1. Introduction
The formation of distant metastases and the dissemination of cancer cells are thought to be the main causes of cancer-related mortality. The metastatic cascade typically initiates in the tumor-draining lymph nodes (tdLNs), also known as the sentinel lymph nodes (SLNs), where locoregional metastases take root [
1,
2]. From the tdLNs, tumor cells can disseminate to distant LNs and organs in the body [
3]. Tumor lymphangiogenesis, involving the proliferation of lymphatic vessels, supports the migration of tumor cells to tdLNs and may be associated with an unfavorable prognosis [
2,
4]. Generally, lymphatic vessels play a crucial role in transporting immune cells and tumor antigens (tAgs) to tdLNs, where both local and systemic immunity are coordinated through innate and specific immune mechanisms. The tdLN serves as the initial site where naïve T cells are primed against tAgs. To evade the induced immune response, the tumor must effectively suppress these primed T cells. Given the high density of dendritic cells (DCs) and T cells in the LNs, costimulatory signals for T and B cells can be provided, often in a bystander manner [
5]. As the tumor progresses, changes occur in the immune composition of the tdLN. For instance, an altered CD4/CD8 T cells ratio and an increase in T regulatory cells (Tregs) become more pronounced, especially following metastatic involvement in melanoma, breast cancer, and cervical cancer [
6,
7,
8]. As metastasis in the tdLN expands, there is a concurrent increase in the recruitment of myeloid-derived suppressor cells (MDSCs), accompanied by the release of immunosuppressive exosomes and soluble mediators [
9]. A pre-metastatic niche may arise as a result of several of these alterations occurring even before the appearance of tumor cells [
10].
Past efforts in therapeutic cancer vaccines primarily targeted tumor-associated antigens (TAAs), yielding modest immune responses due to immune tolerance towards self-antigens. Therefore, neoantigens, not expected to induce T cell tolerance, have attracted attention. Personalized vaccines directed against individual tumor neoantigens, using long peptides or RNA, have shown promising outcomes in mice [
11] and humans [
12,
13]. However, challenges in this approach include identifying individual antigens, rendering epitopes immunogenic in humans, and the substantial tissue required for characterizing the immunopeptidome [
14].
The fact that tumors and their microenvironment offer relatively little support for T cell activation may be the reason for intratumoral immunotherapy comparatively modest level of success as of yet. This is fundamentally different in the LNs, as they are highly specialized structures to effectively activate T cells. Together with the presence of metastatic tumor cells (providing tAgs), LNs represent much better conditions to promote antitumor T cell response than solid tumors. Therefore, intranodal treatments that reverse immune suppression and promote T cell activation hold great promise for harnessing the patient’s immune system for efficient fighting against cancer.
The concept of intralymphatic/intranodal vaccination was initially investigated in the 1970s. Juillard et al. utilized this approach to augment the effectiveness of tumor cell-based vaccination in dogs [
15,
16,
17]. Subsequently, in the 1980s, researchers found that injecting nanograms of proteins via intranodal delivery was enough to elicit a humoral immune response [
18]. A biodistribution study in mice revealed that intranodal injection of antigens resulted in a concentration in the dLNs that was 100-fold higher compared to subcutaneous (s.c.) injection [
16]. Human studies have demonstrated that twenty minutes post-injection into an inguinal lymph node (iLN), the protein had migrated to deeper pelvic LNs, a phenomenon not observed after s.c. injection [
16]. Several pre-clinical and clinical studies have centered on the intralymphatic administration of drugs, immunotherapies, or vaccines. In humans, drugs are typically introduced into an iLN under ultrasound guidance, a routine procedure performed by experienced radiologists [
19]. In the context of cancer immunotherapy, DCs pulsed with antigens and administered intranodally in melanoma patients have shown a propensity to localize in the paracortex of LNs [
20,
21]. While some clinical trials employing intranodal DC therapy have suggested enhanced immune responses, others have failed to demonstrate a significant advantage of the intranodal route compared to intradermal injection [
22,
23,
24].
Virus-like particles (VLPs) are nanoparticles structured in a repetitive icosahedral geometry. Although VLPs resemble their parent viruses, they lack the genetic material necessary for reproduction which contributes to their good safety profile [
25]. A pathogen-associated structural pattern (PASP) that can trigger potent innate and adaptive immune responses is represented by the surface geometry of VLP. In addition to being used and sold as part of preventative vaccinations against the hepatitis B virus (HBV) and human papillomavirus (HPV), VLPs are currently being extensively studied in the area of cancer immunotherapy [
26]. Among others, we have successfully designed and developed different therapeutic cancer vaccines utilizing VLPs and have provided several proofs of concepts in mice [
11,
27,
28,
29]. Recently, we have focused on local delivery of VLPs as effective immunotherapy [
29]. We formulated our plant-derived VLPs (naked VLPs, i.e., do not display any tAgs) with a depot-forming microcrystalline tyrosine adjuvant (MCT) for intratumoral administration in mice harboring the aggressive B16F10 melanoma [
29]. The therapy activated the immune system through multiple immunological pathways resulting in considerable antitumor effects. Importantly, this strategy does not require the knowledge of tAgs, meaning that the same therapy can be used for different cancer types, thus facilitating translation to humans.
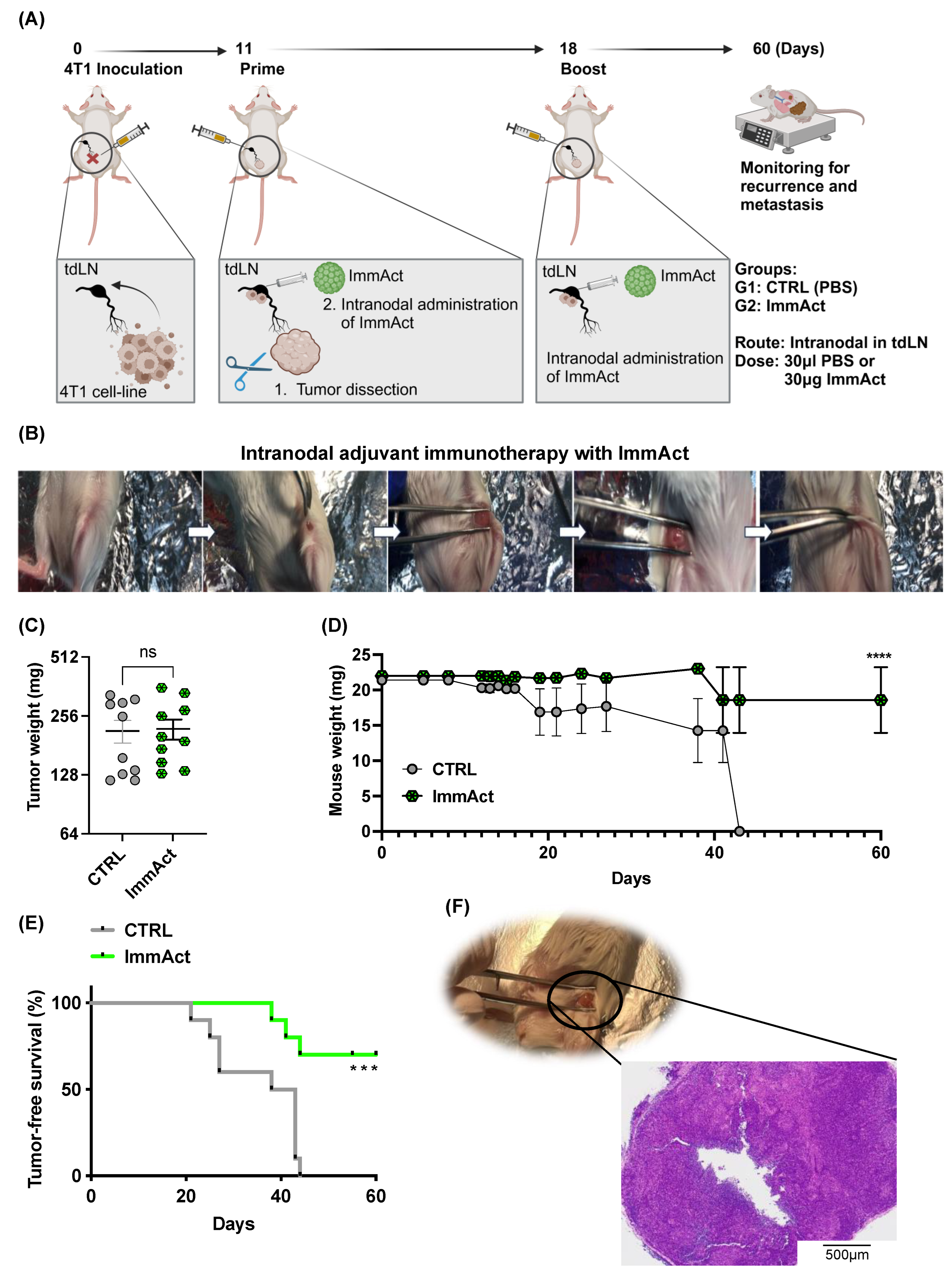
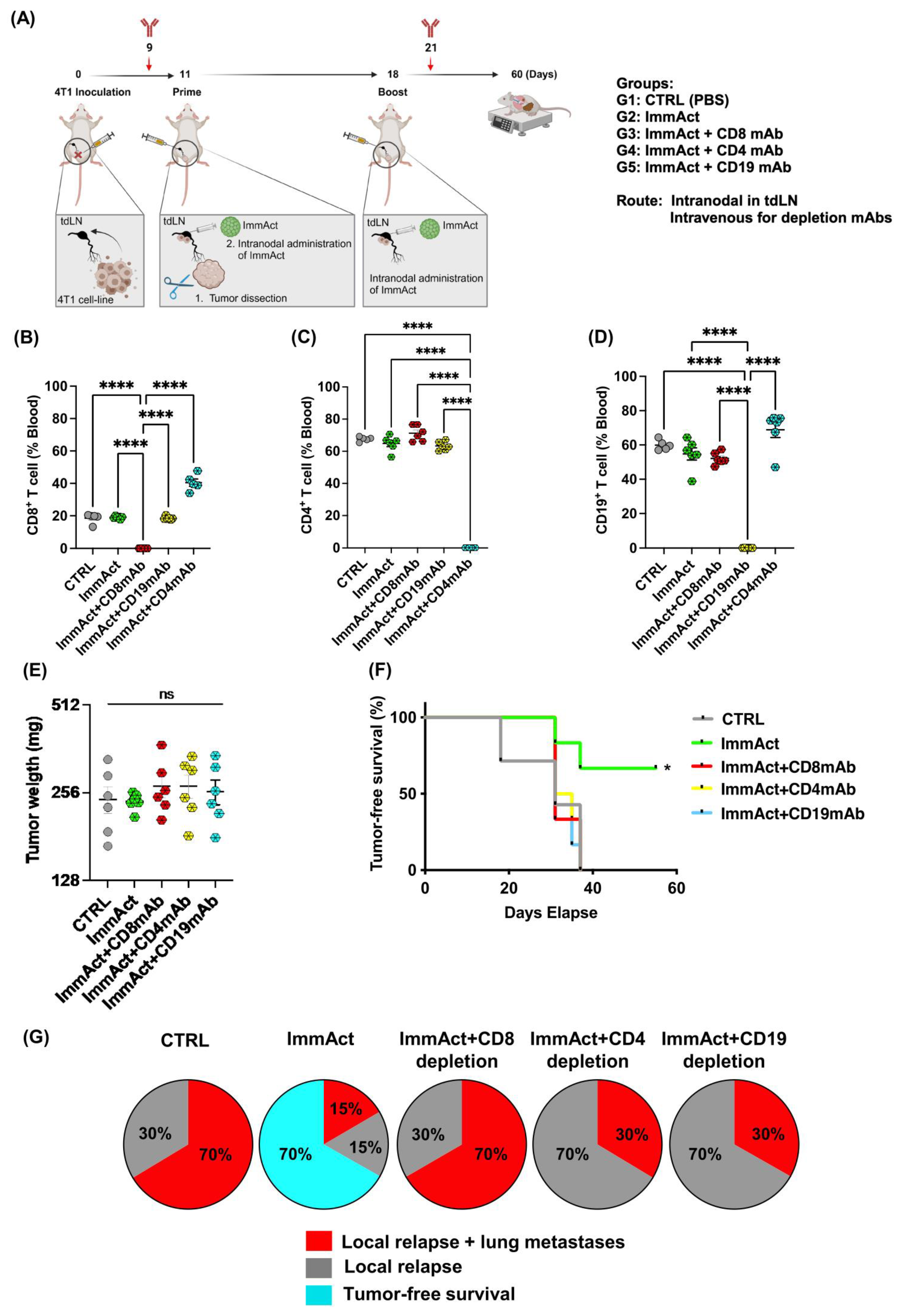
In this study, we evaluated the antitumor efficacy of our plant-derived VLP, henceforth referred to as immune activator (ImmAct) for intranodal administration, directly in the tdLN, aiming to protect mice against local and systemic relapse. We employed an adjuvant approach using the aggressive metastatic murine mammary carcinoma model 4T1.
Our study provides a proof of concept of an effective and novel vaccination strategy with naked VLPs that can readily be implemented for clinical use.
3. Discussion
In this study, we explored the adjuvant antitumor efficacy of plant-derived nanoparticles designed for intranodal injection directly into the tdLN using an aggressive metastatic mammary carcinoma murine model. Our strategy mirrors the in situ/intratumor immunization strategy [
29], aiming to convert immunosuppressed tdLNs into active, hot ones.
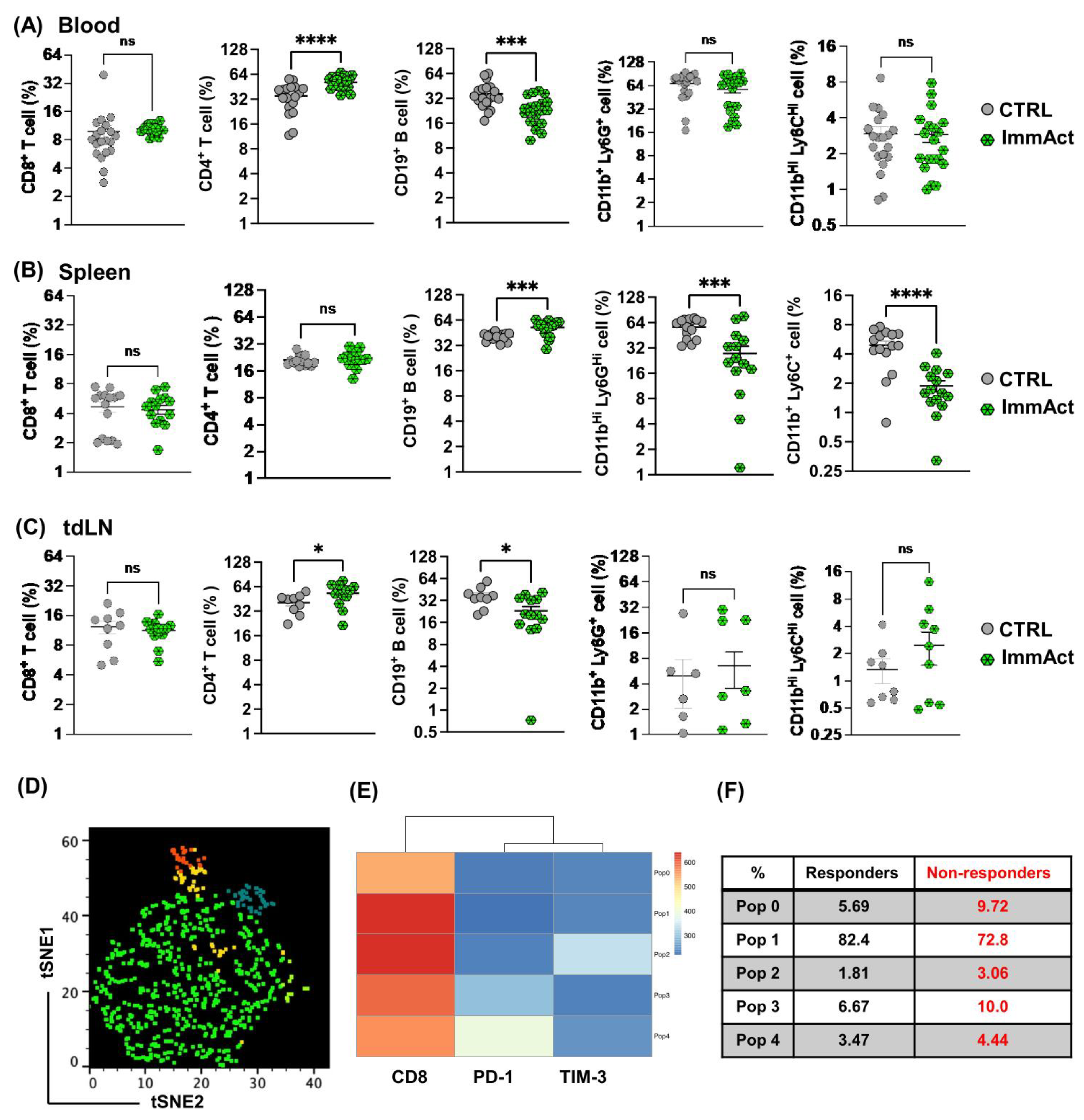
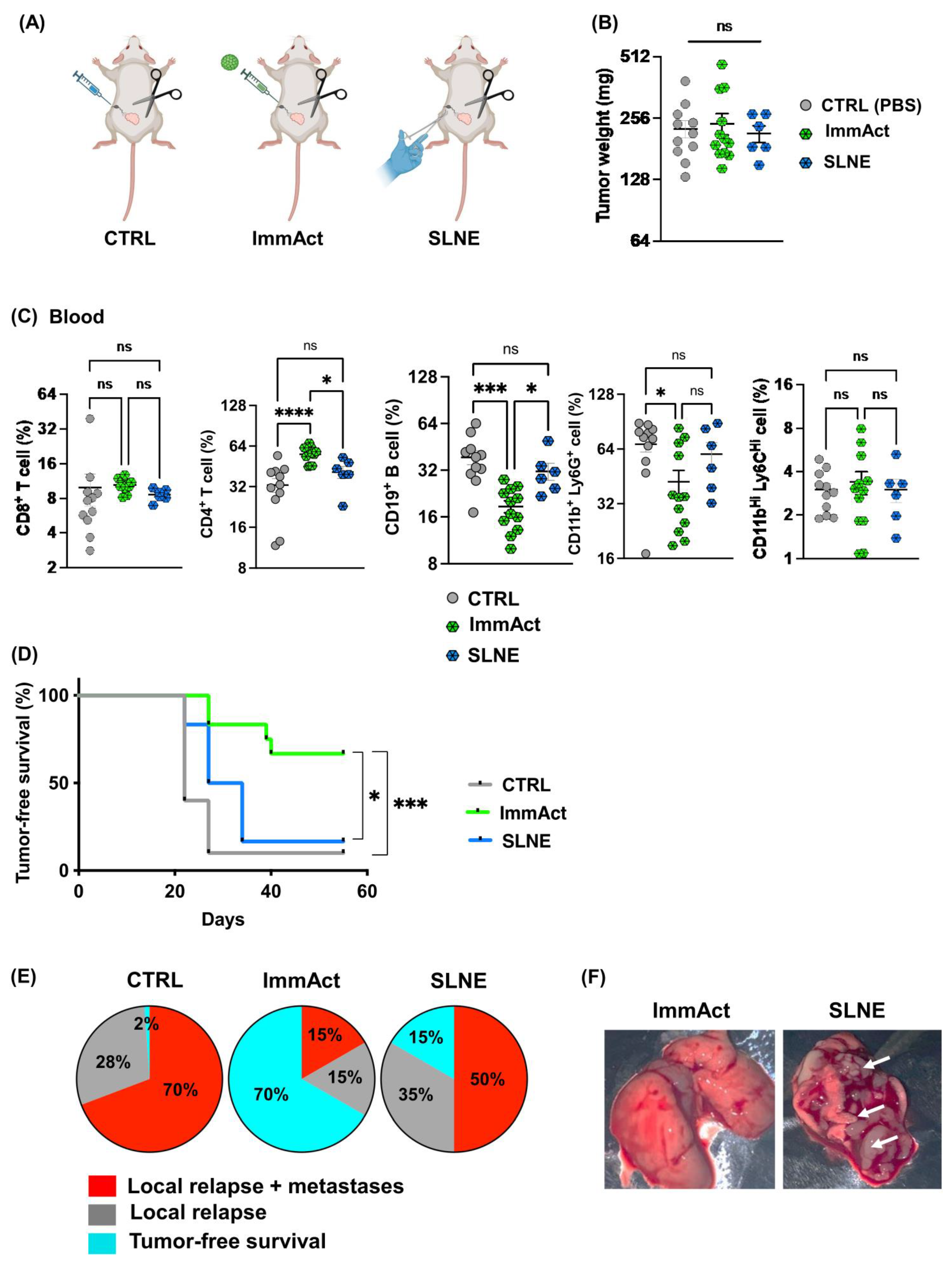
Our results reveal a significant antitumor response evidenced by protection against local and systemic tumor relapse and minimized weight loss. The intranodal administration of ImmAct led to both local and systemic alterations in cellular and humoral immunity, primarily characterized by an enhancement of CD4+ T cells in the blood and the tdLN in the group treated with ImmAct. Furthermore, differentiating between responder and non-responder mice after treatment with ImmAct revealed an increased percentage of CD8+ T cells in the tdLN expressing exhaustion markers such as PD-1 and TIM-3. Indeed, our innovative approach surpassed the gold standard surgery of SLNE.
Our immunotherapy with ImmAct was directly administered into a metastatic tdLN to induce a robust local and systemic immune response and to enhance tumor-free survival. In humans and dogs, intranodal injection of inactivated tumor cells has been tested with promising results [
17,
30,
31]. Additionally, the administration of MHC-I binding peptides into LNs or the spleen has induced robust CD8
+ T cell response, protecting from tumor growth or viral replication in mice [
32]. Furthermore, intranodal immunization has significantly augmented the effects of plasmid DNA [
33,
34] and RNA vaccination in mice [
35,
36]. Intranodal immunotherapy utilizing our ImmAct potentially offers significant benefits compared to other intranodal approaches. The distinct advantage of our method lies in the use of VLPs, which are not only safe, but also equipped with a potent TLR-ligand, coupled with a tetanus toxin (TT) internal epitope. This unique combination is designed to amplify the immune response in future translation into humans, leveraging the widespread immunity to tetanus already established in most individuals. This aspect is particularly advantageous in enhancing the immune responses of elderly patients or those with cancer, who often experience compromised immune functionality.
The advantage of intranodal immunization lies in the high efficiency of reaching both naïve T cells and tumor-infiltrating lymphocytes (TILs) in these LNs, which, have been suppressed by the tumor [
37]. It is well-established that VLPs have an inherent capacity to provide additional T cell help beyond the primary benefits of the VLP itself, as they induce VLP-specific CD4
+ T cells [
38]. This phenomenon likely accounts for the significant increase in CD4
+ T cell response observed in both the bloodstream and the tdLNs within the ImmAct-treated group. Intriguingly, this augmentation in CD4
+ T cells was accompanied by a noteworthy reduction in B cells at these two locations, contrasted by an increase in the spleen. These findings hint at the possibility that activated B cells may be migrating toward and accumulating in the spleen after their activation. This redistribution of B cells may also suggest that ImmAct is altering the homing signals or the environment within the tdLN, making the spleen a more favorable site for B cell activation.
Although there is no discernible rise in CD8
+ T cells observed in the blood, spleen, or tdLN following ImmAct immunization, depletion of this population nullified the antitumor effect, leading to heightened local and distant relapse of approximately 70%. The anti-tumor efficacy of ImmAct was found to be critically dependent on CD8
+ T cells, even though it was partially dependent on CD4
+ T cells and B cells. This was demonstrated by the significantly reduced protection observed upon in vivo depletion of CD8
+ T cells, supporting our previous findings highlighting the critical role of CD8
+ T cells in the s.c. or intratumoral administration of VLP-based vaccines or immune enhancers [
27,
29]. Exploring combination therapy with checkpoint inhibitors may be a strategy to further enhance the efficacy of ImmAct, potentially with synergistic effects for improving immune responses and outcomes in cancer treatment.
Our data demonstrate a significant improvement in tumor-free survival among mice treated with ImmAct compared to the CTRL group. Remarkably, the intranodal administration of ImmAct exhibited superior efficacy over the widely accepted standard medical procedure of SLNE surgery, showing a 70% tumor-free survival rate in contrast to 15% achieved by SLNE alone. This outcome is particularly noteworthy as SLNE has long stood as the gold standard of therapy for preventing metastatic disease in cancer patients. In stage III melanoma patients, treatment has dramatically improved by de-escalating surgery of occult metastases in regional LNs and the introduction of modern immunotherapies [
12]. Recently, Trophy et. al., compared the outcome of adjuvant therapy versus observation in melanoma patients with a positive SLN that did not undergo complete LN dissection [
13]. The study found a significantly better 24 month metastases-free survival in patients undergoing adjuvant therapy (86% vs. 59%), highlighting the importance of this approach [
13].
Remarkably, the analysis of responder and non-responder cohorts post-ImmAct immunotherapy uncovered intriguing insights into CD8 populations. Specifically, the non-responder group exhibited a distinctive rise in exhaustion markers, anti-PD1 and TIM-3. In recent research, findings have indicated that TIM-3 is a component of a complex module encompassing several co-inhibitory receptors, commonly known as checkpoint receptors. These receptors tend to be concurrently expressed and regulated on T cells experiencing dysfunction or exhaustion in the context of chronic viral infections and cancer [
39]. These data may suggest that a higher proportion of non-responders experience T cell dysfunction which could be due to prolonged antigen exposure [
40]. Further exploration of these findings holds promise for optimizing the therapeutic dose of ImmAct to overcome exhaustion and enhance the immunotherapy outcome. We are presently investigating the inclusion of additional T cell exhaustion markers, aiming to enhance the categorization of T cell states.
Adopting an adjuvant approach could pose challenges for future human applications, especially since most patients undergo SLNE during the surgical removal of solid tumors. Consequently, evaluating our ImmAct in a neoadjuvant immunotherapeutic context presents an intriguing avenue and is currently underway in our laboratory.
5. Methods
5.1. CCMVTT-VLPs Cloning, Expression, and Production
The coat protein sequence of the CCMV VLPs can be obtained from the GenBank (accession no. AAA46373). The sequence, cloning, expression, and production of our plant-derived VLPs are detailed in [
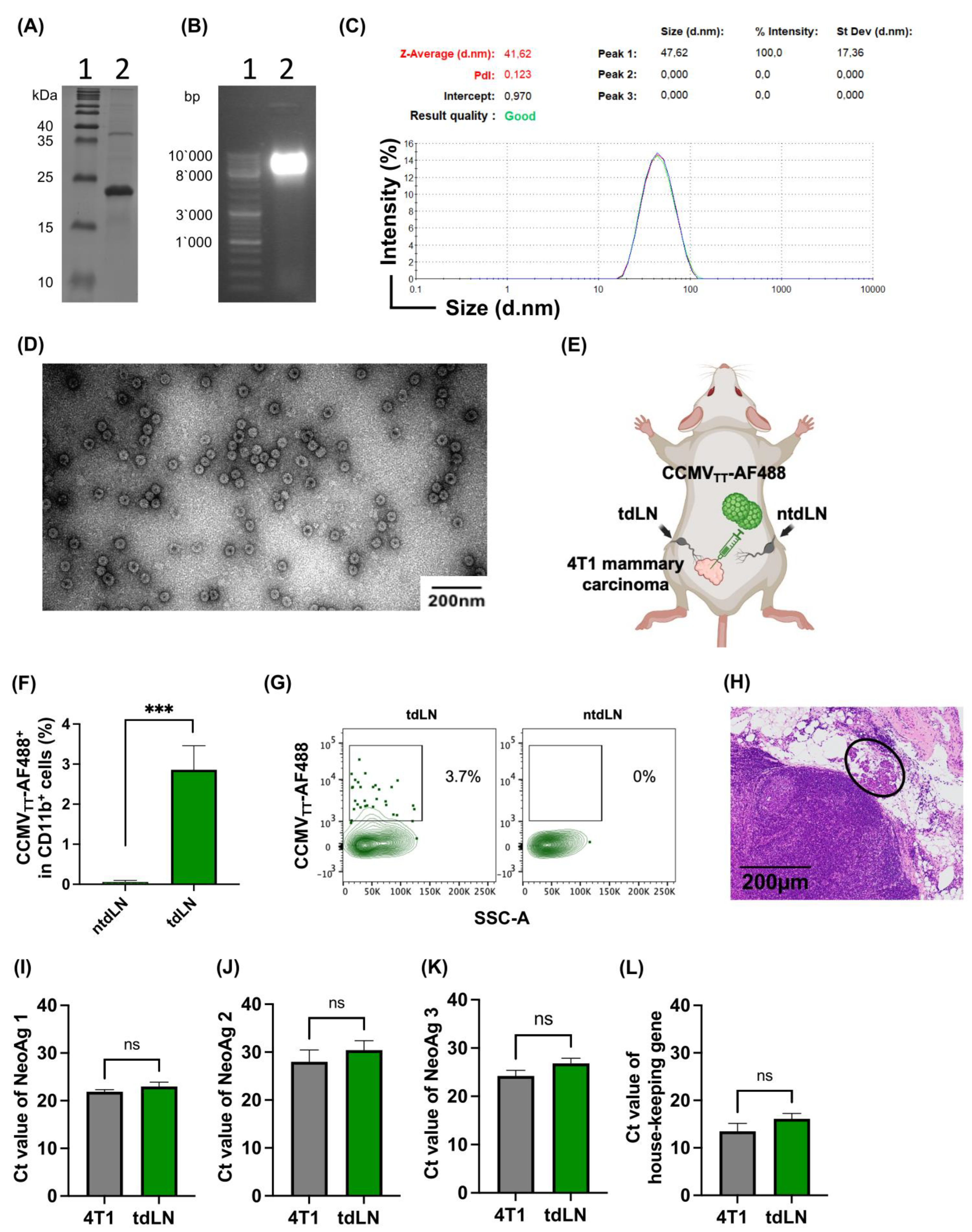
26]. In short, a cloned copy of the CCMV coat protein gene was used in PCR mutagenesis to insert the tetanus toxin coding sequence, resulting in the cloning of CCMVtt.
E. coli C2566 cells were used for cloning and plasmid amplification. Isopropyl-β-D-thiogalactopyranoside (IPTG) was used to initiate the expression, and the pellet was collected using low-speed centrifugation following the incubation period. Using ultracentrifugation in a sucrose gradient ranging from 20% to 60% sucrose, VLPs were isolated from cellular proteins. The fractions were loaded in an SDS-PAGE gel. To eliminate the sucrose, fractions containing CP proteins were mixed and dialyzed. We used an improved version in this work, which is currently undergoing patenting.
5.2. Dynamic Light Scattering Measurement (DLS)
A high-precision micro quartz cuvette (ZEN2112, HellmaAnalytics, sample volume 12 μL) was used to analyze purified, undiluted VLPs at a concentration of 1 mg/mL using Zetasizer Nano ZS equipment (Malvern Instruments Ltd., Malvern, UK). We calculated the average hydrodynamic diameter of VLPs using three consecutive measurements. The Zetasizer software (version 8.01, Malvern Instruments Ltd., Malvern, UK) was used to analyze the results.
5.3. Electron Microscopy
The visualization of purified VLPs involved uranyl acetate negative staining. Initially, 5 µL of the sample at a concentration of 1 mg/mL was adsorbed onto carbon formvar-coated 300 Mesh Copper grids (Agar Scientific, Stansted, UK), with the preparation of 2 grids per sample. After a 3 min incubation, the grids underwent washing with 1 mM ethylenediaminetetraacetic acid (EDTA) and subsequent negative staining using a 0.5% uranyl acetate aqueous solution. Analysis was carried out using a JEM-1230 electron microscope (JEOL, Tokyo, Japan) operating at an accelerating voltage of 100 kV. A minimum of five micrograph pictures were captured for each sample.
5.4. SDS-Page and Gel Electrophoresis
For the evaluation of CCMV
TT-VLPs, 10 μg of the sample was mixed with 4x Laemmli buffer (comprising 100 mM TRIS-HCl pH 7.0, 4% SDS, 5% β-mercaptoethanol (β-ME), 50% glycerine, 0.4% bromphenol blue) and heated at 95 °C for 10 min. Subsequently, this prepared mixture was loaded onto a self-made 12.5% SDS-PAGE gel, following the protocol outlined in [
41], and run for 1 h at 250 V, 20 mA. To eliminate SDS, the gel underwent a 10 min wash with a fixing solution of 10% ethanol and 10% acetic acid, followed by staining with Coomassie G250 dye (composed of 20% (
v/
v) ethanol, 2% (
w/
v) trichloroacetic acid, and 0.5 g/L Coomassie G250) overnight. Page Ruler™ Prestained Plus Protein Ladder (cat. no. 26619, Thermo Fisher Scientific, Waltham, MA, USA) was used as a reference marker. For the gel electrophoresis, 10 μg of the sample was loaded onto a 0.8% agarose gel supplemented with 0.2 μg/mL Ethidium Bromide. A Master Mix DNA ladder (cat. no. SM0331, Thermo Fisher Scientific, Zürich, Switzerland) was used as a reference.
5.5. Labelling CCMVTT-VLPs
CCMVTT-VLPs were labeled with Alexa Fluor 488 as per the manufacturer’s instructions (Thermo Fischer Scientific, Zürich, Switzerland, A10235) and stored at −20 °C. Briefly, 100 μg of CCMVTT-VLPs was mixed with 1 μL of AF488 in an Eppendorf tube (protected from light) on the shaker for 1 h at 400 rpm. ZebaTM spin columns (7k MWCO) were prepared as per the manufacturer’s instruction (Thermo Scientific, Zürich, Switzerland). CCMVTT-AF488 were transferred to Zeba columns and spun for 2 min at 1000× g.
5.6. Cell Culture
Cells (4T1) were grown in Falcon 75 cm2 Flask (Corning, NY, USA, 353136,) with DMEM (GIBCO) medium supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Zürich, Switzerland 16250086), 1% penicillin-streptomycin (p/s) (Merk, Darmstadt, Germany, I9657). Cells were rinsed three times with 1× PBS to eliminate all cell culture medium when they reached 80% confluency. A total of 0.5% Trypsin-EDTA (Thermo Fisher Scientific, Zürich, Switzerland, 15400-054) was added and the flask was incubated for 7 min at 37 °C. Cells were centrifuged at 300× g for 5 min. Cells were resuspended in a culturing medium and kept on ice until further use. The cells were counted using Cellometer mini (Nexcelon Bioscience, Lawrence, MA, USA) and then resuspended in the appropriate amount of medium to inject 105 cells/mouse. Using the Microsart AMP Mycoplasma Kit (Sartorius, Göttigen, Germany, SMB95-1001), mycoplasma contamination was ruled out.
5.7. Mice
All in vivo experiments utilized wild-type female Balb/cOlaHsd mice (8–12 weeks) and were purchased from Harlan. The mice were housed and bred in the pathogen-free animal facility at the University of Bern. All animal procedures were conducted under license BE43/21, adhering to the Swiss Animal Act (455.109.1—5 September 2008).
5.8. In Vivo Drainage of VLPs to tdLN
Under isoflurane anesthesia (Attane, 800-544-7521), 105 4T1 cells were s.c. inoculated in the lower left quadrant of wild-type BALB/cOlaHsd mice. Every two days, mice were monitored to measure the progress of tumors and their overall health. Eleven days post-tumor inoculation, 15 μg of CCMVTT-AF488 was injected directly into the primary palpable tumor under isoflurane anesthesia. Twenty hours later, the tdLN and the collateral non-tdLN were collected and kept on ice. The lymph nodes were treated with collagenase D (Roche, Basel, Switzerland, 11088858001) and DNase I (Roche, 10104159001) in a DMEM medium for 30 min at 37 °C. The ACK buffer (Sigma-Aldrich, Burlington, MA, USA, R7757) was used to lyse red blood cells (RBCs) after the lymph nodes had been smashed through a 70 μm cell strainer. Cells were transferred to a 96 V-shape well plate and stained with Fc block CD16/CD32 (FRC/4G8) (BD Bioscience, Franklin Lake, NJ, USA, 553142) for 5 min at RT in the dark. The plate was centrifuged at 1200 rpm for 5 min and flicked. Cells were then stained with 7-AAD viability staining solution (Thermo Fisher Scientific, 00-6993-50) 1/5000 and anti-mouse CD11b PE-Cy7 clone M1/70 (Thermo Fisher Scientific, 25-0112-82) for 10 min at RT in the dark. The plate was centrifuged at 1200 rpm for 5 min and flicked. After surface staining, cells were washed twice and resuspended in 400 μL FACS buffer. Sample analysis was performed using a BD LSRII and evaluated with Flojo (V.10) and GraphPad Prism (V10). The gating strategy involved a sequential selection of singlets, live/dead followed by monocytes and further categorized into CD11b+ AF488+ cells.
5.9. Real-Time Quantitative PCR
We assessed four different neoantigens in the tdLNs and ntLNs in mice harboring 4T1 cells. A total of 105 4T1 cells were s.c. inoculated in the lower left quadrant of wild-type BALB/cOlaHsd mice. The tdLN and the ntdLN were dissected on day 11, stored in RNA (Merk, Darmstadt, Germany), and kept at −20 °C. For RNA extraction, we followed the manufacturer’s instructions (NucleoSpin RNA Kit) and the RNA content was measured by Nanodrop. The cDNA was produced using a High-Capacity RNA-to-cDNA Kit as per the manufacturer’s instructions and the cDNA content was measured with Nanodrop. To amplify the mutated region, three primer pairs of about 24 bp each were created, resulting in three amplicons of about 200 bp each. After the PCR products were electrophoresed and gel-extracted, the Zymoclean Gel DNA Recovery Kit was used to purify the amplicons that were predicted in size. Following PCR amplification, amplicons were Sanger sequenced using the primers. The housekeeping gene was included.
5.10. Intranodal Administration of ImmAct
Under isoflurane anesthesia (Attane, 800-544-7521), 105 4T1 cells were s.c. inoculated in the lower left quadrant of wild-type BALB/cOlaHsd mice. Mice were followed every 2 days to assess tumor growth and general health score. Eleven days later, the primary tumor was surgically dissected as follows: the weight of the mice was recorded, and 50 μL/25 g of narcotic mixture was injected s.c. The narcotic mixture consisted of Dormitor (Medetomidine Hydrochloride) at a concentration of 1 mg/mL and a dosage of 0.4 mg/kg, Dormicum (Midazolam) at a concentration of 5 mg/mL and a dosage of 4 mg/kg, and Fentanyl at a concentration of 0.05 mg/mL and a dosage of 0.04 mg/kg. Ophthalmic ointment (Paralube®) was applied to the mouse’s eyes to avoid the drying of the cornea. Ten minutes later, the mice were assessed for reflex responses, confirming their state of full sedation. Mice were positioned on their backs to expose the iLN, the skin of this area was shaved using a trimmer, and disinfection was carried out with Betadine. The hip joint was bent to an approximately 90° angle and a small incision of <5 mm through the skin was made using sterile curved micro-dissecting forceps and a surgical scissor. The incision was widened using the tip of the surgical scissor which enlarged the incision to about 10 mm. The iLN was localized and immobilized using the curved forceps and the surgical scissor. The iLN appeared greyish within the surrounding fat tissue. Using an insulin syringe, 0.5 mL, 30 μg, and 10 μL of the naked CCMVTT-VLPs (ImmAct) or Phosphate buffer reagent (PBS) was directly injected into the iLN. If the lymph node swelled, the injection was deemed successful. The incision was sealed using sterile 9 mm wound clips inserted into a 9 mm auto-clip applier. Clips were removed after 10–12 days. The mice were boosted with the same method after one week. Mice were monitored closely as per our license BE43/2021.
5.11. Histology
The collected iLNs were fixed with 4% buffered formalin (Formafix GmbH, Zürich, Switzerland) for at least 24 h and routinely processed and stained with Hematoxylin and Eosin (HE) for histological examination. The iLN tissue sections were evaluated for the presence of neoplastic cells and any other histopathological changes by a board-certified veterinary pathologist (SdB).
5.12. Blood, Spleen, and Lymph Node Processing
On day 22, blood from the tail vein of mice was collected in Eppendorf tubes containing 0.5 M EDTA in 1× PBS. For all the steps, centrifugation for 5 min at 1200 rpm was used. Blood was centrifuged and the supernatant was aspirated. Red blood cells (RBCs) were lysed using AmmoniumChloride—Potassium (ACK) buffer on ice for 5 min. Cells were centrifuged for 5 min at 1200 rpm and washed twice with FACS buffer (1× PBS containing 0.1% Bovine Serum Albumin (BSA)). After the final wash, the cells were suspended in FACS buffer, transferred to a 96 U-shape well plate, and kept on ice for staining. The spleens were collected on ice and smashed using 70 μm cell strainers, and RBCs were lysed with ACK buffer. Cells were centrifuged and washed as summarized above. A total of 106 splenocytes were transferred to a 96 U-shape well plate and kept on ice for staining. For lymph node processing, tdLNs were collected on ice and then treated with collagenase D (Roche, 11088858001) and DNase I (Roche, 10104159001) in a DMEM medium for 30 min at 37 °C. The lymph nodes were smashed using 70 μm cell strainers, and RBCs were lysed with ACK buffer. All cells were transferred to a 96 U-shape well plate and kept on ice for the staining. Cells were stained as follows: first with Fc block CD16/CD32 (FRC/4G8) (BD Bioscience, 553142) for 15 min at RT in the dark. Cells were centrifuged and washed with FACS buffer and next stained with anti-mouse: CD8α clone 53-6, CD4 clone RM4-5 (BioLegend), CD19 clone 1D3 (BD Biosciences), CD11b clone M/170 (BioLegend), Ly6G clone 1A8 (BioLegend), Ly6C clone HK1.4 (BioLegend), PD-1 clone 29F.1A12 (BioLegend), and TIM-3 clone RMT3-23 (BioLegend) markers were used as exhaustion markers for CD8+ T cells. Sample analysis was performed using a BD LSRII and evaluated with Flojo (V.10) and GraphPad Prism (V10). The gating strategy involved a sequential selection of singlets, live/dead followed by lymphocytes or monocytes, and further categorized into CD8+, CD4+ T cells, CD19+ B cells, CD11b+ Ly6C+, and CD11b+ Ly6G+ MDSCs.
5.13. High Dimensional Analysis of Flow Cytometry Data
High dimensional analysis of flow cytometry data was performed using t-distributed stochastic neighbor embedding (tSNE) and FlowSOM clustering and a visualizing technique. FCS files of TILs were used following compensation and gating on single cells and live cells using FlowJo X version 10.4.2. FCS files (4 FCS files from each group) were exported and then concatenated; each used file included a consistent number of events (180 events), and the total number of concatenated events was 720. The following markers were used: CD8, PD-1, and TIM-3. In FlowSOM analysis, five clusters were applied, as indicated in the figure legends. Histogram count was used to confirm marker expression.
5.14. Sentinel Lymph Node Excision (SLNE)
Under isoflurane anesthesia (Attane, 800-544-7521), 105 4T1 cells were s.c. inoculated in the lower left quadrant of wild-type BALB/cOlaHsd mice. Mice were followed every 2 days to assess tumor growth and general health score. Eleven days later, the primary tumor was surgically dissected as follows: the weight of the mice was recorded, and 50 μL/25 g of narcotic mixture was injected s.c. Ophthalmic ointment (Paralube®) was applied to each mouse’s eyes to avoid the drying of the cornea. Ten minutes later, the mice were assessed for reflex responses, confirming their state of full sedation. Mice were positioned on their backs to expose the iLN, the skin of this area was shaved using a trimmer, and disinfection was carried out with Betadine. The hip joint was bent to an approximately 90° angle and a small incision of <5 mm through the skin was made using sterile curved micro-dissecting forceps and a surgical scissor. The incision was widened using the tip of the surgical scissor which enlarged the incision to about 10 mm. The iLN was localized and completely excised. The incision was sealed using sterile 9 mm wound clips inserted into a 9 mm auto-clip applier. Clips were removed after 10–12 days. Mice were monitored closely as per our license BE43/2021.
5.15. In Vivo Cell Depletion Studies
For the depletion of CD8+, CD4+, and CD19+ cells, 300 μg of anti-CD8mAb clone CD8β Lyt3.2 (BioXcell, BE0223), anti-CD4mAb clone GK1.5 (eBioscience, 14-0041-82), and anti-CD19 mAb clone 1D3 (BioXcell, BE0150) was administered intravenously (i.v) on day 9 and 200 µg on day 21. Depletion efficiency was ~99% as determined by flow cytometry on day 14.
5.16. Statistical Analysis
The data were analyzed and presented as mean ± SEM using GraphPad (V.8.4.2, Boston, MA, USA,
www.graphpad.com) (464). Statistical comparisons among more than two groups employed one-way analysis of variance (ANOVA), while comparisons between two groups utilized the Student’s
t-test. The survival rate was analyzed by log-rank (Mantel-Cox) test. Exact
p-values were provided, and significance levels were denoted as ****
p < 0.0001, ***
p < 0.001, **
p < 0.01, and *
p < 0.05.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




