M.G.; Stynenbosch, L.F.; Kerpershoek, G.; van Persijn van Meerten, E.L.; van den Hende, M.; Löwik, M.J.; Berends-van der Meer, D.M.; Fathers, L.M.; et al. HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. J. Transl. Med. 2013, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Slingluff, C.L.; Slingluff, C.L., Jr. The Present and Future of Peptide Vaccines for Cancer. Cancer J. 2011, 17, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.; Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.; McClaren, J.L.; Oguin, T.H.; Rothwell, W.; Todd, B.; Morris, M.Y.; Becksfort, J.; Reynolds, C.; Brown, S.A.; Doherty, P.C.; et al. Paired analysis of TCRα and TCRβ chains at the single-cell level in mice. J. Clin. Investig. 2011, 121, 288–295. [Google Scholar] [CrossRef]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for comprehensive adaptive immunity profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Shugay, M.; Bagaev, D.V.; Turchaninova, M.A.; Bolotin, D.A.; Britanova, O.V.; Putintseva, E.V.; Pogorelyy, M.V.; Nazarov, V.I.; Zvyagin, I.V.; Kirgizova, V.I.; et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 2015, 11, e1004503. [Google Scholar] [CrossRef]
- Pogorelyy, M.V.; Shugay, M. A Framework for Annotation of Antigen Specificities in High-Throughput T-Cell Repertoire Sequencing Studies. Front. Immunol. 2019, 10, 2159. [Google Scholar] [CrossRef]
- Pogorelyy, M.V.; Minervina, A.A.; Shugay, M.; Chudakov, D.M.; Lebedev, Y.B.; Mora, T.; Walczak, A.M. Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol. 2019, 17, e3000314. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Shin, S.; Yin, J.H.; Park, J.; Jung, S.-H.; Chung, Y.-J. Single-cell RNA sequencing reveals the existence of pro-metastatic subpopulation within a parental B16 murine melanoma cell line. Biochem. Biophys. Res. Commun. 2022, 613, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.F.; Schoonderwoerd, M.; Knopf, P.; Camps, M.G.M.; Hawinkels, L.J.A.C.; Kneilling, M.; van Hall, T.; Ossendorp, F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. J. Clin. Investig. 2018, 3, e124507. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Philip, H.; Snir, T.; Gordin, M.; Shugay, M.; Zilberberg, A.; Efroni, S. A T cell repertoire timestamp is at the core of responsiveness to CTLA-4 blockade. iScience 2021, 24, 102100. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.S.; Kallarakal, M.A.; Jayaraman, S.; Ibukun, F.I.; Tong, K.P.; Orzolek, L.D.; Larman, H.B.; Krummey, S.M. Transplantation elicits a clonally diverse CD8+ T cell response that is comprised of potent CD43+ effectors. Cell Rep. 2023, 42, 112993. [Google Scholar] [CrossRef] [PubMed]
- Kohanim, Y.K.; Tendler, A.; Mayo, A.; Friedman, N.; Alon, U. Endocrine Autoimmune Disease as a Fragility of Immune Surveillance against Hypersecreting Mutants. Immunity 2020, 52, 872–884.e5. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, D.; Li, W.; Huang, Y.; Ye, X.; Waite, J.; Barry, T.; Edelmann, K.H.; Levenkova, N.; Guo, C.; et al. Evaluation of the capacities of mouse TCR profiling from short read RNA-seq data. PLoS ONE 2018, 13, e0207020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef]
- A Spidale, N.; Malhotra, N.; Frascoli, M.; Sylvia, K.; Miu, B.; Freeman, C.; Stadinski, B.D.; Huseby, E.; Kang, J. Neonatal-derived IL-17 producing dermal γδ T cells are required to prevent spontaneous atopic dermatitis. eLife 2020, 9, e51188. [Google Scholar] [CrossRef]
- Quandt, J.; Schlude, C.; Bartoschek, M.; Will, R.; Cid-Arregui, A.; Schölch, S.; Reissfelder, C.; Weitz, J.; Schneider, M.; Wiemann, S.; et al. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. OncoImmunology 2018, 7, e1500671. [Google Scholar] [CrossRef]
- Brezar, V.; Godot, V.; Cheng, L.; Su, L.; Lévy, Y.; Seddiki, N. T-Regulatory Cells and Vaccination “Pay Attention and Do Not Neglect Them”: Lessons from HIV and Cancer Vaccine Trials. Vaccines 2016, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Wong, W.-I.; Wang, Y.-L.; Hsieh, M.-P.; Lu, C.-W.; Liang, C.-Y.; Jui, S.-H.; Wu, F.-Y.; Chen, P.-J.; Yang, H.-C. Vaccine-induced antigen-specific regulatory T cells attenuate the antiviral immunity against acute influenza virus infection. Mucosal Immunol. 2018, 11, 1239–1253. [Google Scholar] [CrossRef]
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017, 547, 89–93. [Google Scholar] [CrossRef]
- Goncharov, M.M.; Bryushkova, E.A.; Sharayev, N.I.; Skatova, V.D.; Baryshnikova, A.M.; Sharonov, G.V.; Karnaukhov, V.; Vakhitova, M.T.; Samoylenko, I.V.; Demidov, L.V.; et al. Pinpointing the tumor-specific T cells via TCR clusters. eLife 2022, 11, e77274. [Google Scholar] [CrossRef]
- Izraelson, M.; Nakonechnaya, T.O.; Moltedo, B.; Egorov, E.S.; Kasatskaya, S.A.; Putintseva, E.V.; Mamedov, I.Z.; Staroverov, D.B.; Shemiakina, I.I.; Zakharova, M.Y.; et al. Comparative analysis of murine T-cell receptor repertoires. Immunology 2018, 153, 133–144. [Google Scholar] [CrossRef]
- McDonnell, W.J.; Koethe, J.R.; Mallal, S.A.; Pilkinton, M.A.; Kirabo, A.; Ameka, M.K.; Cottam, M.A.; Hasty, A.H.; Kennedy, A.J. High CD8 T-Cell Receptor Clonality and Altered CDR3 Properties Are Associated with Elevated Isolevuglandins in Adipose Tissue During Diet-Induced Obesity. Diabetes 2018, 67, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.C.; Rodríguez, E.G. Vaccine adjuvants revisited. Vaccine 2007, 25, 3752–3762. [Google Scholar] [CrossRef] [PubMed]
- Aichele, P.; Brduscha-Riem, K.; Zinkernagel, R.M.; Hengartner, H.; Pircher, H. T cell priming versus T cell tolerance induced by synthetic peptides. J. Exp. Med. 1995, 182, 261–266. [Google Scholar] [CrossRef]
- Toes, R.E.; Offringa, R.; Blom, R.J.; Melief, C.J.; Kast, W.M. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc. Natl. Acad. Sci. USA 1996, 93, 7855–7860. [Google Scholar] [CrossRef]
- Rapaka, R.R.; Cross, A.S.; McArthur, M.A. Using Adjuvants to Drive T Cell Responses for Next-Generation Infectious Disease Vaccines. Vaccines 2021, 9, 820. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.-F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef]
- Aloulou, M.; Carr, E.J.; Gador, M.; Bignon, A.; Liblau, R.S.; Fazilleau, N.; Linterman, M.A. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat. Commun. 2016, 7, 10579. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-I.; Barrios, K.; Lee, Y.-R.; Linowski, A.K.; Celis, E. BiVax: A peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol. Immunother. 2013, 62, 787–799. [Google Scholar] [CrossRef]
- Kemmler, C.B.; Clambey, E.T.; Kedl, R.M.; Slansky, J.E. Elevated Tumor-Associated Antigen Expression Suppresses Variant Peptide Vaccine Responses. J. Immunol. 2011, 187, 4431–4439. [Google Scholar] [CrossRef]
- Kenison, J.E.; Stevens, N.A.; Quintana, F.J. Therapeutic induction of antigen-specific immune tolerance. Nat. Rev. Immunol. 2023, 1–20. [Google Scholar] [CrossRef]
- Perica, K.; Bieler, J.G.; Schütz, C.; Varela, J.C.; Douglass, J.; Skora, A.; Chiu, Y.L.; Oelke, M.; Kinzler, K.; Zhou, S.; et al. Enrichment and Expansion with Nanoscale Artificial Antigen Presenting Cells for Adoptive Immunotherapy. ACS Nano 2015, 9, 6861–6871. [Google Scholar] [CrossRef]
- Li, M.; Itoh, A.; Xi, J.; Yu, C.; Wu, Y.; Ridgway, W.M.; Liu, H. Enhancing Antigen Presentation and Inducing Antigen-Specific Immune Tolerance with Amphiphilic Peptides. J. Immunol. 2021, 207, 2051–2059. [Google Scholar] [CrossRef]
- Gangaplara, A.; Massilamany, C.; Lasrado, N.; Steffen, D.; Reddy, J. Evidence for Anti-Viral Effects of Complete Freund’s Adjuvant in the Mouse Model of Enterovirus Infection. Vaccines 2020, 8, 364. [Google Scholar] [CrossRef]
- Roy, D.G.; Geoffroy, K.; Marguerie, M.; Khan, S.T.; Martin, N.T.; Kmiecik, J.; Bobbala, D.; Aitken, A.S.; de Souza, C.T.; Stephenson, K.B.; et al. Adjuvant oncolytic virotherapy for personalized anti-cancer vaccination. Nat. Commun. 2021, 12, 26261. [Google Scholar] [CrossRef] [PubMed]
- Ritvo, P.-G.; Saadawi, A.; Barennes, P.; Quiniou, V.; Chaara, W.; El Soufi, K.; Bonnet, B.; Six, A.; Shugay, M.; Mariotti-Ferrandiz, E.; et al. High-resolution repertoire analysis reveals a major bystander activation of Tfh and Tfr cells. Proc. Natl. Acad. Sci. USA 2018, 115, 9604–9609. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.P.; Ruiter, B.; Virkud, Y.V.; Tu, A.A.; Monian, B.; Moon, J.J.; Love, J.C.; Shreffler, W.G. Identification of antigen-specific TCR sequences based on biological and statistical enrichment in unselected individuals. J. Clin. Investig. 2021, 6, e140028. [Google Scholar] [CrossRef] [PubMed]
- Madi, A.; Poran, A.; Shifrut, E.; Reich-Zeliger, S.; Greenstein, E.; Zaretsky, I.; Arnon, T.; Van Laethem, F.; Singer, A.; Lu, J.; et al. T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. eLife 2017, 6, e22057. [Google Scholar] [CrossRef] [PubMed]
- Yuzhakova, D.V.; Volchkova, L.N.; Pogorelyy, M.V.; Serebrovskaya, E.O.; Shagina, I.A.; Bryushkova, E.A.; Nakonechnaya, T.O.; Izosimova, A.V.; Zavyalova, D.S.; Karabut, M.M.; et al. Measuring Intratumoral Heterogeneity of Immune Repertoires. Front. Oncol. 2020, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Aran, A.; Lázaro, G.; Marco, V.; Molina, E.; Abancó, F.; Peg, V.; Gión, M.; Garrigós, L.; Pérez-García, J.; Cortés, J.; et al. Analysis of tumor infiltrating CD4+ and CD8+ CDR3 sequences reveals shared features putatively associated to the anti-tumor immune response. Front. Immunol. 2023, 14, 1227766. [Google Scholar] [CrossRef] [PubMed]
- Gordin, M.; Philip, H.; Zilberberg, A.; Gidoni, M.; Margalit, R.; Clouser, C.; Adams, K.; Vigneault, F.; Cohen, I.R.; Yaari, G.; et al. Breast cancer is marked by specific, Public T-cell receptor CDR3 regions shared by mice and humans. PLoS Comput. Biol. 2021, 17, e1008486. [Google Scholar] [CrossRef]
- Vanderlugt, C.L.; Miller, S.D. Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.-W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the Tcr Repertoire of Tumor-Infiltrating t Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Schober, K.; Fuchs, P.; Mir, J.; Hammel, M.; Fanchi, L.; Flossdorf, M.; Busch, D.H. The CMV-Specific CD8+ T Cell Response Is Dominated by Supra-Public Clonotypes with High Generation Probabilities. Pathogens 2020, 9, 650. [Google Scholar] [CrossRef]
- Collier, J.L.; Pauken, K.E.; Lee, C.A.A.; Patterson, D.G.; Markson, S.C.; Conway, T.S.; Fung, M.E.; France, J.A.; Mucciarone, K.N.; Lian, C.G.; et al. Single-Cell Profiling Reveals Unique Features of Diabetogenic T Cells in Anti-PD-1-Induced Type 1 Diabetes Mice. J. Exp. Med. 2023, 220, e20221920. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK+ CD8+ T Cells as Conserved Hallmark of Inflammaging. Immunity 2021, 54, 99–115.e12. [Google Scholar] [CrossRef]
- Alli, R.; Zhang, Z.M.; Nguyen, P.; Zheng, J.J.; Geiger, T.L. Rational Design of T Cell Receptors with Enhanced Sensitivity for Antigen. PLoS ONE 2011, 6, e18027. [Google Scholar] [CrossRef]
- Wei, S.C.; Sharma, R.; Anang, N.-A.A.S.; Levine, J.H.; Zhao, Y.; Mancuso, J.J.; Setty, M.; Sharma, P.; Wang, J.; Pe’er, D.; et al. Negative Co-Stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. Immunity 2019, 50, 1084–1098.e10. [Google Scholar] [CrossRef]
- Bagaev, D.V.; Vroomans, R.M.A.; Samir, J.; Stervbo, U.; Rius, C.; Dolton, G.; Greenshields-Watson, A.; Attaf, M.; Egorov, E.S.; Zvyagin, I.V.; et al. VDJdb in 2019: Database Extension, New Analysis Infrastructure and a T-Cell Receptor Motif Compendium. Nucleic Acids Res. 2019, 48, D1057–D1062. [Google Scholar] [CrossRef] [PubMed]
- Tickotsky, N.; Sagiv, T.; Prilusky, J.; Shifrut, E.; Friedman, N. McPAS-TCR: A Manually Curated Catalogue of Pathology-Associated T Cell Receptor Sequences. Bioinformatics 2017, 33, 2924–2929. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | TRBV | CDR3 | Frequencies, × 10−3 | Occurrence, Reference | |||
|---|---|---|---|---|---|---|---|
| dLN1 | ndLN1 | dLN2 | ndLN2 | ||||
| p20 | 17 | CASSPRQGANTEVFF | 2.7 | 6 | - | - | Public, [22] |
| p20 | 16 | CASSLDDINTEVFF | - | - | 3.2 | 2.9 | |
| p20 | 14 | CASSFSWGGDTQYF | - | - | 2.1 | 4.2 | |
| p20 | 20 | CGALTGENTLYF | 4.1 | 1.24 | - | - | |
| p20 | 12-2 | CASSLVGGYEQYF | - | 0.05 | 3.45 | 1.5 | Public, [23] |
| p30 | 17 | CASSRRNYAEQFF | 7.2 | 6.4 | - | - | Public, enriched after a CTLA4 treatment, [19,21] |
| p30 | 29 | CASSPGQANTEVFF | 8.5 | 3.3 | 0.3 | 0.01 | MC38 colon cancer CTLA4 response, [19] |
| p30 | 29 | CASSPGQSAETLYF | 1.35 | 0.8 | 8.3 | - | Public, [22] SOX13KO, [24] |
| p30 | 15 | CASSDRVEQYF | 0.16 | - | 8.7 | 1 | Allogenic skin graft, [20] |
| p30 | 13-2 | CASGDARNTLYF | - | 0.05 | 8.5 | - | Public, [22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izosimova, A.V.; Shabalkina, A.V.; Myshkin, M.Y.; Shurganova, E.V.; Myalik, D.S.; Ryzhichenko, E.O.; Samitova, A.F.; Barsova, E.V.; Shagina, I.A.; Britanova, O.V.; et al. Local Enrichment with Convergence of Enriched T-Cell Clones Are Hallmarks of Effective Peptide Vaccination against B16 Melanoma. Vaccines 2024, 12, 345. https://doi.org/10.3390/vaccines12040345
Izosimova AV, Shabalkina AV, Myshkin MY, Shurganova EV, Myalik DS, Ryzhichenko EO, Samitova AF, Barsova EV, Shagina IA, Britanova OV, et al. Local Enrichment with Convergence of Enriched T-Cell Clones Are Hallmarks of Effective Peptide Vaccination against B16 Melanoma. Vaccines. 2024; 12(4):345. https://doi.org/10.3390/vaccines12040345
Chicago/Turabian StyleIzosimova, Anna Vyacheslavovna, Alexandra Valerievna Shabalkina, Mikhail Yurevich Myshkin, Elizaveta Viktorovna Shurganova, Daria Sergeevna Myalik, Ekaterina Olegovna Ryzhichenko, Alina Faritovna Samitova, Ekaterina Vladimirovna Barsova, Irina Aleksandrovna Shagina, Olga Vladimirovna Britanova, and et al. 2024. "Local Enrichment with Convergence of Enriched T-Cell Clones Are Hallmarks of Effective Peptide Vaccination against B16 Melanoma" Vaccines 12, no. 4: 345. https://doi.org/10.3390/vaccines12040345