
Cellular and Molecular Immunity to Influenza Viruses and Vaccines



Abstract
:1. Introduction
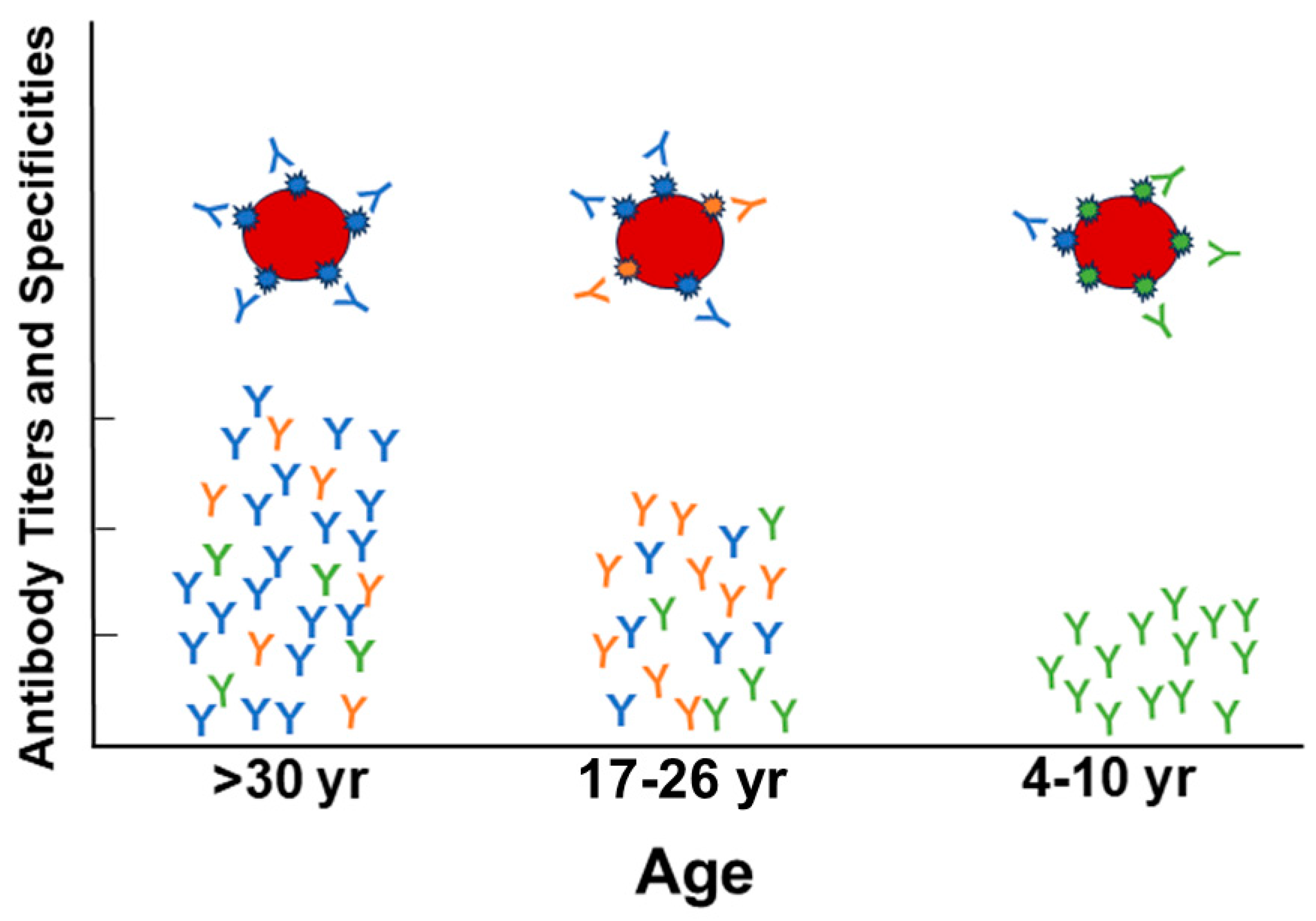
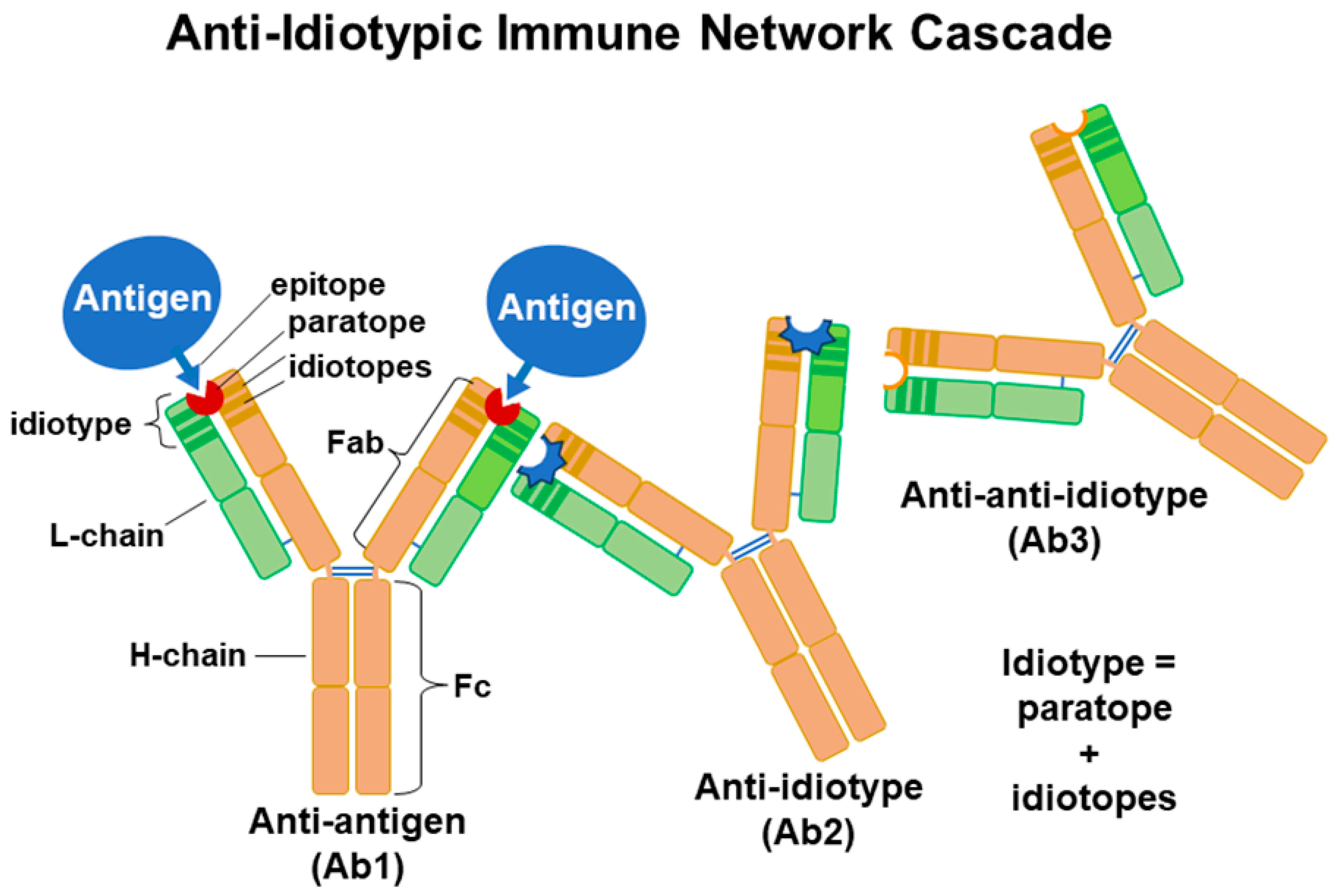
2. Theories of Ab Generation
3. Infection vs. Vaccination and Immune Response
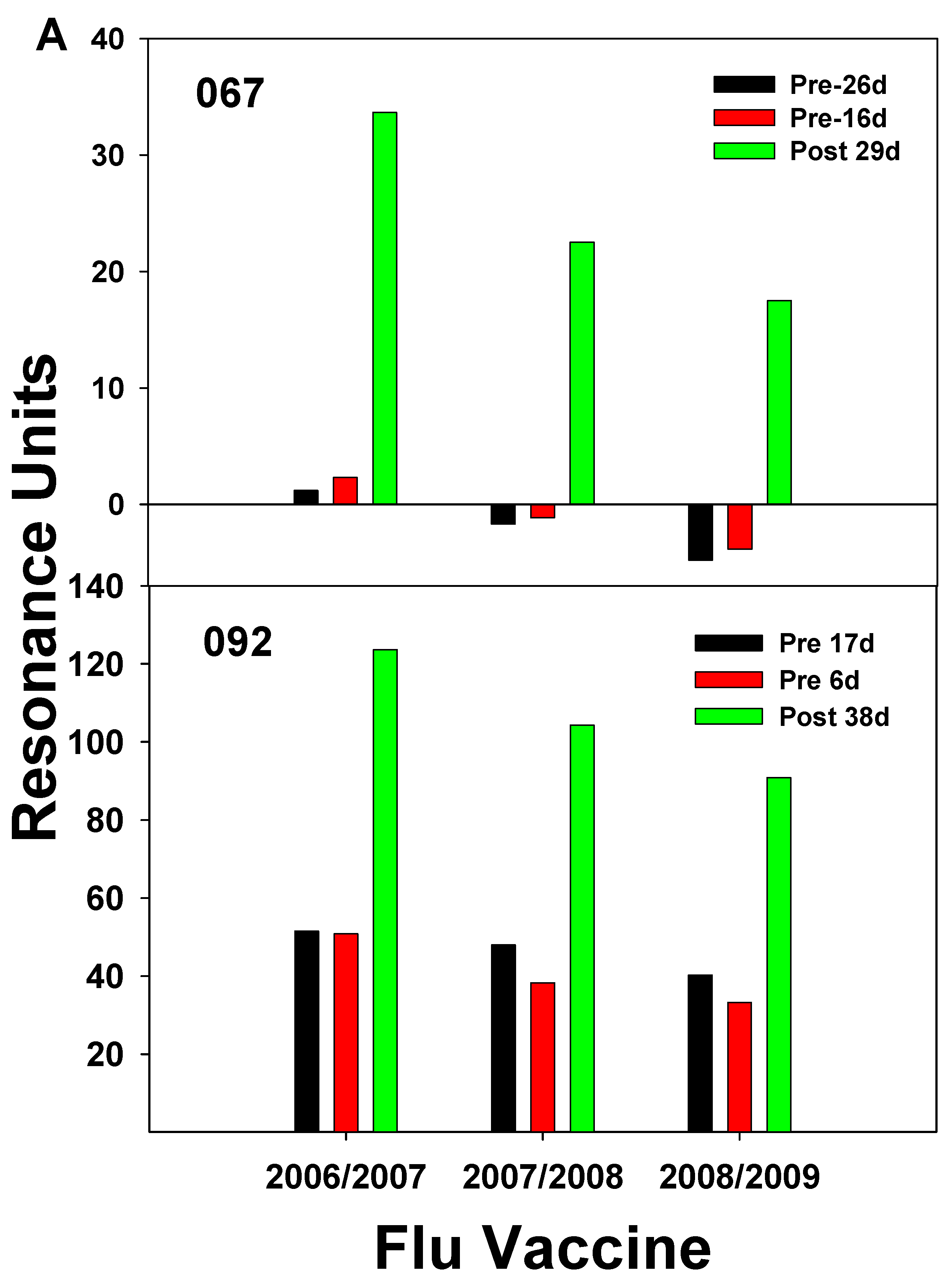
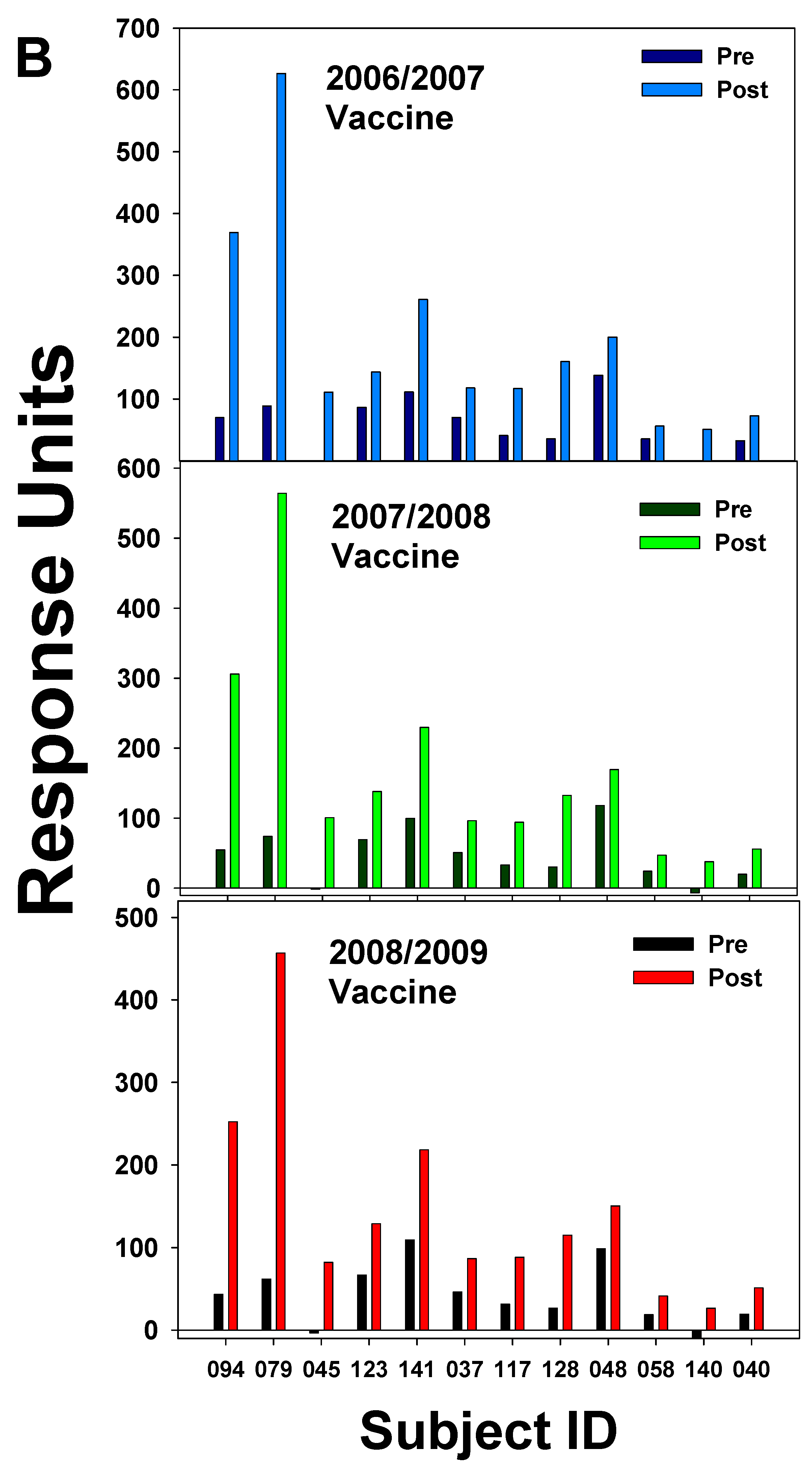
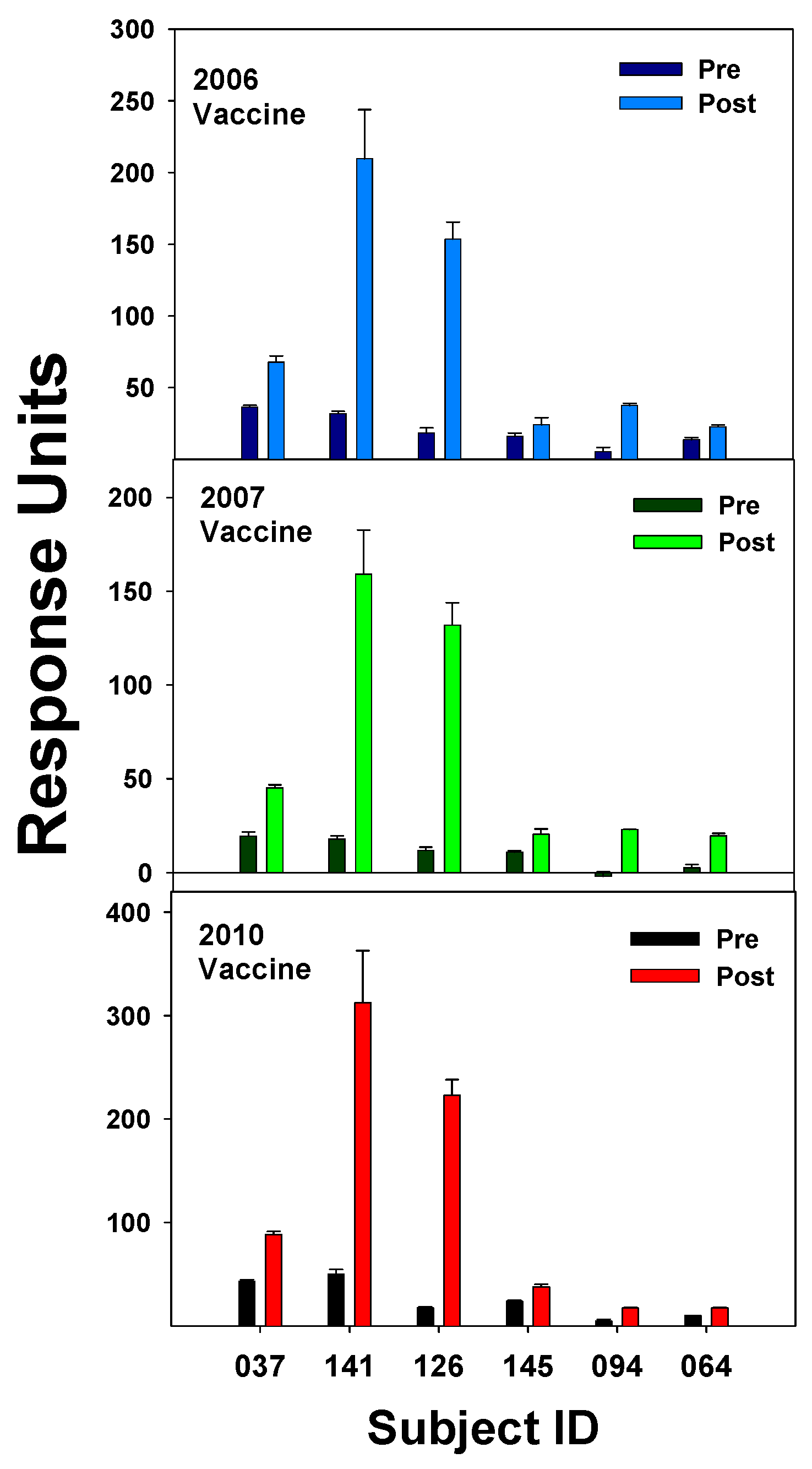
3.1. Pre- and Post-Vaccination Abs to Flu Proteins
3.2. Study of the 2009 Pandemic Strain and Ab Response
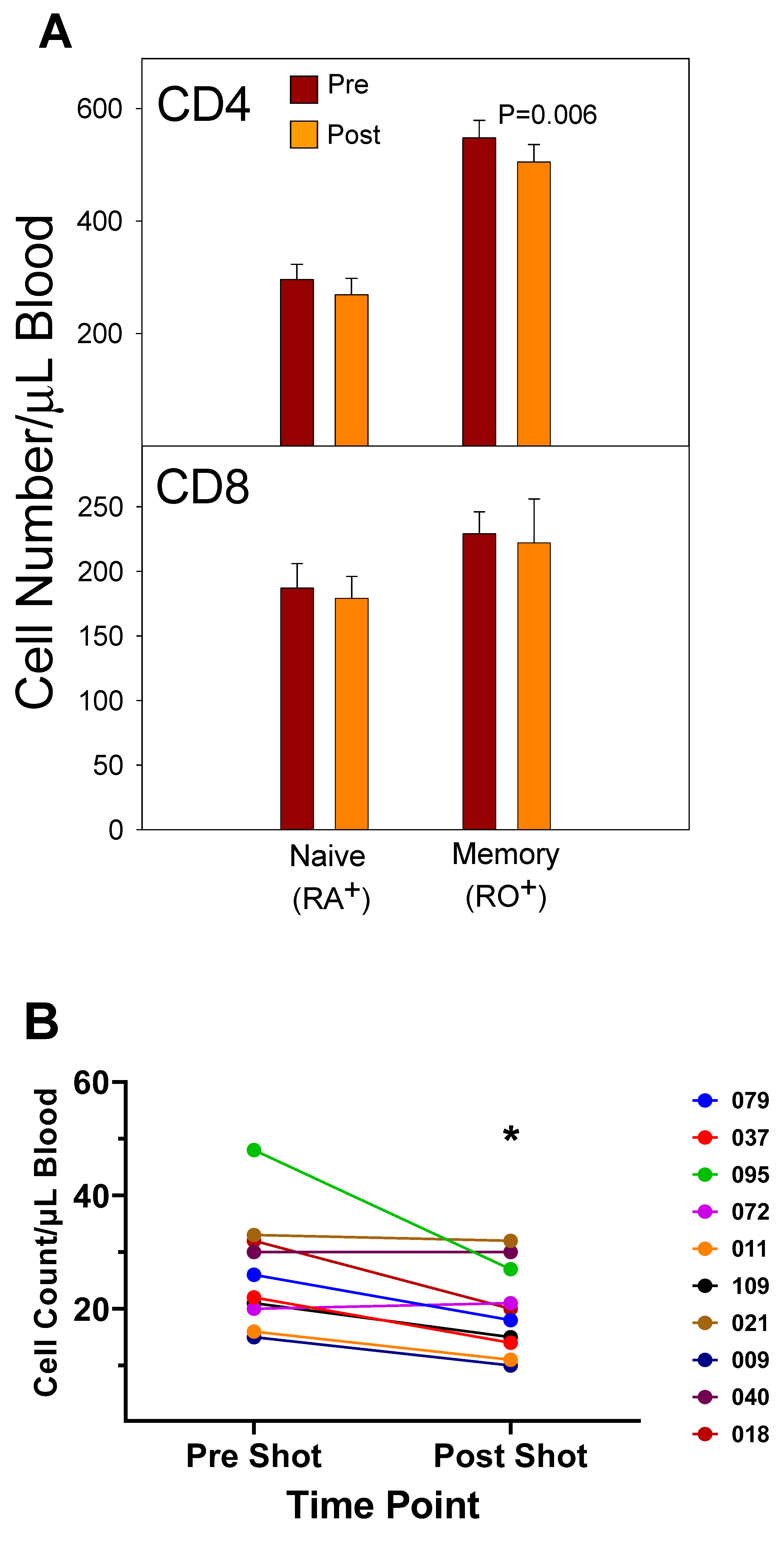
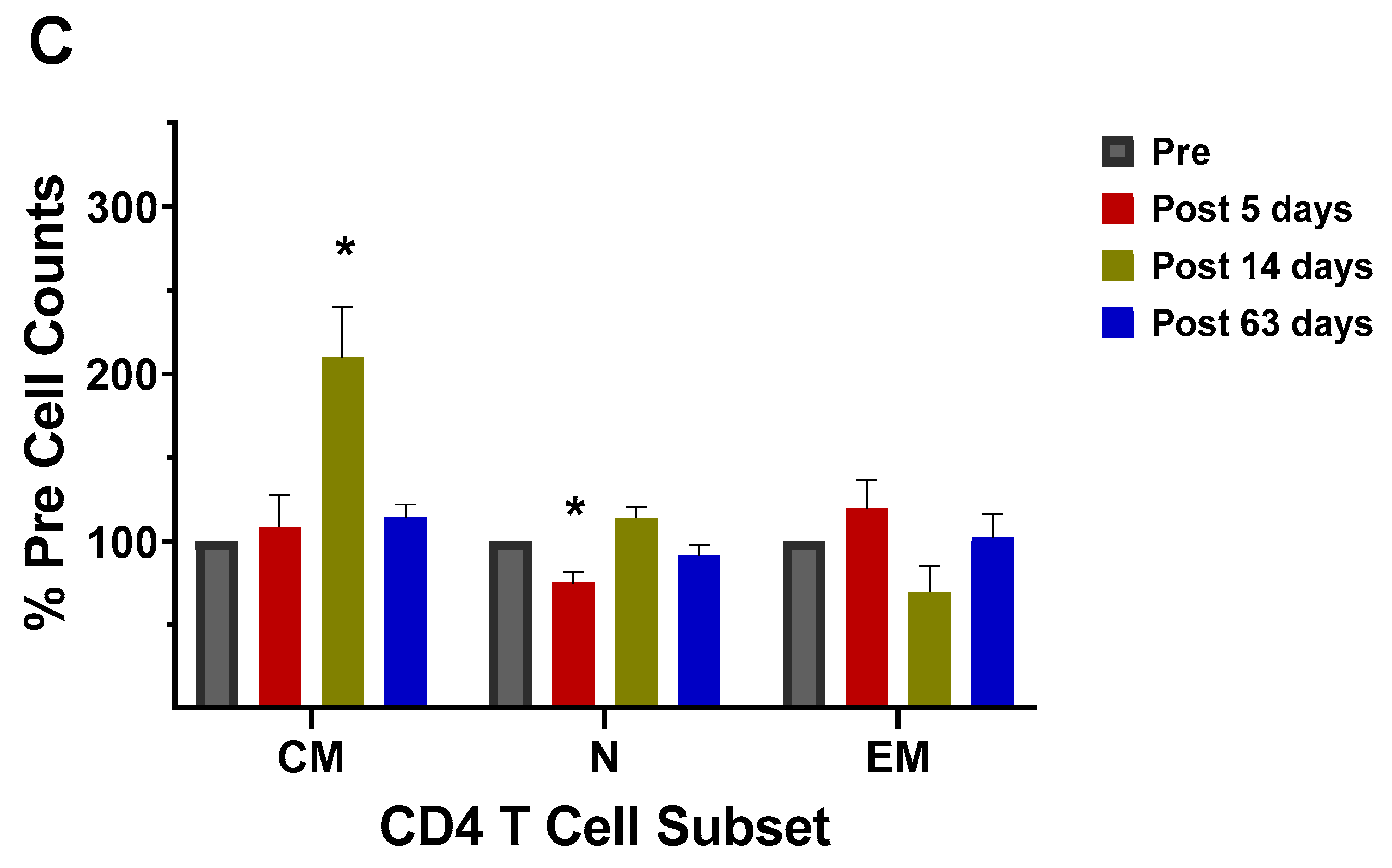
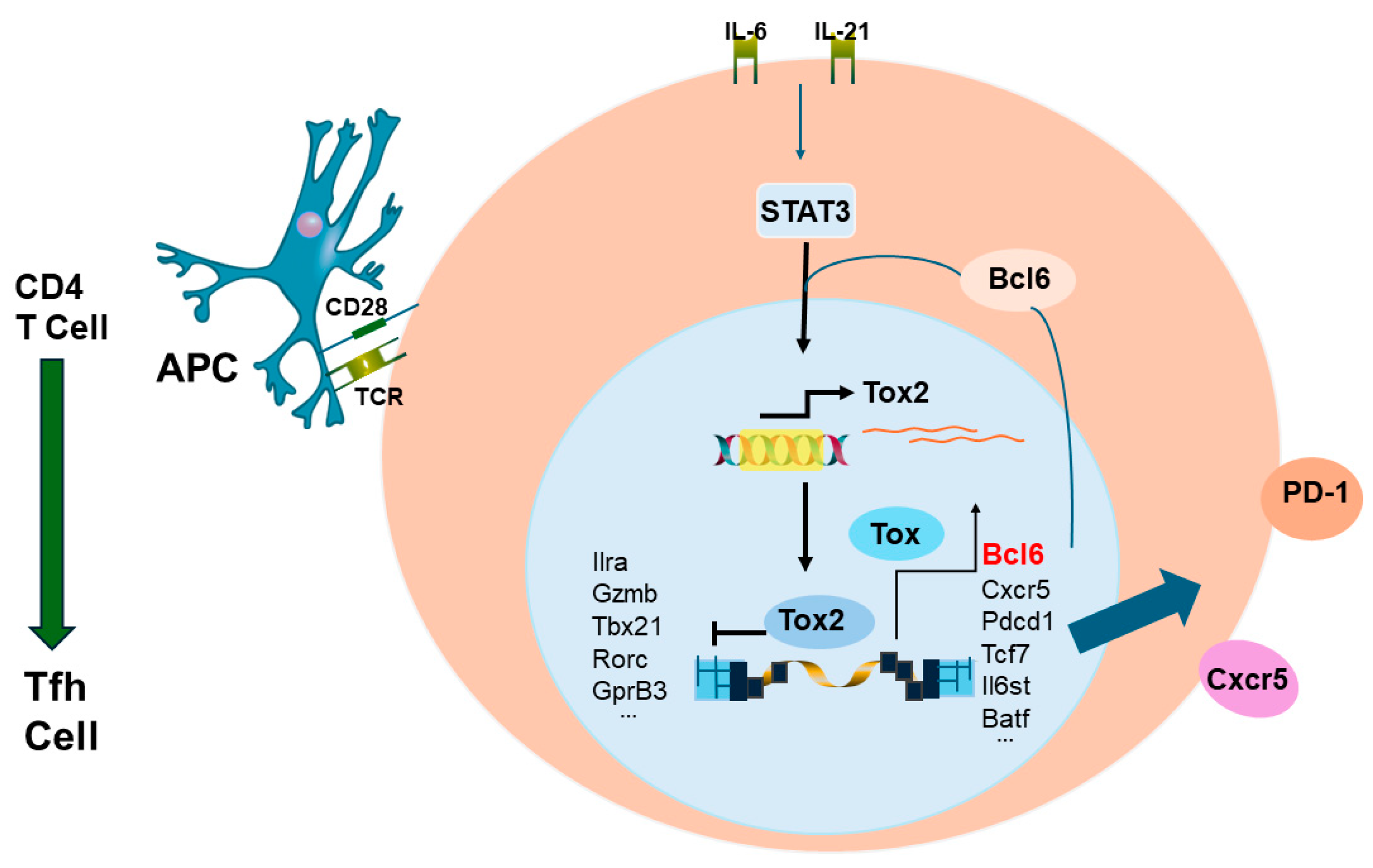
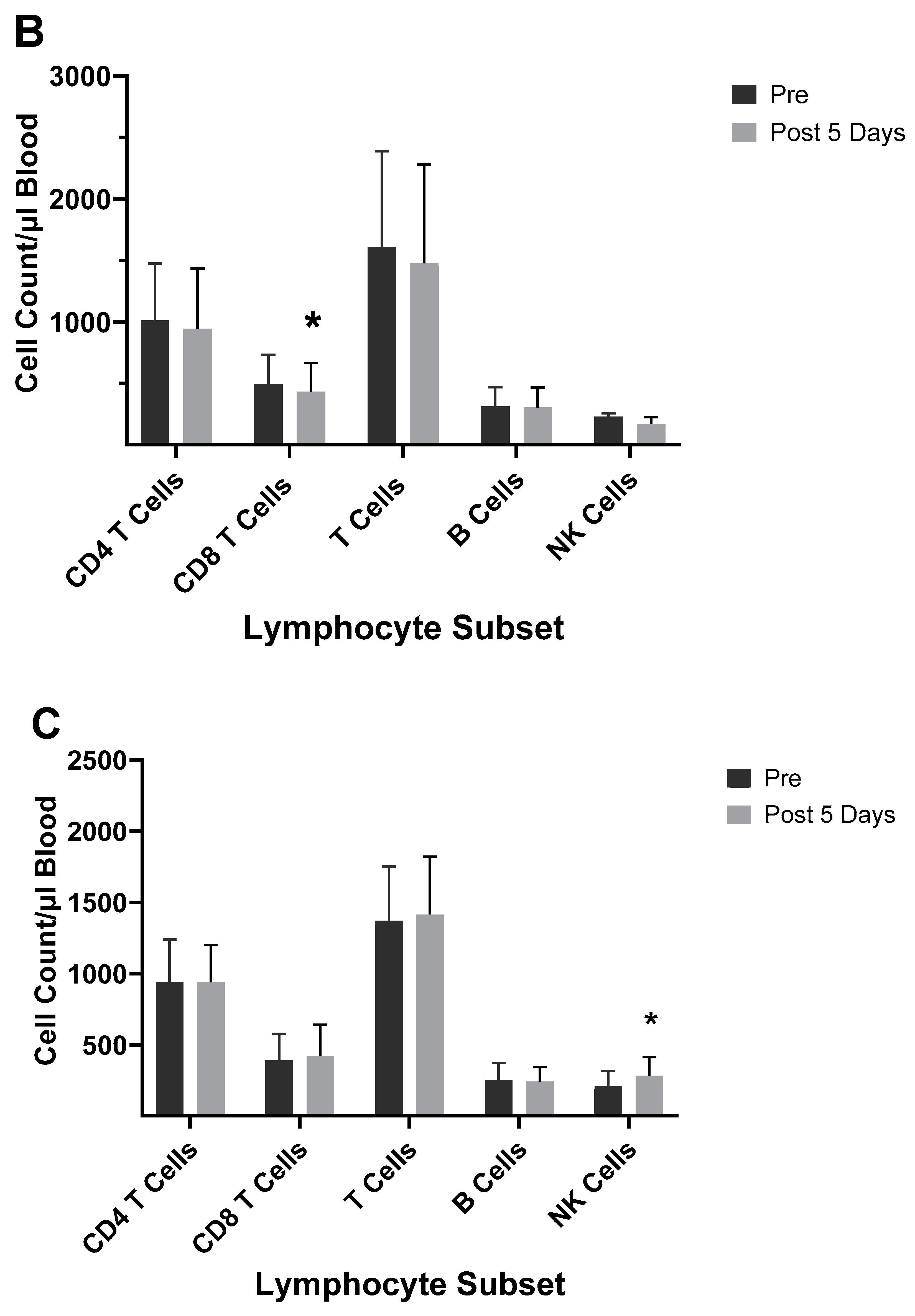
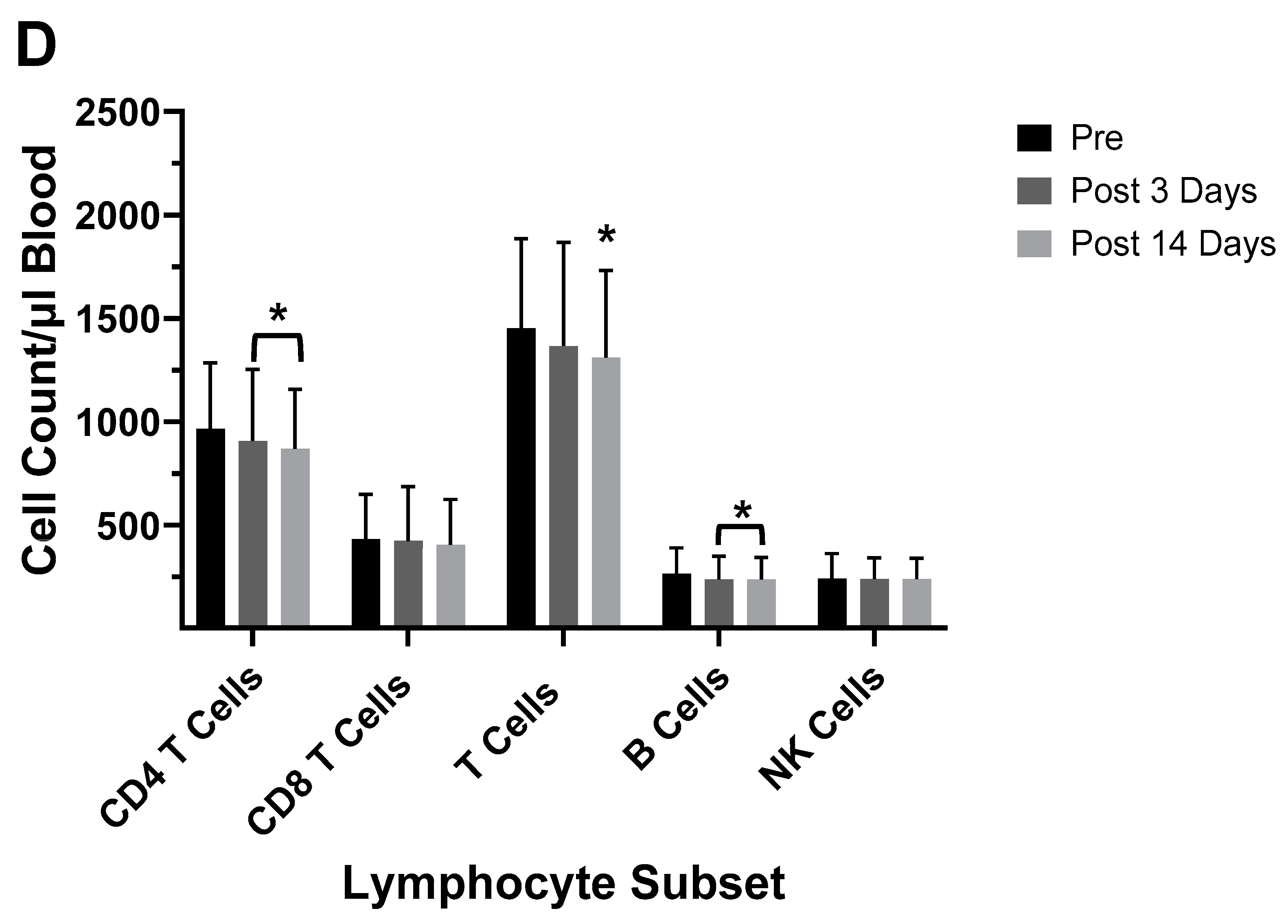
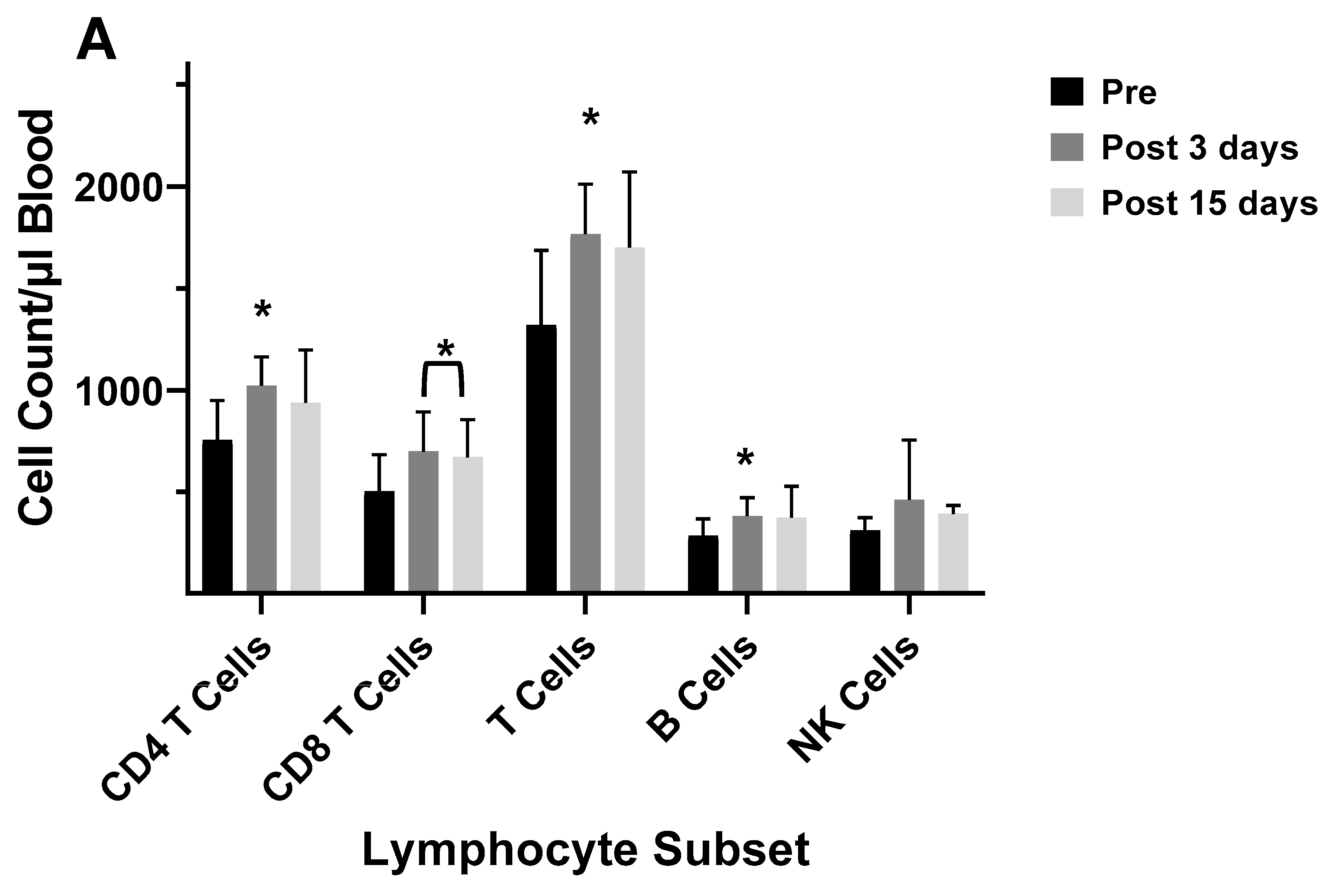
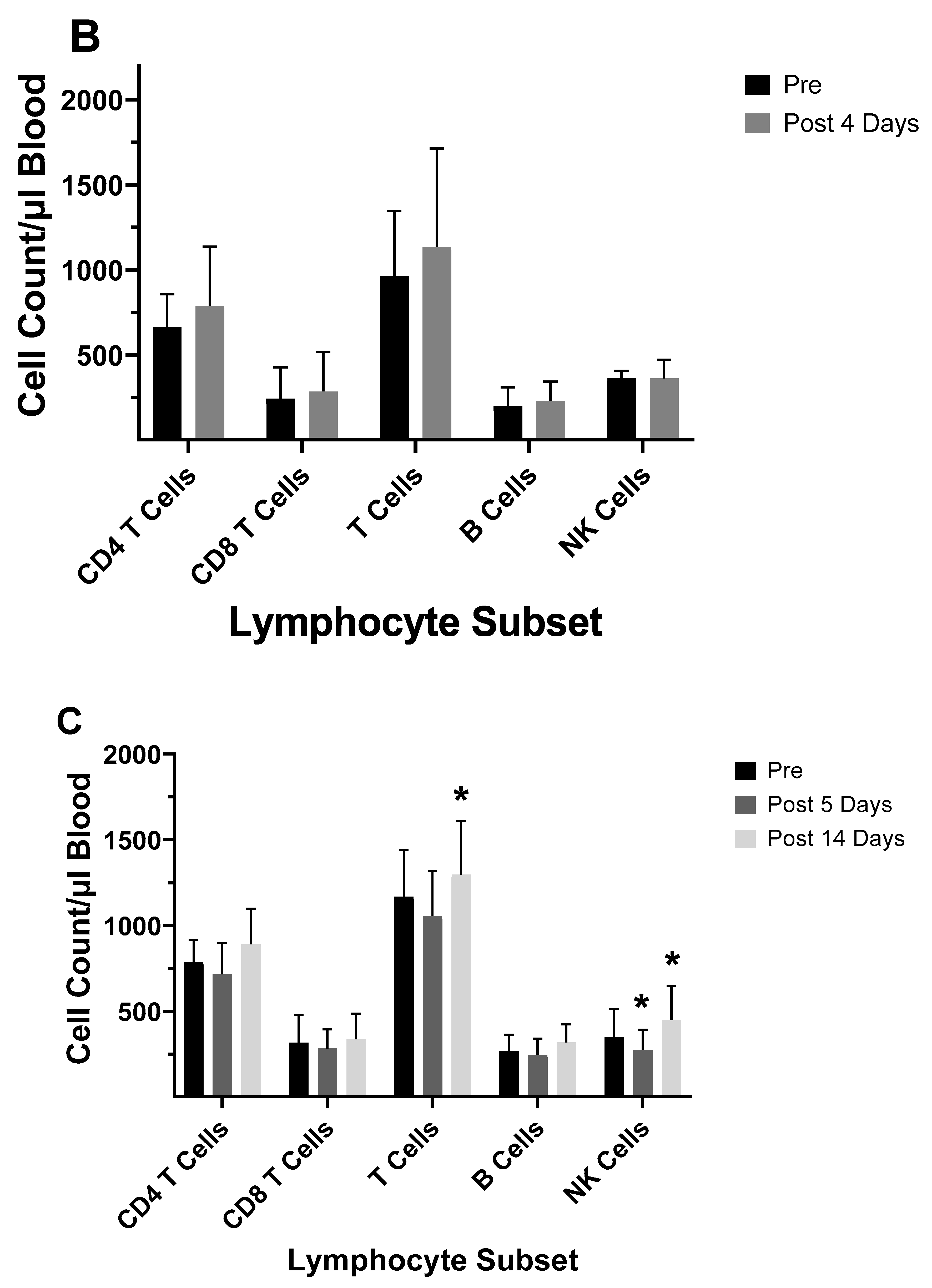
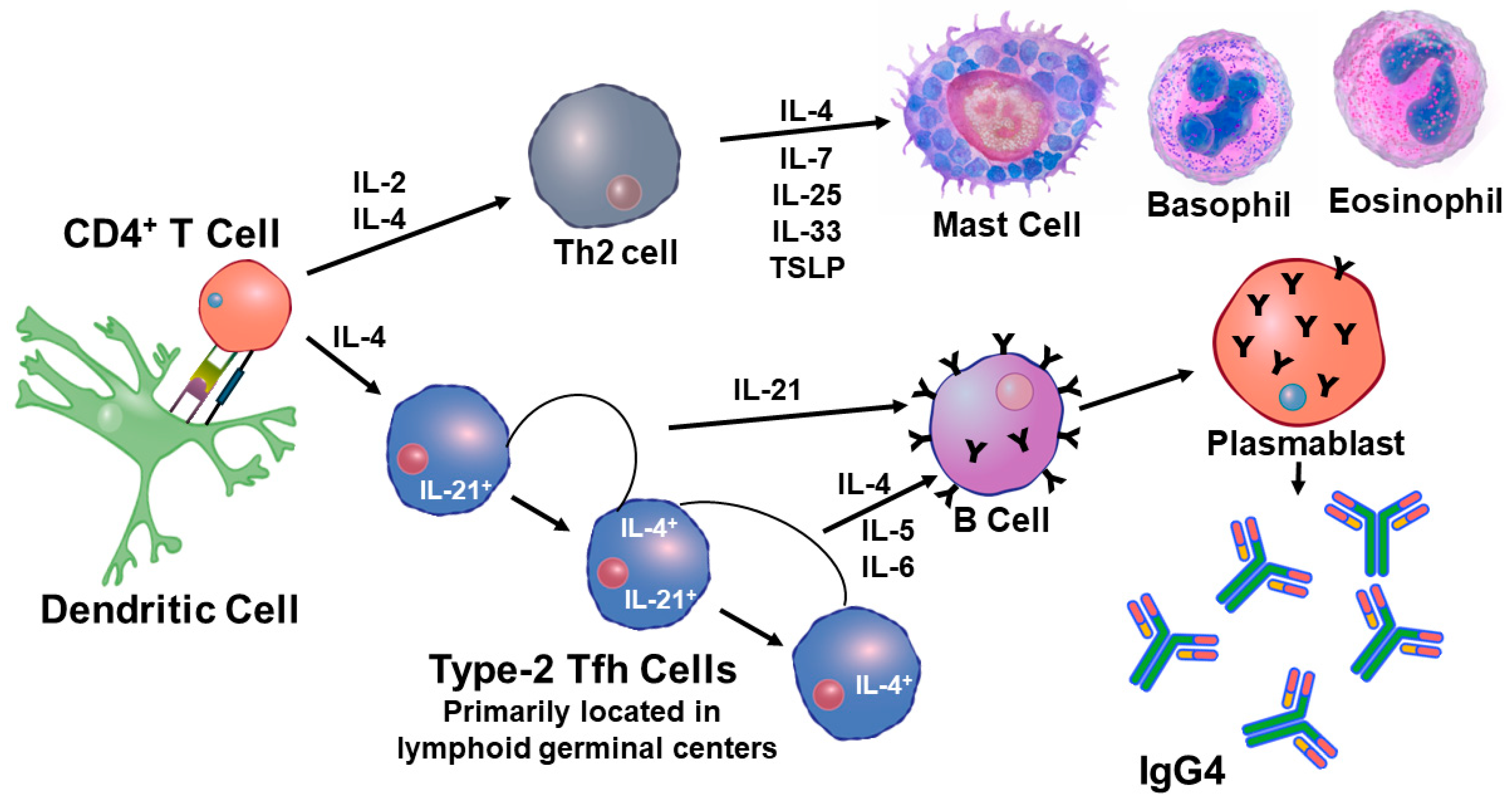
3.3. CD4+ T Cell Memory to Influenza Virus Infection/Vaccination
3.4. CD8+ T-Cell Response to Influenza Infection/Vaccination
3.5. NK Cells and Response to Influenza Virus Infection/Vaccination
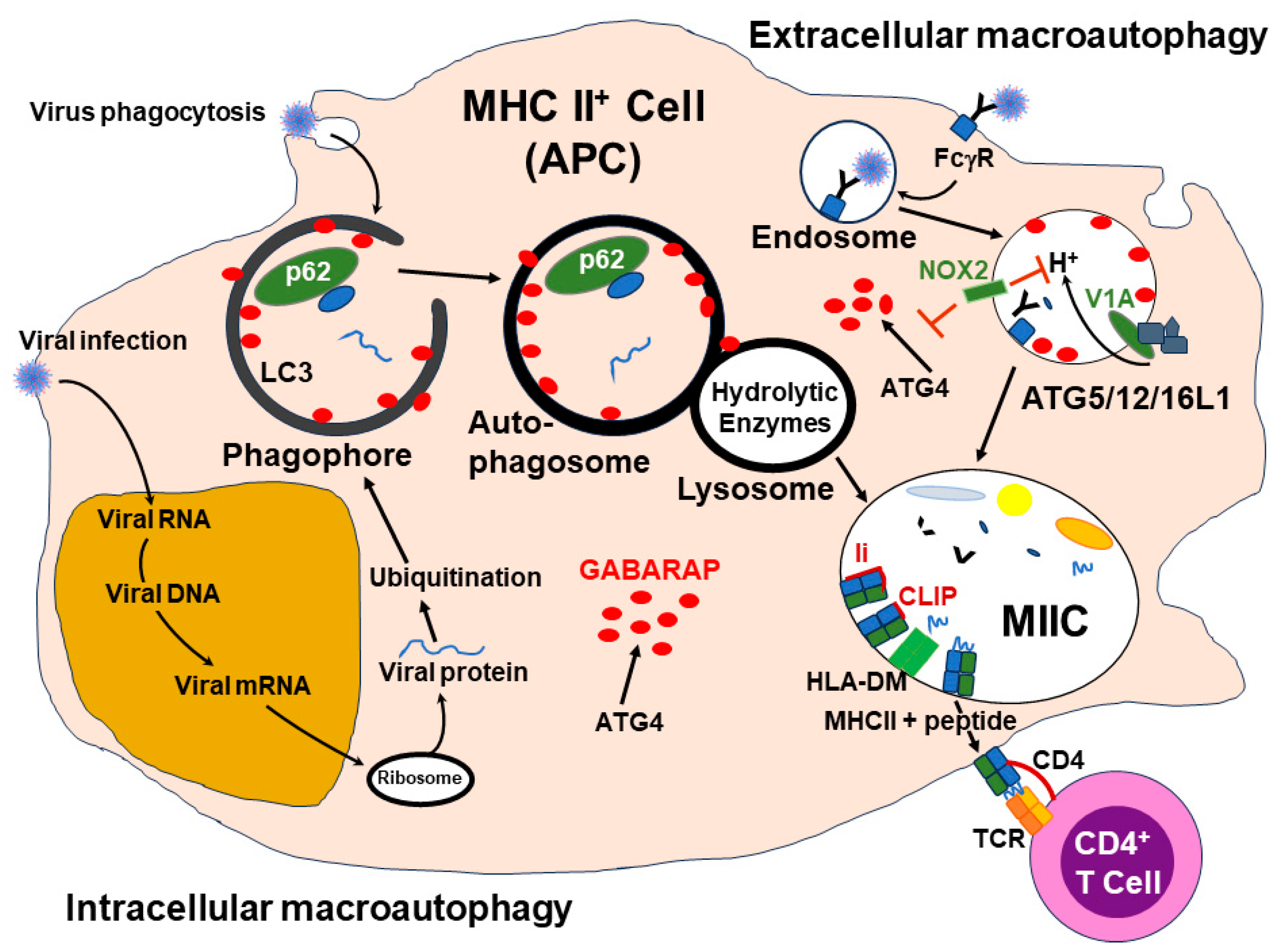
3.6. Viral Antigen Presentation by MHC Molecules—The Role of Autophagy
3.7. The Role of Macrophages in Influenza Infection/Vaccination
3.8. Flu vaccine development
3.9. Imprinting and vaccine boosting
3.10. Concluding remarks
4. Materials and Methods
4.1. Reference Search Method
4.2. Characteristics of the Study Populations
4.3. Isolation of Human IgG
4.4. Antibody Binding Analysis by Surface Plasmon Resonance
4.5. TBNK Immunophenotyping
4.6. Memory Cell Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deutsch, S.; Bussard, A.E. Original antigenic sin at the cellular level 1. Antibodies produced by individual cells against cross-reacting haptens. Eur. J. Immunol. 1972, 2, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Vatti, A.; Monsalve, D.M.; Pacheco, Y.; Chang, C.; Anaya, J.-M.; Gershwin, M.E. Original antigenic sin: A comprehensive review. J. Autoimmun. 2017, 83, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Nachbagauer, R.; Choi, A.; Hirsh, A.; Margina, I.; Lida, S.; Barrera, A.; Ferres, M.; Albrecht, R.A.; Garcia-Sastro, A.; Bouvier, N.M.; et al. Defining the antibody cross-reactomes against the influenza virus surface glycoproteins. Nat. Immunol. 2017, 18, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Francis, T. On the doctrine of original antigenic sin. Proc. Am. Phil. Soc. 1960, 104, 572–578. [Google Scholar]
- de St. Groth, F.; Webster, R.G. Disquisitions of original antigenic sin: I. Evidence in man. J. Exp. Med. 1966, 124, 331–345. [Google Scholar] [CrossRef]
- Monto, A.; Malosh, R.E.; Petrie, J.G.; Martin, E.T. The doctrine of original antigenic sin: Separating good from evil. J. Infect. Dis. 2017, 215, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Davenport, F.M.; Hennessy, A.V. Predetermination by infection and by vaccination of antibody response to influenza virus vaccines. J. Exp. Med. 1957, 106, 835–850. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; Van de Velde, L.-A.; Allison, K.J.; Branum, K.C.; Webby, R.J.; Flynn, P.M. Recipients of vaccine against the 1976 “Swine Flu” have enhanced neutralization responses to the 2009 novel H1N1 influenza virus. Clin. Infect. Dis. 2010, 56, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Gostic, K.M.; Ambrose, M.; Worobey, M.; Lloyd-Smith, J.O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 2016, 354, 722–726. [Google Scholar] [CrossRef]
- Guthmiller, J.J.; Wilson, P.C. Harnessing immune history to combat influenza viruses. Curr. Opin. Immunol. 2018, 53, 187–195. [Google Scholar] [CrossRef]
- Fearon, D.T.; Locksley, R.M. The instructive role of innate immunity in the acquired immune response. Science 1996, 272, 50–53. [Google Scholar] [CrossRef]
- Lambkin, R.; Novelli, P.; Oxford, J.; Gelder, C. Human genetics and responses to influenza vaccination. Am. J. Pharmacogenetics 2004, 4, 293–298. [Google Scholar] [CrossRef]
- Smith, A.J.P.; Humphries, S.E. Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine Growth Factor Rev. 2009, 20, 43–59. [Google Scholar] [CrossRef]
- Breinl, F.; Haurowitz, F. Chemishe untersachugen des präezipitates aus häemoglobin und antihäemoglobulin-serum und hemerkungen über batur der antiköerper. Z. Physiol. Chem. 1930, 192, 45–57. [Google Scholar] [CrossRef]
- Pauling, L. A theory of the structure and process of formation of antibodies. J. Amer. Chem. Soc. 1940, 62, 2643–2657. [Google Scholar] [CrossRef]
- Burnet, F.M.; Fenner, F. The Production of Antibodies: Monograph of the Walter and Eliza Hall Institute of Research in Pathology and Medicine, 2nd ed.; Macmillan: Melbourne, Australia, 1949. [Google Scholar]
- Burnet, F.M. The Clonal Selection Theory of Acquired Immunity. Abraham Flexner lecture 1958; Vanderbilt University Press: Nashville, TN, USA, 1959. [Google Scholar]
- Jerne, N.K. Towards a network theory of the immune system. Ann. Immunol. 1974, 125, 373–389. [Google Scholar]
- Nakamura, Y.; Moi, M.L.; Shiina, T.; Tadasu, S.-I.; Suzuki, R. Idiotope-driven T-cell/B-cell collaboration-based T-cell epitope prediction using B-cell receptor repertoire sequences in infectious diseases. Viruses 2023, 15, 1186. [Google Scholar] [CrossRef]
- Kohler, H.; Pashov, A.; Kieber-Emmons, T. The promise of anti-idiotype revisited. Front. Immunol. 2019, 10, 608. [Google Scholar] [CrossRef]
- Henry, C.; Palm, A.-K.E.; Krammer, F.; Wilson, P.C. From original antigenic sin to the universal influenza virus vaccine. Trends Immunol. 2018, 39, 70–78. [Google Scholar] [CrossRef]
- Oxford, J.S.; Gill, D. Unanswered questions about the 1918 influenza pandemic: Origin, pathology, and the virus itself. Lancet Infect. 2018, 18, e348–e354. [Google Scholar] [CrossRef]
- Zhang, A.; Stacey, H.D.; Mullarkey, C.E.; Miller, M.S. Original antigenic sin: How first exposure shapes lifelong anti-influenza virus immune responses. J. Immunol. 2019, 202, 335–340. [Google Scholar] [CrossRef]
- Worobey, M.; Plotkin, S.; Hensley, S.E. Influenza vaccines delivered in early childhood could turn antigenic sin in to antigenic blessings. Cold Spring Harb. Perspect. Med. 2020, 10, a038471. [Google Scholar] [CrossRef]
- Andrews, S.F.; Huang, Y.; Kaur, K.; Popova, L.I.; Ho, I.Y.; Pauli, N.T.; Dunand, C.J.H.; Taylor, W.M.; Lim, S.; Huang, M.; et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 2015, 7, 192. [Google Scholar] [CrossRef]
- Andrews, S.F.; Kaur, K.; Pauli, N.T.; Huang, M.; Huang, Y.; Wilson, P.C. High preexisting serological antibody levels correlate with diversification of the influenza vaccine response. J. Virol. 2015, 89, 3308–3317. [Google Scholar] [CrossRef]
- Openshaw, P.J.M.; Dunning, J. Influenza vaccination: Lessons learned from the pandemic (H1N1) 2009 influenza outbreak. Mucosal Immunol. 2010, 34, 422–424. [Google Scholar] [CrossRef]
- Huang, R.-Y.A.; Li, C.K.-F.; Clutterbuck, E.; Choi, C.; Wilkinson, T.; Gilbert, A.; Oxford, J.; Lambkin-Williams, R.; Liu, T.-Y.; McMicheal, A.J.; et al. Virus-specific antibody secreting cell, memory B-cell, and sero-antibody responses in the human influenza challenge model. J. Infect. Dis. 2014, 209, 1354–1361. [Google Scholar] [CrossRef]
- Lam, J.H.; Baumgarth, N. The multifaceted B cell response to influenza virus. J. Immunol. 2019, 202, 351–359. [Google Scholar] [CrossRef]
- Langley, W.A.; Bradley, K.C.; Li, Z.-N.; Talekar, G.R.; Galloway, S.E.; Steinhauer, D.A. The effects of preexisting immunity to influenza on responses to influenza vectors in mice. Vaccine 2010, 28, 6305–6313. [Google Scholar] [CrossRef]
- Singh, R.A.K.; Rodgers, J.R.; Barry, M.A. The role of T cell antagonism and original antigenic sin in genetic immunization. J. Immunol. 2002, 169, 6779–6786. [Google Scholar] [CrossRef]
- Wang, T.T.; Palese, P. Universal epitopes of influenza virus hemagglutinins? Nat. Struct. Molec. Biol. 2009, 16, 233–234. [Google Scholar] [CrossRef]
- Sridhar, S. Heterosubtypic T-cell immunity to influenza in humans: Challenges for universal T-cell influenza vaccines. Front. Immunol. 2016, 7, 195. [Google Scholar] [CrossRef]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza virus: Dealing with a drifting and shifting pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Ghedin, E.; Sengamalay, N.A.; Shumway, M.; Zaborsky, J.; Feldblyum, T.; Subbu, V.; Spiro, D.J.; Siz, J.; Koo, H.; Bolotov, P.; et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 2005, 437, 1162–1166. [Google Scholar] [CrossRef]
- Zheng, N.-Y.; Wilson, K.; Jared, M.; Wilson, P.C. Intricate targeting of immunoglobulin somatic hypermutation maximizes the efficiency of affinity maturation. J. Exp. Med. 2005, 201, 1467–1478. [Google Scholar] [CrossRef]
- Li, G.-M.; Chiu, C.; Wrammert, J.; McCausland, M.; Andrews, S.; Zheng, N.-Y.; Lee, J.-H.; Huang, M.; Qu, X.; Edupuganti, S.; et al. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9047–9052. [Google Scholar] [CrossRef]
- Co, M.D.T.; Orphin, L.; Cruz, J.; Pazoles, P.; Rothman, A.L.; Ennis, F.A.; Terajima, M. Discordance between antibody and T cell responses in recipients of trivalent inactivated influenza vaccine. Vaccine 2008, 26, 1990–1998. [Google Scholar] [CrossRef]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zhang, N.-Y.; Mays, I.; Garman, L.; Helmes, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–673. [Google Scholar] [CrossRef]
- Haliiley, J.; Kyu, S.; Kobie, J.J.; Walsh, E.E.; Falsey, A.R.; Randall, T.D.; Treanor, J.; Feng, C.; Sanz, I.; Lee, F.E.-H. Peak frequencies of circulating human influenza-specific antibody secreting cells correlate with serum antibody response after immunization. Vaccine 2010, 28, 3582–3587. [Google Scholar] [CrossRef]
- Grandea, A.G., III; Olsen, O.A.; Cox, T.C.; Renshaw, M.; Hammond, P.W.; Chan-Hui, P.-Y.; Mitcham, J.L.; Cieplak, W.; Stewart, S.M.; Grantham, M.L.; et al. Human antibodies reveal a protective epitope that is highly conserved among human and nonhuman influenza A viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12658–12663. [Google Scholar] [CrossRef]
- Wrammert, J.; Koutsonanos, D.; Li, G.-M.; Edupuganti, S.; Sui, J.; Morrissey, M.; McCausland, M.; Skountzou, I.; Hornig, M.; Lipkin, W.I.; et al. Broadly cross-reactive antibodies dominate the B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011, 208, 181–193. [Google Scholar] [CrossRef]
- Eliebedy, A.H.; Jackson, K.J.L.; Kissick, H.T.; Nakaya, H.I.; Davis, C.W.; Roskin, K.M.; McElroy, A.K.; Oshansky, C.M.; Elbein, R.; Thomas, S.; et al. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat. Immunol. 2016, 17, 1226–1236. [Google Scholar] [CrossRef]
- Fonville, J.M.; Wilks, S.H.; James, S.L.; Fox, A.; Ventresca, M.; Aban, M.; Xua, I.; Jones, T.C.; Le, N.M.H.; Pham, Q.T.; et al. Antibody landscapes after influenza virus infection or vaccination. Science 2014, 346, 996–1000. [Google Scholar] [CrossRef]
- Baas, D.C.; Koopmans, M.P.; de Bruin, E.; ten Hulscher, H.I.; Buisman, A.M.; Hendrikx, L.H.; van Beek, J.; Godeke, G.-J.; Reimerink, J.; van Binnendijk, R.S. Detection of influenza A virus homo-and heterosubtype-specific memory B-cells using a novel protein microarray-based analysis tool. J. Med. Virol. 2013, 85, 809–909. [Google Scholar] [CrossRef]
- Corti, D.; Suguitan, A.L.; Pinna, D.; Silacci, C.; Fernandez-Rodriguez, B.M.; Vanzetta, F.; Santos, C.; Luke, C.J.; Torres-Velez, F.J.; Temperton, N.J.; et al. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J. Clin. Investig. 2010, 120, 1663–1673. [Google Scholar] [CrossRef]
- Hoa, L.N.M.; Mai, L.Q.; Bryant, J.E.; Thai, P.Q.; Hang, N.L.K.; Yen, N.T.T.; Duong, T.N.; Thoang, D.D.; Horby, P.; Werheim, H.F.L.; et al. Association between hemagglutinin stem-reactive antibodies and influenza A/H1N1 virus infection during the 2009 pandemic. J. Virol. 2016, 90, 6549–6556. [Google Scholar] [CrossRef]
- Xu, W.; Han, L.; Lin, Z. Screening of random peptide library of hemagglutinin from pandemic 2009 A(H1N1) influenza virus reveals unexpected antigenically important regions. PLoS ONE 2011, 6, e18016. [Google Scholar] [CrossRef]
- Gaiotto, T.; Hufton, S.E. Cross-neutralizing nanobodies bind to a conserved pocket in the hemagglutinin stem region identified using yeast display and deep mutational scanning. PLoS ONE 2016, 11, e0164296. [Google Scholar] [CrossRef]
- Peng, B.; Peng, N.; Zhang, Y.; Zhang, F.; Li, X.; Chang, H.; Fang, F.; Wang, F.; Lu, F.; Chen, Z. Comparison of the protective efficacy of neutralizing epitopes of 2009 pandemic H1N1 influenza hemagglutinin. Front. Immunol. 2017, 6, 1070. [Google Scholar] [CrossRef]
- Hu, W.; Chen, A.; Miao, Y.; Xia, S.; Ling, Z.; Xu, K.; Wang, T.; Xu, Y.; Cui, J.; Wu, H.; et al. Fully human broadly neutralizing monoclonal antibodies against influenza A viruses generated from the memory B cells of a 2009 pandemic H1N1 influenza vaccine recipient. Virology 2013, 435, 320–328. [Google Scholar] [CrossRef]
- Thomson, C.A.; Wang, V.; Jackson, L.M.; Olson, M.; Wang, W.; Liavonchanka, A.; Keleta, L.; Silva, V.; Diederich, S.; Jones, R.B.; et al. Pandemic H1N1 influenza infection and vaccination in humans induces cross-protective antibodies that target the hemagglutinin stem. Front. Immunol. 2012, 3, 87. [Google Scholar] [CrossRef]
- Ahmed, R.; Gray, D. Immunological memory and protective immunity: Understanding their relation. Science 1996, 272, 54–60. [Google Scholar] [CrossRef]
- Farber, D.L. Remembrance of antigens past: New insights into memory T cells. Scand. J. Immunol. 2003, 58, 145–154. [Google Scholar] [CrossRef]
- Swain, S.L. Regulation of the generation and maintenance of T-cell memory: A direct, default pathway from effectors to memory cells. Microb. Infect. 2003, 5, 213–219. [Google Scholar] [CrossRef]
- Zens, K.D.; Farber, D.L. Memory CD4 T cells in influenza. Curr. Top. Microbiol. Immunol. 2015, 386, 399–421. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Bautista, B.; Zhang, W.; Kuang, Y.; Cooper, A.M.; Swain, S.L. Effector CD4 T cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat. Commun. 2015, 5, 5377. [Google Scholar] [CrossRef]
- Causi, E.L.; Parikh, S.C.; Chudley, L.; Layfield, D.M.; Ottensmeier, C.H.; Stevenson, F.K.; DiGenova, G. Vaccination expands antigen-specific CD4+ memory T cells and mobilizes bystander central memory T cells. PLoS ONE 2015, 10, e0136717. [Google Scholar] [CrossRef]
- Wilkinson, T.M.; Li, C.K.F.; Chui, C.S.C.; Huang, A.K.Y.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–281. [Google Scholar] [CrossRef]
- Rufer, N.; Heig, C.; Chapuis, B.; Roosnek, E. Human memory T cells: Lessons from stem cell transplantation. Trends Immunol. 2001, 22, 36–140. [Google Scholar] [CrossRef]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef]
- DiPiazza, A.; Richards, K.A.; Knowlden, Z.A.G.; Nayak, J.L.; Sant, A.J. The role of CD4 T cell memory in generating protective immunity to novel and potentially pandemic strains of influenza. Front. Immunol. 2016, 7, 10. [Google Scholar] [CrossRef]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015, 6, 16. [Google Scholar] [CrossRef]
- Song, W.; Craft, J. T follicular helper cell heterogeneity: Time, space, and function. Immunol. Rev. 2019, 288, 85–96. [Google Scholar] [CrossRef]
- Valkenburg, S.A.; Li, O.T.W.; Bull, M.; Waldmann, T.A.; Perera, L.P.; Peiris, M.; Poon, L.L.M. Protection by universal influenza vaccine is mediated by memory CD4 T cells. Vaccine 2018, 36, 4198–4206. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhao, X.; Wang, X.; Feng, H.; Gou, M.; Wang, X.; Liu, X.; Dong, C. The transcription factor Tox2 drives T follicular helper cell development via regulating chromatin accessibility. Immunity 2019, 53, 826–839. [Google Scholar] [CrossRef]
- Wild, K.; Smits, M.; Killmer, S.; Strohmeier, S.; Neumann-Haefelin, C.; Bengsch, B.; Krammer, F.; Schwemmle, M.; Hofmann, M.; Thimme, R.; et al. Pre-existing immunity and vaccine history determine hemagglutinin-specific CD4 T cell and IgG response following seasonal influenza vaccination. Nat. Commun. 2021, 12, 6720. [Google Scholar] [CrossRef]
- Freyn, A.W.; da Silva, J.R.; Rosado, V.C.; Bliss, C.M.; Pine, M.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; de Souza Ferreira, L.C.; Weissman, D.; et al. A multi-targeting, nucleoside-modified mRNA influenza virus vaccine provides broad protection in mice. Molec. Ther. 2020, 28, 1569–1584. [Google Scholar] [CrossRef]
- Wu, N.C.; Eliebedy, A.H. Targeting neuraminidase: The next frontier for broadly protective influenza vaccines. Trends Immunol. 2023. [CrossRef]
- Do, T.H.T.; Wheatly, A.K.; Kent, S.J.; Koutsakos, M. Influenza B virus neuraminidase: A potential target for next-generation vaccines? Exp. Rev. Vaccines 2024, 23, 39–48. [Google Scholar] [CrossRef]
- Lei, R.; Kim, W.; Ly, H.; Mou, Z.; Scherm, M.I.; Schmitz, A.I.; Turner, J.S.; Tan, T.I.C.; Wang, Y.; Ouyang, W.O.; et al. Leveraging vaccination-induced protective antibodies to define conserved epitopes on influenza N2 neuraminidase. Immunity 2023, 56, 2621–2634.e6. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Liu, D.; Li, J.; Zhang, X.; Chen, X.; Hou, S.; Peng, L.; Xu, C.; Liu, W.; et al. Follicular T helper cell recruitment governed by bystander B cells and ICOS- driven motility. Nature 2013, 496, 523–527. [Google Scholar] [CrossRef]
- He, X.-S.; Holmes, T.H.; Sasaki, S.; Jaimes, M.C.; Kemble, G.W.; Dekker, C.L.; Arvin, A.M.; Greenberg, H.B. Baseline levels of influenza-specific CD4 memory T-cells affect T-cell responses to influenza vaccines. PLoS ONE 2008, 3, e2574. [Google Scholar] [CrossRef]
- He, X.-S.; Holmes, T.H.; Mahmood, K.; Kemble, G.W.; Dekker, C.L.; Arvin, A.M.; Greenburg, H.B. Phenotypic changes in influenza specific CD8+ T cells after immunization of children and adults with influenza vaccines. J. Infect. Dis. 2008, 197, 803–811. [Google Scholar] [CrossRef]
- Jagaskanda, S.; Luka, C.; Hickman, H.D.; Sangster, M.Y.; Wieland-Alter, W.F.; McBride, J.M.; Yewdeli, J.W.; Wright, P.F.; Treanor, J.; Rosenberger, C.M.; et al. Generation and protective ability of influenza virus-specific antibody-dependent cellular cytotoxicity in humans elicited by vaccination, natural infection, and experimental challenge. J. Infect. Dis. 2016, 214, 945–952. [Google Scholar] [CrossRef]
- Bihl, F.; Germain, C.; Luci, C.; Braud, V.M. Mechanisms of NK cell activation: CD4+ T cells enter the scene. Cell. Molec. Life Sci. 2011, 68, 3457–3467. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef]
- Frank, K.; Paust, S. Dynamic natural killer cell and T cell responses to influenza infection. Front. Cell. Infect. Microbiol. 2020, 10, 425. [Google Scholar] [CrossRef]
- He, X.-S.; Draghi, M.; Mahmood, K.; Holmes, T.H.; Kemble, G.W.; Dekker, C.L.; Arvin, A.M.; Parham, P.; Greenberg, H.B. T cell-dependent production of IFN-γ by NK cells in response to influenza A virus. J. Clin. Investig. 2004, 114, 1812–1819. [Google Scholar] [CrossRef]
- Horowitz, A.; Behrens, R.H.; Okell, L.; Fooks, A.R.; Riley, E.M. NK cells as effectors of acquired immune responses: Effector CD4+ T cell-dependent activation of NK cells following vaccination. J. Immunol. 2010, 185, 2808–2818. [Google Scholar] [CrossRef]
- Jost, S.; Quiliay, H.; Reardon, E.; Peterson, E.; Simmons, R.P.; Parry, B.A.; Bryant, N.N.P.; Binder, W.D.; Altfeld, M. Changes in cytokine levels and NK cell activation associated with influenza. PLoS ONE 2011, 6, e25060. [Google Scholar] [CrossRef] [PubMed]
- Kronstad, L.M.; Seiler, C.; Vergara, R.; Holmes, S.P.; Blish, C.A. Differential induction of IFN-α and modulation of CD112 and CD54 expression govern the magnitude of NK cell IFN-γ response to influenza A viruses. J. Immunol. 2018, 201, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Mooney, J.P.; Qendro, T.; Keith, M.; Philbey, A.W.; Groves, H.T.; Tregoning, J.S.; Goodier, M.R.; Riley, E.M. Natural killer cells dampen the pathogenic features of recall responses to influenza infection. Front. Immunol. 2020, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Long, B.R.; Michaelsson, J.; Loo, C.P.; Ballan, W.M.; Vu, B.-A.N.; Hecht, F.M.; Lanier, L.L.; Chapman, J.M.; Nixon, D.F. Elevated frequency of gamma interferon-producing NK cells in healthy adults vaccinated against influenza virus. Clin. Vaccine Immunol. 2008, 15, 120–130. [Google Scholar] [CrossRef]
- Achdout, H.; Manaster, I.; Mandelboim, O. Influenza virus infection augments NK cell Inhibition through reorganization of major histocompatibility complex class I proteins. J. Virol. 2008, 82, 8030–8037. [Google Scholar] [CrossRef] [PubMed]
- Guillonneau, C.; Mintern, A.D.; Hubert, F.-X.; Hurt, A.C.; Besra, G.S.; Porcelli, S.; Barr, I.G.; Doherty, P.C.; Godfrey, D.I.; Turner, S.J. Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.-W.; Yuan, S.; Poon, K.-M.; Yang, D.; Sun, Z.; Li, C.; Hu, M.; Shuai, H.; Zhou, J.; Zhang, M.-Y.; et al. Antibody-dependent cell-mediated cytotoxicity epitopes on the hemagglutinin head region of pandemic H1N1 influenza virus play detrimental roles in H1N1-infected mice. Front. Immunol. 2017, 8, 317. [Google Scholar] [CrossRef]
- Münz, C. Canonical and non-canonical functions of the autophagy machinery in MHC restricted antigen presentation. Front. Immunol. 2022, 13, 868888. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Sarango, C.; Richetta, C.; Pereira, M.; Kuman, A.; Ghosh, M.; Bertrand, L.; Pionneau, C.; Le Gall, M.; Grégoire, S.; Jeger-Madiot, R.; et al. The autophagy receptor TAX1BP1 (T6BP) improves antigen presentation by MHC-II molecules. EMBO Rep. 2022, 23, e55470. [Google Scholar] [CrossRef]
- Murera, D.; Arbogast, F.; Arnold, J.; Bouis, D.; Muller, S.; Gros, F. CD4 T cell autophagy is integral to memory maintenance. Sci. Rep. 2018, 8, 5951. [Google Scholar] [CrossRef]
- Alsaleh, G.; Panse, I.; Swadling, L.; Zhang, H.; Richter, F.C.; Meyer, A.; Lord, J.; Barnes, E.; Klenerman, P.; Green, C.; et al. Autophagy in T cells from aged donors is maintained by spermidine and correlates with function and vaccine responses. eLife 2020, 9, e57950. [Google Scholar] [CrossRef]
- Crotzer, V.I.; Blum, J.S. Autophagy and adaptive immunity. Immunology 2010, 131, 9–17. [Google Scholar] [CrossRef]
- Mintern, J.D.; Macri, C.; Chin, W.J.; Panozza, S.E.; Segura, E.; Patterson, N.I.; Zelier, P.; Bourges, D.; Bedoui, S.; McMillan, P.J.; et al. Differential use of autophagy by primary dendritic cells specialized in cross presentation. Autophagy 2015, 116, 906–917. [Google Scholar] [CrossRef]
- Bettosini, F.; Fiorillo, M.T.; Magnacca, A.; Leone, L.; Torrisi, M.R.; Sorrentino, R. The C terminus of the nucleoprotein of influenza A virus delivers antigens transduced by Tat to the trans-Golgi network and promotes an efficient presentation through HLA class I. J. Virol. 2005, 79, 15537–15546. [Google Scholar] [CrossRef]
- Cline, T.D.; Beck, D.; Bianchini, E. Influenza virus replication in marcrophages: Balancing protection and pathogenesis. J. Gen Virol. 2017, 98, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Hoeve, M.A.; Nash, A.A.; Jackson, D.; Randall, R.E.; Dransfield, I. Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. PLoS ONE 2012, 7, e29443. [Google Scholar] [CrossRef]
- Meischel, T.; Villalon-Letelier, F.; Saunders, P.M.; Reading, P.C.; Londrigan, S.L. Influenza A virus interactions with macrophages: Lessons from epithelial cells. Cell. Microbiol. 2020, 5, e13170. [Google Scholar] [CrossRef]
- Baudon, E.M.; Bajenoff, M. Tingible body macrophages: Gargantuan chameleons of the germinal center. J. Exp. Med. 2023, 220, e20230250. [Google Scholar] [CrossRef]
- Kamperdijk, E.W.A.; Raaymakers, E.M.; DeLeeuw, J.H.S.; Hoefsmit, E.C.M. Lymph node macrophages and reticulum cells in the immune response. Cell Tiss. Res. 1978, 192, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Lister, A.M.; Tew, J.G.; Szakal, A.K. Kinetics of the tingible body macrophage response in mouse germinal center development and its depression with age. Anat. Rec. 1991, 229, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Burton, G.F.; Tew, J.G.; Szakal, A.K. Tingible body macrophages in regulation of germinal center reactions. Devel. Immun. 1998, 6, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.S.M.; Shao, W.-H.; Khan, T.N.; Zhen, Y.; Cohen, P.L. Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J Immunol. 2010, 185, 5859–5868. [Google Scholar] [CrossRef] [PubMed]
- Gurwicz, N.; Stoler-Barak, L.; Schwan, N.; Bandyopadhyay, A.; Meyer-Hermann, M.; Shulman, Z. Tingible body macrophages arise from lymph node-resident precursors and uptake B cells by dendrites. J. Exp. Med. 2023, 220, e20222173. [Google Scholar] [CrossRef] [PubMed]
- Grootveld, A.K.; Kyaw, W.; Panova, V.; Lau, A.W.Y.; Ashwin, E.; Seuzaret, G.; Dhenni, R.; Bhattacharyya, N.D.; Khoo, W.H.; Biro, M.; et al. Apoptotic cell fragments locally activate tingible body macrophages in the germinal center. Cell 2023, 186, 1144–1161. [Google Scholar] [CrossRef]
- Li, S.; Rouphael, N.; Duraisingham, S.; Romero-Stainer, S.; Presnell, S.; Davis, C.; Schmidt, D.S.; Johnson, S.E.; Milton, A.; Rajam, G.; et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014, 15, 195–204. [Google Scholar] [CrossRef]
- Galassie, A.C.; Link, A.J. Proteomic contributions to our understanding of vaccine and immune responses. Proteomics Clin. Appl. 2015, 9, 972–989. [Google Scholar] [CrossRef]
- Kumar, A.; Meldgaard, T.S.; Bertholet, S. Novel platforms for the development of a universal influenza vaccine. Front. Immunol. 2018, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Mullarkey, C.E.; Duty, A.; Moran, T.M.; Palese, P.; Miller, M.S. Broadly neutralizing anti-influenza virus antibodies: Enhancement of neutralizing potency in polyclonal mixtures and IgA backbones. J. Virol. 2015, 89, 3610–3618. [Google Scholar] [CrossRef]
- Alexander, J.; Bilsel, P.; del Guercio, M.-F.; Stewart, S.; Marinkovic-Petrovic, A.; Southwood, S.; Crimi, C.; Vang, L.; Walker, L.; Ishioka, G.; et al. Universal influenza DNA vaccine encoding conserved CD4+ T cell epitopes protects against lethal viral challenge in HLA-DR transgenic mice. Vaccine 2010, 28, 664–672. [Google Scholar] [CrossRef]
- Xu, H.; Cai, L.; Hufnagel, S.; Cui, Z. Intranasal vaccine: Factors to consider in research and development. Intl. J. Pharm. 2021, 609, 121180. [Google Scholar] [CrossRef] [PubMed]
- Ennis, F.A.; Dowdle, W.R.; Barry, D.W.; Hockstein, H.D.; Wright, P.F.; Karzon, D.T.; Marine, W.M.; Meyer, H.M., Jr. Endotoxin content and clinical reactivity to influenza vaccines. J. Biol. Stand. 1977, 5, 165–172. [Google Scholar] [CrossRef]
- Nogales, A.; DeDiego, M.L.; Topham, D.J.; Martinez-Sobrido, L. Rearrangement of influenza virus spliced segments for the development of live-attenuated vaccines. J. Virol. 2016, 90, 6291–6302. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Christensen, J.P.; Korsholm, K.S.; Isling, L.K.; Erneholm, K.; Thomsen, A.R.; Andersen, P. Seasonal influenza split vaccines confer partial cross-protection against heterologous influenza virus in ferrets when combined with the CAF01 adjuvant. Front. Immunol. 2017, 8, 1928. [Google Scholar] [CrossRef] [PubMed]
- Langley, J.M.; Frenette, L.; Ferguson, L.; Riff, D.; Sheldon, E.; Risi, G.; Johnson, C.; Li, P.; Kenney, R.; Innis, B.; et al. Safety and cross-reactive immunogenicity of candidate AS03-adjuvanted prepandemic H5N1 influenza vaccines: A randomized controlled phase1/2 trial in adults. J. Infect. Dis. 2010, 201, 1644–1663. [Google Scholar] [CrossRef]
- Eliebedy, A.H.; Nachbagauer, R.; Jackson, K.J.L.; Dai, Y.-N.; Han, J.; Alsoussi, W.; Davis, C.W.; Stadlbauer, D.; Rouphael, N.; Chromikova, V.; et al. Adjuvanted H5N1 influenza vaccine enhances both cross-reactive memory B cell and strain-specific naïve B cell responses in humans. Proc. Natl. Acad. Sci. USA 2020, 117, 17957–17964. [Google Scholar] [CrossRef] [PubMed]
- Nachbagauer, R.; Palese, P. Is a universal influenza virus vaccine possible? Ann. Rev. Med. 2020, 71, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Nachbagauer, R.; Feser, J.; Naficy, A.; Bernstein, D.J.; Guptill, J.; Walter, E.B.; Berlanda-Scorza, F.; Stadbauer, D.; Wilson, P.C.; Aydillo, T.; et al. A Chimeric hemagglutinin-based universal influenza virus vaccine approach induces broad and long-lasting immunity in a randomized, placebo-controlled phase 1 trial. Nat. Med. 2021, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.F.; Cominsky, L.Y.; Shimberg, G.D.; Gillespie, R.A.; Gorman, J.; Raab, J.E.; Brand, J.; Creanga, A.; Gajjala, S.R.; Narpala, S.; et al. An influenza H1 hemagglutinin stem-only immunogen elicits a broadly cross-reactive B cell response in humans. Sci. Transl. Med. 2023, 15, eade4976. [Google Scholar] [CrossRef]
- Widge, A.T.; Hofstetter, A.R.; Houser, K.V.; Awan, S.F.; Chen, G.L.; Burgos Florez, M.C.; Berkowitz, N.M.; Mendoza, F.; Hendel, C.S.; Holman, L.A.; et al. An Influenza hemagglutinin stem nanoparticle vaccine induces cross-group 1 neutralizing antibodies in healthy adults. Sci. Transl. Med. 2023, 15, eade4790. [Google Scholar] [CrossRef]
- Kelvin, A.A.; Falzarano, D. The influenza universe in an mRNA vaccine: An mRNA-lipid nanoparticle vaccine protests animals from 20 influenza lineages. Science 2022, 378, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, C.; Bolton, M.J.; La Sage, V.; Ye, N.; Furey, C.; Muramatsu, H.; Alameh, M.-G.; Pardi, N.; Drapeau, E.M.; Parkhouse, K.; et al. A multivalent nucleoside-modified mRNA vaccine against all known influenza virus subtypes. Science 2022, 378, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Loo, G.; Liu, R.; Feng, J.; Long, F.; Peng, T. The race toward a universal influenza vaccine: Front runners and the future directions. Antivir. Res. 2023, 200, 105505. [Google Scholar] [CrossRef]
- Lasrado, N.; Collier, A.Y.; Miller, J.; Hachmann, N.P.; Liu, J.; Anand, T.; Bondzie, E.A.; Fisher, J.L.; Mazurek, C.R.; Patio, R.C.; et al. Waning immunity and IgG4 responses following bivalent mRNA boosting. Sci. Adv. 2024, 10, eadj9945. [Google Scholar] [CrossRef] [PubMed]
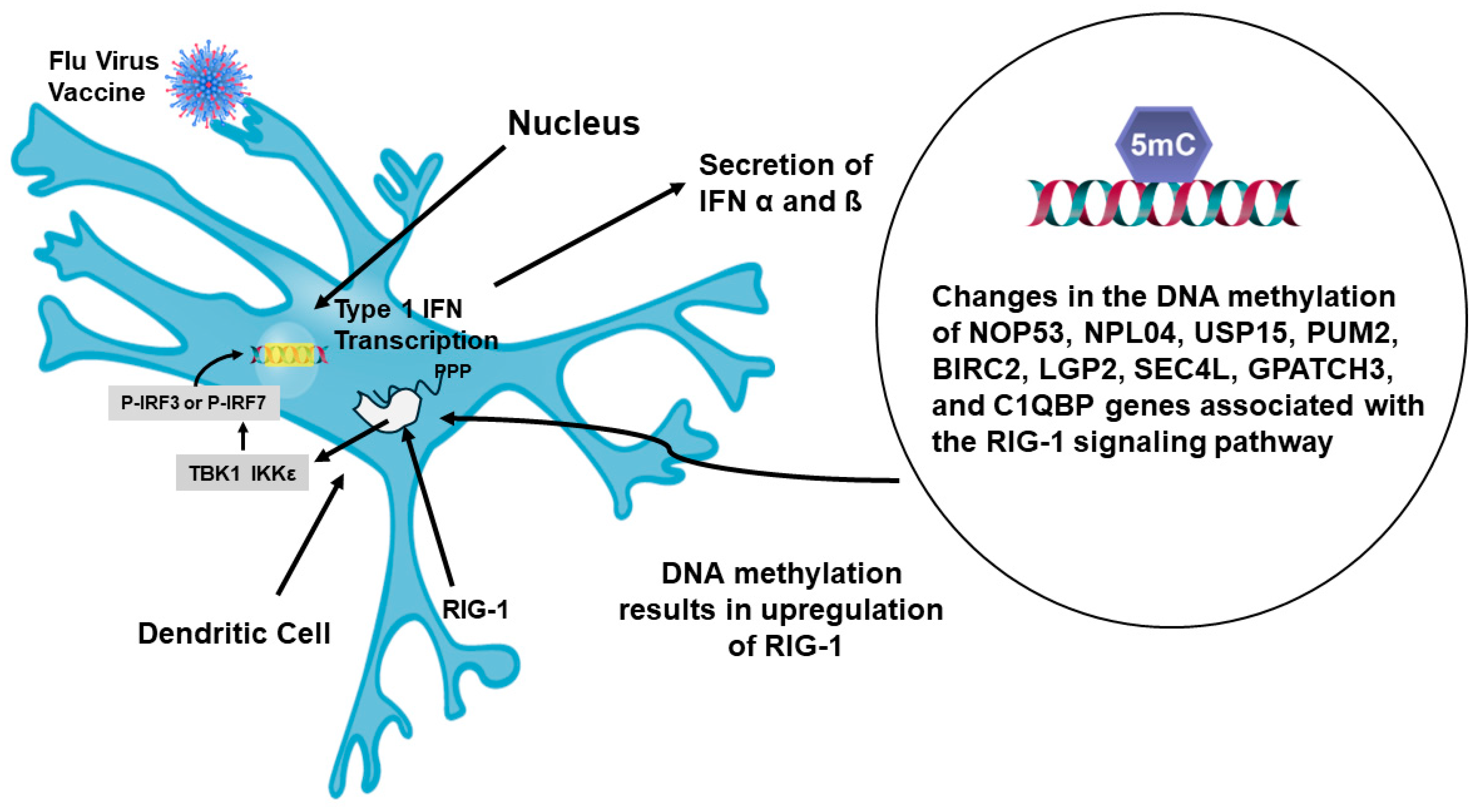
- Fu, H.; Pickering, H.; Rubbi, L.; Ross, T.M.; Reed, E.F.; Pellegrini, M. Longitudinal analysis of influenza vaccination implicates regulation of RIG-1 signaling by DNA methylation. Sci. Rep. 2024, 14, 1455. [Google Scholar] [CrossRef]
- Kulkarni, R.R.; Rasheed, M.A.U.; Bhaumik, K.; Ranjan, P.; Cao, W.; David, C.; Marisetti, K.; Thomas, S.; Gangappa, S.; Sambhara, S.; et al. Activation of the RIG-1 pathway during influenza vaccination enhances the germinal center reaction, promotes T follicular helper cell induction, and provides a dose-sparing effect on protective immunity. J. Virol. 2014, 88, 13990–14001. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2003–2004 vaccine |
| A/New Caledonia (H1N1), A/Panama (H3N2), and B/Hong Kong |
| 2005–2006 vaccine |
| A/New Caledonia (H1N1), A/California (H3N2), and B/Shanghai |
| 2006–2007 vaccine |
| A/New Caledonia/20/99 (H1N1), A/Wisconsin/67/2005 (H3N2), and B/Malaysia/2506/2004 |
| 2007–2008 vaccine |
| A/Solomon Islands/3/2006 (H1N1), A/Wisconsin/67/2005 (H3N2), and B/Malaysia/2506/2004 |
| 2008–2009 vaccine |
| A/Brisbane/59/2007 (H1N1), A/Brisbane/10/2007 (H3N2), and B/Florida/4/2006 |
| 2009–2010 vaccine |
| A/Brisbane/59/2007, IVR-148 (H1N1), A/Uruguay/716/2007, NYMC X-175C (H3N2), B/Brisbane/60/2008 (3,11) |
| 2010–2011 vaccine |
| A/California/07/2009 (HINI), A/Perth /16/2009 (H3N2), B/Brisbane/60/2008 |
| 2011–2012 vaccine |
| A/California/07/2009 (H1N1), A/Perth/16/2009 (H3N2), B/Brisbane/60/2008 |
| 2014–2015 vaccine |
| A/California/07/2009 (H1N1), A/Texas/50/2012 (H3N2), B/Massachusetts/02/2012 |
| 2016–2017 vaccine |
| A/ California/07/2009 X-179A (H1N1), A/Hong Kong/4801/2014 X-263-B (H3N2), B/Brisbane/60/2008 (B Victoria Lineage) |
| 2017–2018 vaccine |
| A/Michigan/45/2015 (H1N1) pdm09-like virus, A/Hong Kong/4801/2014 (H3N2)-like virus, B/Brisbane/60/2008-like virus (B/Victoria lineage), B/Phuket/3073/2013-like virus (B/Yamagata lineage) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasten-Jolly, J.; Lawrence, D.A. Cellular and Molecular Immunity to Influenza Viruses and Vaccines. Vaccines 2024, 12, 389. https://doi.org/10.3390/vaccines12040389
Kasten-Jolly J, Lawrence DA. Cellular and Molecular Immunity to Influenza Viruses and Vaccines. Vaccines. 2024; 12(4):389. https://doi.org/10.3390/vaccines12040389
Chicago/Turabian StyleKasten-Jolly, Jane, and David A. Lawrence. 2024. "Cellular and Molecular Immunity to Influenza Viruses and Vaccines" Vaccines 12, no. 4: 389. https://doi.org/10.3390/vaccines12040389
APA StyleKasten-Jolly, J., & Lawrence, D. A. (2024). Cellular and Molecular Immunity to Influenza Viruses and Vaccines. Vaccines, 12(4), 389. https://doi.org/10.3390/vaccines12040389