
Impact of Recombinant VSV-HIV Prime, DNA-Boost Vaccine Candidates on Immunogenicity and Viremia on SHIV-Infected Rhesus Macaques
 , , , , , , , , ,
, , , , , , , , ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
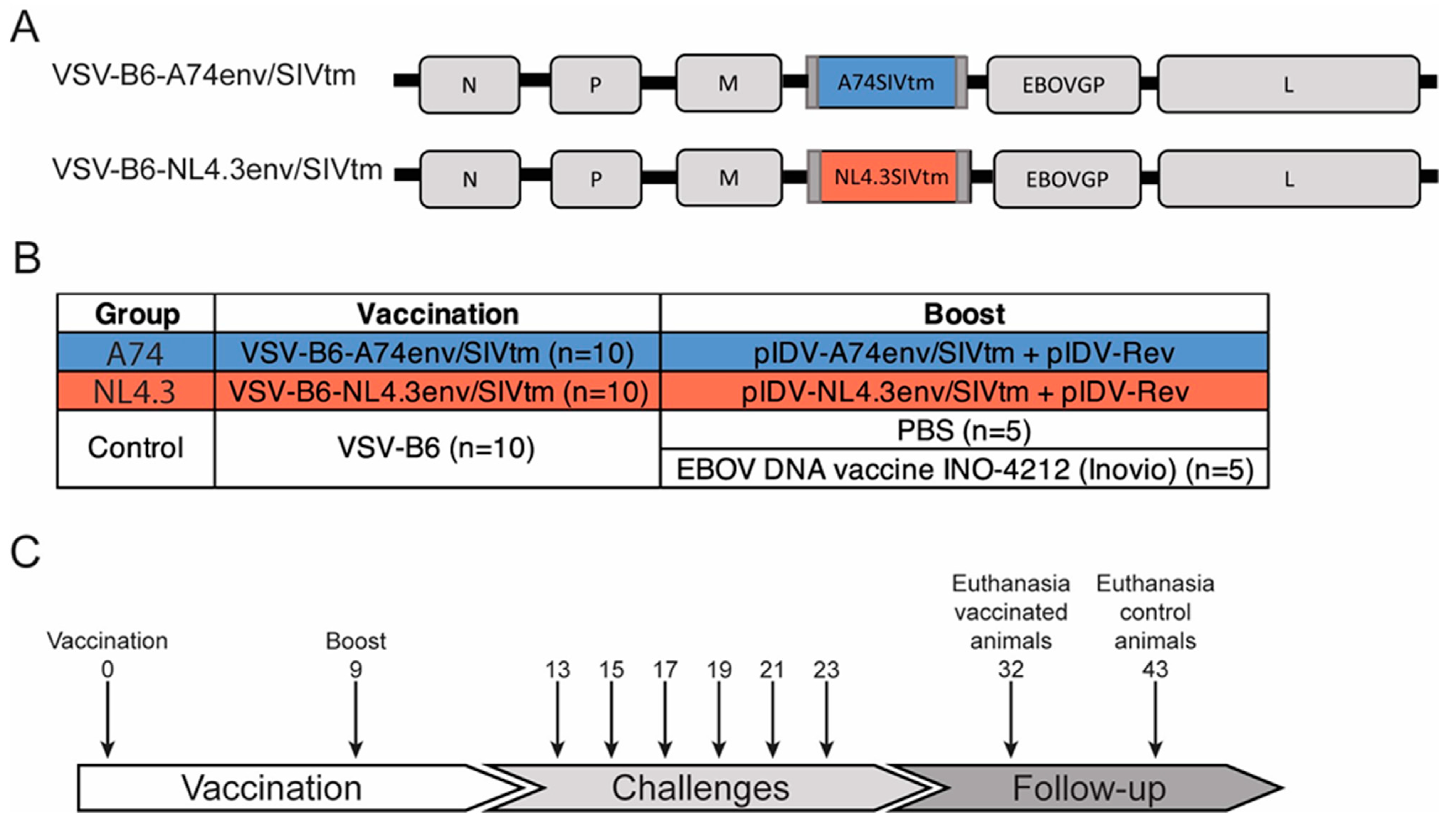
2.1. Vaccine Candidates
2.2. Animals, Vaccinations, and Challenges
2.3. ELISA
2.4. Virus Neutralization Assay
2.5. Plasma Viral Load and Viral Rectal Shedding
2.6. T-Cell Responses
2.7. Statistical Analysis
3. Results
3.1. Recombinant VSV-HIV Vaccines Induce Strong Humoral Responses
3.2. Plasma Viral Load Trends and Correlation with Rectal Viral Shedding
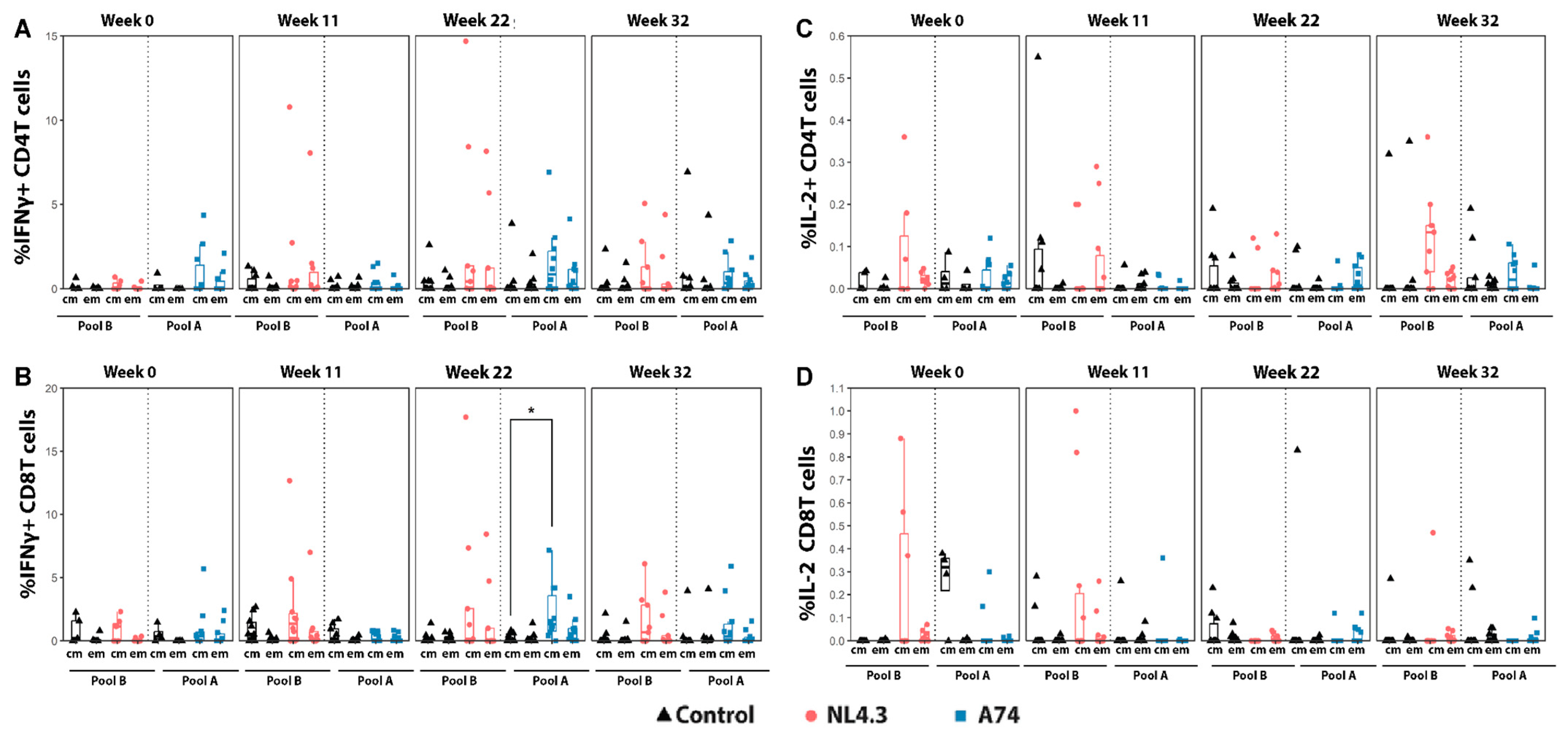
3.3. Cytokine Production and Activation Markers by Central and Effector Memory T-Cells
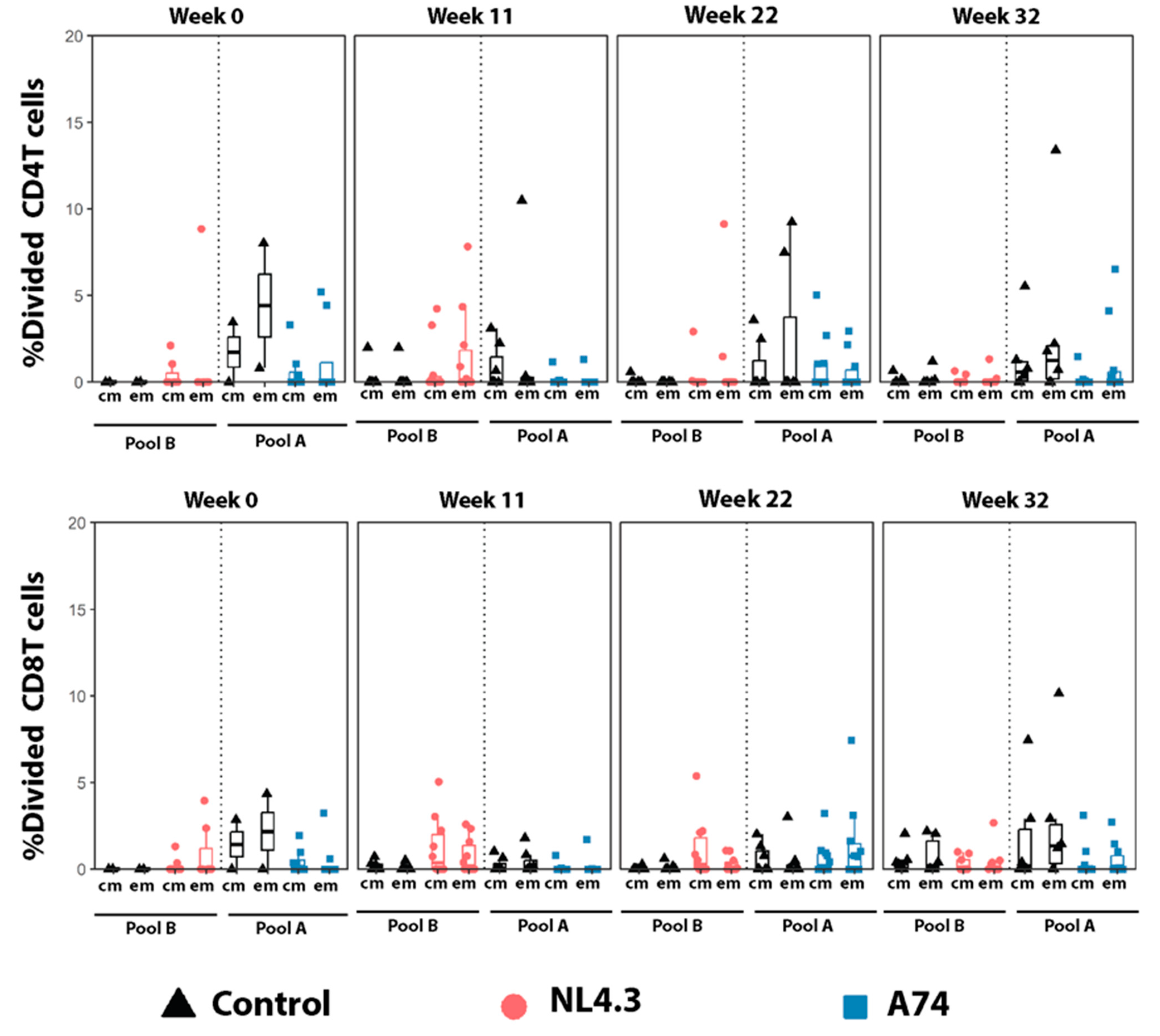
3.4. Proliferating Central- and Effector Memory T-Cells
3.5. Development of an AIDS-like Syndrome in a Control-Vaccinated Animal
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burton, D.R. Advancing an HIV vaccine; advancing vaccinology. Nat. Rev. Immunol. 2019, 19, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to Prevent HIV-1 Infection in Thailand. N. Engl. J. Med. 2009, 361, 2209. [Google Scholar] [CrossRef]
- Robinson, H.L. HIV/AIDS Vaccines: 2018. Clin. Pharmacol. Ther. 2018, 104, 1062. [Google Scholar] [CrossRef] [PubMed]
- Robb, M.L.; Rerks-Ngarm, S.; Nitayaphan, S.; Pitisuttithum, P.; Kaewkungwal, J.; Kunasol, P.; Khamboonruang, C.; Thongcharoen, P.; Morgan, P.; Kim, J.H.; et al. Ad Hoc Analysis of Behavior and Time as Co-Variates of the Thai Phase III Efficacy Trial: RV 144. Lancet Infect. Dis. 2012, 12, 531–537. [Google Scholar] [CrossRef]
- Barnett, S.W.; Burke, B.; Sun, Y.; Kan, E.; Legg, H.; Lian, Y.; Bost, K.; Zhou, F.; Goodsell, A.; zur Megede, J.; et al. Antibody-Mediated Protection against Mucosal Simian-Human Immunodeficiency Virus Challenge of Macaques Immunized with Alphavirus Replicon Particles and Boosted with Trimeric Envelope Glycoprotein in MF59 Adjuvant. J. Virol. 2010, 84, 5975–5985. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.B.; Rose, N.F.; Bahl, K.; Diller, K.; Buonocore, L.; Hunter, M.; Marx, P.A.; Gambhira, R.; Tang, H.; Montefiori, D.C.; et al. Significant Protection against High-Dose Simian Immunodeficiency Virus Challenge Conferred by a New Prime-Boost Vaccine Regimen. J. Virol. 2011, 85, 5764–5772. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound Early Control of Highly Pathogenic SIV by an Effector Memory T-Cell Vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef]
- Lai, L.; Kwa, S.; Kozlowski, P.A.; Montefiori, D.C.; Ferrari, G.; Johnson, W.E.; Hirsch, V.; Villinger, F.; Chennareddi, L.; Earl, P.L.; et al. Prevention of Infection by a Granulocyte-Macrophage Colony-Stimulating Factor Co-Expressing DNA/Modified Vaccinia Ankara Simian Immunodeficiency Virus Vaccine. J. Infect. Dis. 2011, 204, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Jalah, R.; Kulkarni, V.; Valentin, A.; Rosati, M.; Alicea, C.; von Gegerfelt, A.; Huang, W.; Guan, Y.; Keele, B.F.; et al. DNA and Virus Particle Vaccination Protects against Acquisition and Confers Control of Viremia upon Heterologous Simian Immunodeficiency Virus Challenge. Proc. Natl. Acad. Sci. USA 2013, 110, 2975–2980. [Google Scholar] [CrossRef]
- Barouch, D.H.; Liu, J.; Li, H.; Maxfield, L.F.; Abbink, P.; Lynch, D.M.; Iampietro, M.J.; SanMiguel, A.; Seaman, M.S.; Ferrari, G.; et al. Vaccine Protection against Acquisition of Neutralization-Resistant SIV Challenges in Rhesus Monkeys. Nature 2012, 482, 89–93. [Google Scholar] [CrossRef]
- Flatz, L.; Cheng, C.; Wang, L.; Foulds, K.E.; Ko, S.-Y.; Kong, W.-P.; Roychoudhuri, R.; Shi, W.; Bao, S.; Todd, J.-P.; et al. Gene-Based Vaccination with a Mismatched Envelope Protects against Simian Immunodeficiency Virus Infection in Nonhuman Primates. J. Virol. 2012, 86, 7760–7770. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Tomaka, F.L.; Wegmann, F.; Stieh, D.J.; Alter, G.; Robb, M.L.; Michael, N.L.; Peter, L.; Nkolola, J.P.; Borducchi, E.N.; et al. Evaluation of a Mosaic HIV-1 Vaccine in a Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 1/2a Clinical Trial (APPROACH) and in Rhesus Monkeys (NHP 13-19). Lancet 2018, 392, 232. [Google Scholar] [CrossRef] [PubMed]
- Pauthner, M.G.; Nkolola, J.P.; Havenar-Daughton, C.; Murrell, B.; Reiss, S.M.; Bastidas, R.; Prévost, J.; Nedellec, R.; von Bredow, B.; Abbink, P.; et al. Vaccine-Induced Protection from Homologous Tier 2 SHIV Challenge in Nonhuman Primates Depends on Serum-Neutralizing Antibody Titers. Immunity 2019, 50, 241–252.e6. [Google Scholar] [CrossRef] [PubMed]
- Felber, B.K.; Lu, Z.; Hu, X.; Valentin, A.; Rosati, M.; Remmel, C.A.; Weiner, J.A.; Carpenter, M.C.; Faircloth, K.; Stanfield-Oakley, S.; et al. Co-Immunization of DNA and Protein in the Same Anatomical Sites Induces Superior Protective Immune Responses against SHIV Challenge. Cell Rep. 2020, 31, 107624. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Alter, G.; Broge, T.; Linde, C.; Ackerman, M.E.; Brown, E.P.; Borducchi, E.N.; Smith, K.M.; Nkolola, J.P.; Liu, J.; et al. Protective Efficacy of Adenovirus/Protein Vaccines against SIV Challenges in Rhesus Monkeys. Science 2015, 349, 320. [Google Scholar] [CrossRef] [PubMed]
- Hessell, A.J.; Poignard, P.; Hunter, M.; Hangartner, L.; Tehrani, D.M.; Bleeker, W.K.; Parren, P.W.H.I.; Marx, P.A.; Burton, D.R. Effective, Low-Titer Antibody Protection against Low-Dose Repeated Mucosal SHIV Challenge in Macaques. Nat. Med. 2009, 15, 951. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-Correlates Analysis of an HIV-1 Vaccine Efficacy Trial. New Engl. J. Med. 2012, 366, 1275. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Finak, G.; Ushey, K.; Seshadri, C.; Hawn, T.R.; Frahm, N.; Scriba, T.J.; Mahomed, H.; Hanekom, W.; Bart, P.-A.; et al. COMPASS Identifies T-Cell Subsets Correlated with Clinical Outcomes. Nat. Biotechnol. 2015, 33, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and Effectiveness of an RVSV-Vectored Vaccine in Preventing Ebola Virus Disease: Final Results from the Guinea Ring Vaccination, Open-Label, Cluster-Randomised Trial (Ebola Ça Suffit!). Lancet 2017, 389, 505. [Google Scholar] [CrossRef]
- Clarke, D.K.; Xu, R.; Matassov, D.; Latham, T.E.; Ota-Setlik, A.; Gerardi, C.S.; Luckay, A.; Witko, S.E.; Hermida, L.; Higgins, T.; et al. Safety and Immunogenicity of a Highly Attenuated RVSVN4CT1-EBOVGP1 Ebola Virus Vaccine: A Randomised, Double-Blind, Placebo-Controlled, Phase 1 Clinical Trial. Lancet Infect. Dis. 2020, 20, 455. [Google Scholar] [CrossRef]
- Agnandji, S.T.; Huttner, A.; Zinser, M.E.; Njuguna, P.; Dahlke, C.; Fernandes, J.F.; Yerly, S.; Dayer, J.-A.; Kraehling, V.; Kasonta, R.; et al. Phase 1 Trials of RVSV Ebola Vaccine in Africa and Europe. N. Engl. J. Med. 2016, 374, 1647–1660. [Google Scholar] [CrossRef]
- Huttner, A.; Dayer, J.-A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J.; Eickmann, M.; Finckh, A.; Goncalves, A.R.; Hooper, J.W.; et al. The Effect of Dose on the Safety and Immunogenicity of the VSV Ebola Candidate Vaccine: A Randomised Double-Blind, Placebo-Controlled Phase 1/2 Trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Fast, P.E.; Modjarrad, K.; Clarke, D.K.; Martin, B.K.; Fusco, J.; Nichols, R.; Heppner, D.G.; Simon, J.K.; Dubey, S.; et al. RVSVΔG-ZEBOV-GP (Also Designated V920) Recombinant Vesicular Stomatitis Virus Pseudotyped with Ebola Zaire Glycoprotein: Standardized Template with Key Considerations for a Risk/Benefit Assessment. Vaccine X 2019, 1, 100009. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, I.C.; Nguyen, H.T.; Kemelman, M.; Lindsay, R.W.; Yuan, M.; Wright, K.J.; Arendt, H.; Back, J.W.; DeStefano, J.; Hoffenberg, S.; et al. The Stem of Vesicular Stomatitis Virus G Can Be Replaced with the HIV-1 Env Membrane-Proximal External Region without Loss of g Function or Membrane-Proximal External Region Antigenic Properties. AIDS Res. Hum. Retroviruses 2014, 30, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Azizi, H.; Knapp, J.P.; Li, Y.; Berger, A.; Lafrance, M.-A.; Pedersen, J.; de la Vega, M.-A.; Racine, T.; Kang, C.-Y.; Mann, J.F.S.; et al. Optimal Expression, Function, and Immunogenicity of an HIV-1 Vaccine Derived from the Approved Ebola Vaccine, RVSV-ZEBOV. Vaccines 2023, 11, 977. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.; Yuan, M.; Coleman, J.; Destefano, J.; Zhang, X.; Yates, N.; Barouch, D.; Ackerman, M.; Decamp, A.; Alter, G.; et al. Protection from Rectal SHIV Infection Induced by Mucosal Vaccination with a Replication-Competent VSV-HIV Chimera Delivering Env Trimers. In AIDS Research and Human Retroviruses; Mary Ann Liebert, Inc.: New Rochelle, NY, USA, 2016. [Google Scholar]
- Egan, M.A.; Chong, S.Y.; Rose, N.F.; Megathi, S.; Lopez, K.J.; Schadeck, E.B.; Johnson, J.E.; Masood, A.; Piacente, P.; Druilhet, R.E.; et al. Immunogenicity of Attenuated Vesicular Stomatitis Virus Vectors Expressing HIV Type 1 Env and SIV Gag Proteins: Comparison of Intranasal and Intramuscular Vaccination Routes. AIDS Res. Hum. Retroviruses 2004, 20, 989. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.F.; Marx, P.A.; Luckay, A.; Nixon, D.F.; Moretto, W.J.; Donahoe, S.M.; Montefiori, D.; Roberts, A.; Buonocore, L.; Rose, J.K. An Effective AIDS Vaccine Based on Live Attenuated Vesicular Stomatitis Virus Recombinants. Cell 2001, 106, 539. [Google Scholar] [CrossRef]
- Bresk, C.A.; Hofer, T.; Wilmschen, S.; Krismer, M.; Beierfuß, A.; Effantin, G.; Weissenhorn, W.; Hogan, M.J.; Jordan, A.P.; Gelman, R.S.; et al. Induction of Tier 1 HIV Neutralizing Antibodies by Envelope Trimers Incorporated into a Replication Competent Vesicular Stomatitis Virus Vector. Viruses 2019, 11, 159. [Google Scholar] [CrossRef]
- Wong, G.; Qiu, X.; Ebihara, H.; Feldmann, H.; Kobinger, G.P. Characterization of a Bivalent Vaccine Capable of Inducing Protection Against Both Ebola and Cross-Clade H5N1 Influenza in Mice. J. Infect. Dis. 2015, 212, S435. [Google Scholar] [CrossRef]
- Jelinski, J.; Kowatsch, M.M.; Lafrance, M.-A.; Berger, A.; Pedersen, J.; Azizi, H.; Li, Y.; Scholte, F.; Gomez, A.; Hollett, N.; et al. Rhesus Macaques Show Increased Resistance to Repeated SHIV Intrarectal Exposure Following a Heterologous Regimen of RVSV Vector Vaccine Expressing HIV Antigen. Emerg. Microbes Infect. 2023, 12, 2251595. [Google Scholar] [CrossRef]
- Mangion, M.; Gélinas, J.-F.; Gashti, A.B.Z.; Azizi, H.; Kiesslich, S.; Nassoury, N.; Chahal, P.S.; Kobinger, G.; Gilbert, R.; Garnier, A.; et al. Evaluation of Novel HIV Vaccine Candidates Using Recombinant Vesicular Stomatitis Virus Vector Produced in Serum-Free Vero Cell Cultures. Vaccine 2020, 38, 7949–7955. [Google Scholar] [CrossRef]
- Ratcliff, A.N.; Shi, W.; Arts, E.J. HIV-1 Resistance to Maraviroc Conferred by a CD4 Binding Site Mutation in the Envelope Glycoprotein Gp120. J. Virol. 2013, 87, 923–934. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of Acquired Immunodeficiency Syndrome-Associated Retrovirus in Human and Nonhuman Cells Transfected with an Infectious Molecular Clone. J. Virol. 1986, 59, 284. [Google Scholar] [CrossRef]
- Urbanowicz, R.A.; McClure, C.P.; Sakuntabhai, A.; Sall, A.A.; Kobinger, G.; Müller, M.A.; Holmes, E.C.; Rey, F.A.; Simon-Loriere, E.; Ball, J.K. Human Adaptation of Ebola Virus during the West African Outbreak. Cell 2016, 167, 1079. [Google Scholar] [CrossRef]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.M.; Jones, S.M.; Wagner, R.; Volchkov, V.E.; Klenk, H. Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses. J. Virol. 2004, 78, 5458. [Google Scholar] [CrossRef]
- Babuadze, G.G.; Echanove, J.; Lamarre, C.; deLaVega, M.-A.; Fausther-Bovendo, H.; Racine, T.; Gomez, A.M.; Azizi, H.; Wade, M.; Kozak, R.; et al. A Novel DNA Platform Designed for Vaccine Use with High Transgene Expression and Immunogenicity. Vaccine 2021, 39, 7175–7181. [Google Scholar] [CrossRef]
- Gomez, A.M.; Babuadze, G.; Plourde-Campagna, M.A.; Azizi, H.; Berger, A.; Kozak, R.; de La Vega, M.A.; Xiii, A.; Naghibosadat, M.; Nepveu-Traversy, M.E.; et al. A Novel Intradermal Tattoo-Based Injection Device Enhances the Immunogenicity of Plasmid DNA Vaccines. NPJ Vaccines 2022, 7, 172. [Google Scholar] [CrossRef]
- Seaman, M.S.; Janes, H.; Hawkins, N.; Grandpre, L.E.; Devoy, C.; Giri, A.; Coffey, R.T.; Harris, L.; Wood, B.; Daniels, M.G.; et al. Tiered Categorization of a Diverse Panel of HIV-1 Env Pseudoviruses for Assessment of Neutralizing Antibodies. J. Virol. 2010, 84, 1439. [Google Scholar] [CrossRef]
- Thermofisher, BestProtocols: Staining Intracellular Antigens for Flow Cytometry. Available online: https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/staining-intracellular-antigens-flow-cytometry.html (accessed on 30 September 2020).
- Murphy, K. Janeway’s Immuno Biology, 8th ed.; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Ratcliff, A.N.; Venner, C.M.; Olabode, A.S.; Knapp, J.; Pankrac, J.; Derecichei, I.; Gibson, R.M.; Finzi, A.; Li, Y.; Mann, J.F.S.; et al. Enhancement of CD4 Binding, Host Cell Entry, and Sensitivity to CD4bs Antibody Inhibition Conferred by a Natural but Rare Polymorphism in the HIV-1 Envelope. J. Virol. 2022, 96, e0185121. [Google Scholar] [CrossRef]
- McGettigan, J.P.; Sarma, S.; Orenstein, J.M.; Pomerantz, R.J.; Schnell, M.J. Expression and Immunogenicity of Human Immunodeficiency Virus Type 1 Gag Expressed by a Replication-Competent Rhabdovirus-Based Vaccine Vector. J. Virol. 2001, 75, 8724–8732. [Google Scholar] [CrossRef]
- Temchura, V.; Überla, K. Intrastructural Help: Improving the HIV-1 Envelope Antibody Response Induced by Virus-like Particle Vaccines. Curr. Opin. HIV AIDS 2017, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Marasini, B.; Vyas, H.K.; Lakhashe, S.K.; Hariraju, D.; Akhtar, A.; Ratcliffe, S.J.; Ruprecht, R.M. Mucosal AIDS Virus Transmission Is Enhanced by Antiviral IgG Isolated Early in Infection. AIDS 2021, 35, 2423. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Fernández, M.; de la Fuente, H.; Martín, P.; Cibrián, D.; Sánchez-Madrid, F. Unraveling Cd69 Signaling Pathways, Ligands And Laterally Associated Molecules. EXCLI J. 2023, 22, 334. [Google Scholar] [PubMed]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From Activation Marker to Metabolic Gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Chen, H.; Qin, Y.; Chou, M.; Cyster, J.G.; Li, X. Transmembrane Protein CD69 Acts as an S1PR1 Agonist. Elife 2023, 12, e88204. [Google Scholar] [CrossRef] [PubMed]
- Kahle, E.M.; Bolton, M.; Hughes, J.P.; Donnell, D.; Celum, C.; Lingappa, J.R.; Ronald, A.; Cohen, C.R.; de Bruyn, G.; Fong, Y.; et al. Plasma Cytokine Levels and Risk of HIV Type 1 (HIV-1) Transmission and Acquisition: A Nested Case-Control Study among HIV-1-Serodiscordant Couples. J. Infect. Dis. 2015, 211, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Naranbhai, V.; Karim, S.S.A.; Altfeld, M.; Samsunder, N.; Durgiah, R.; Sibeko, S.; Karim, Q.A.; Carr, W.H. Innate Immune Activation Enhances HIV Acquisition in Women, Diminishing the Effectiveness of Tenofovir Microbicide Gel. J. Infect. Dis. 2012, 206, 993–1001. [Google Scholar] [CrossRef]
- Van Kooten, G.; Banchereau, J. CD40-CD40 Ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Grewal, I.S.; Flavell, R.A. CD40 and CD154 in Cell-Mediated Immunity. Annu. Rev. Immunol. 1998, 16, 111–135. [Google Scholar] [CrossRef]
- Kopycinski, J.; Yang, H.; Hancock, G.; Pace, M.; Kim, E.; Frater, J.; Stöhr, W.; Hanke, T.; Fidler, S.; Dorrell, L. Therapeutic Vaccination Following Early Antiretroviral Therapy Elicits Highly Functional T Cell Responses against Conserved HIV-1 Regions. Sci. Rep. 2023, 13, 17155. [Google Scholar] [CrossRef]
- Llano, A.; Parera, M.; Lopez, M.; Oriol-Tordera, B.; Ruiz-Riol, M.; Coll, J.; Perez, F.; Leselbaum, A.R.; McGowan, I.; Sengupta, D.; et al. Safety, Immunogenicity and Effect on Viral Rebound of HTI Vaccines in Early Treated HIV-1 Infection: A Randomized, Placebo-Controlled Phase 1 Trial. Nat. Med. 2022, 28, 2611–2621. [Google Scholar]
- Weaver, E.A.; Nehete, P.N.; Nehete, B.P.; Yang, G.; Buchl, S.J.; Hanley, P.W.; Palmer, D.; Montefiori, D.C.; Ferrari, G.; Ng, P.; et al. Comparison of Systemic and Mucosal Immunization with Helper-Dependent Adenoviruses for Vaccination against Mucosal Challenge with SHIV. PLoS ONE 2013, 8, e67574. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.B.; Bahl, K.; Folta-Stogniew, E.; Rose, N.; Buonocore, L.; Marx, P.A.; Gambhira, R.; Rose, J.K. Antigenic Requirement for Gag in a Vaccine That Protects against High-Dose Mucosal Challenge with Simian Immunodeficiency Virus. Virology 2015, 476, 405–412. [Google Scholar] [CrossRef]
- Gautam, R.; Iyer, A.; Hunter, M.; Das, A.; Williams, T.; Dufour, J.; Apetrei, C.; Kousoulas, K.G.; Marx, P.A. Vesicular Stomatitis Virus-Simian Retrovirus Type 2 Vaccine Protects Macaques from Detectable Infection and B-Cell Destruction. J. Virol. 2011, 85, 5889–5896. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berger, A.; Pedersen, J.; Kowatsch, M.M.; Scholte, F.; Lafrance, M.-A.; Azizi, H.; Li, Y.; Gomez, A.; Wade, M.; Fausther-Bovendo, H.; et al. Impact of Recombinant VSV-HIV Prime, DNA-Boost Vaccine Candidates on Immunogenicity and Viremia on SHIV-Infected Rhesus Macaques. Vaccines 2024, 12, 369. https://doi.org/10.3390/vaccines12040369
Berger A, Pedersen J, Kowatsch MM, Scholte F, Lafrance M-A, Azizi H, Li Y, Gomez A, Wade M, Fausther-Bovendo H, et al. Impact of Recombinant VSV-HIV Prime, DNA-Boost Vaccine Candidates on Immunogenicity and Viremia on SHIV-Infected Rhesus Macaques. Vaccines. 2024; 12(4):369. https://doi.org/10.3390/vaccines12040369
Chicago/Turabian StyleBerger, Alice, Jannie Pedersen, Monika M. Kowatsch, Florine Scholte, Marc-Alexandre Lafrance, Hiva Azizi, Yue Li, Alejandro Gomez, Matthew Wade, Hugues Fausther-Bovendo, and et al. 2024. "Impact of Recombinant VSV-HIV Prime, DNA-Boost Vaccine Candidates on Immunogenicity and Viremia on SHIV-Infected Rhesus Macaques" Vaccines 12, no. 4: 369. https://doi.org/10.3390/vaccines12040369