
Feasibility of Using a Type I IFN-Based Non-Animal Approach to Predict Vaccine Efficacy and Safety Profiles


Abstract
1. Introduction
2. Interferons (IFNs)
2.1. Type I IFNs
2.2. Type I IFN Signaling Pathways
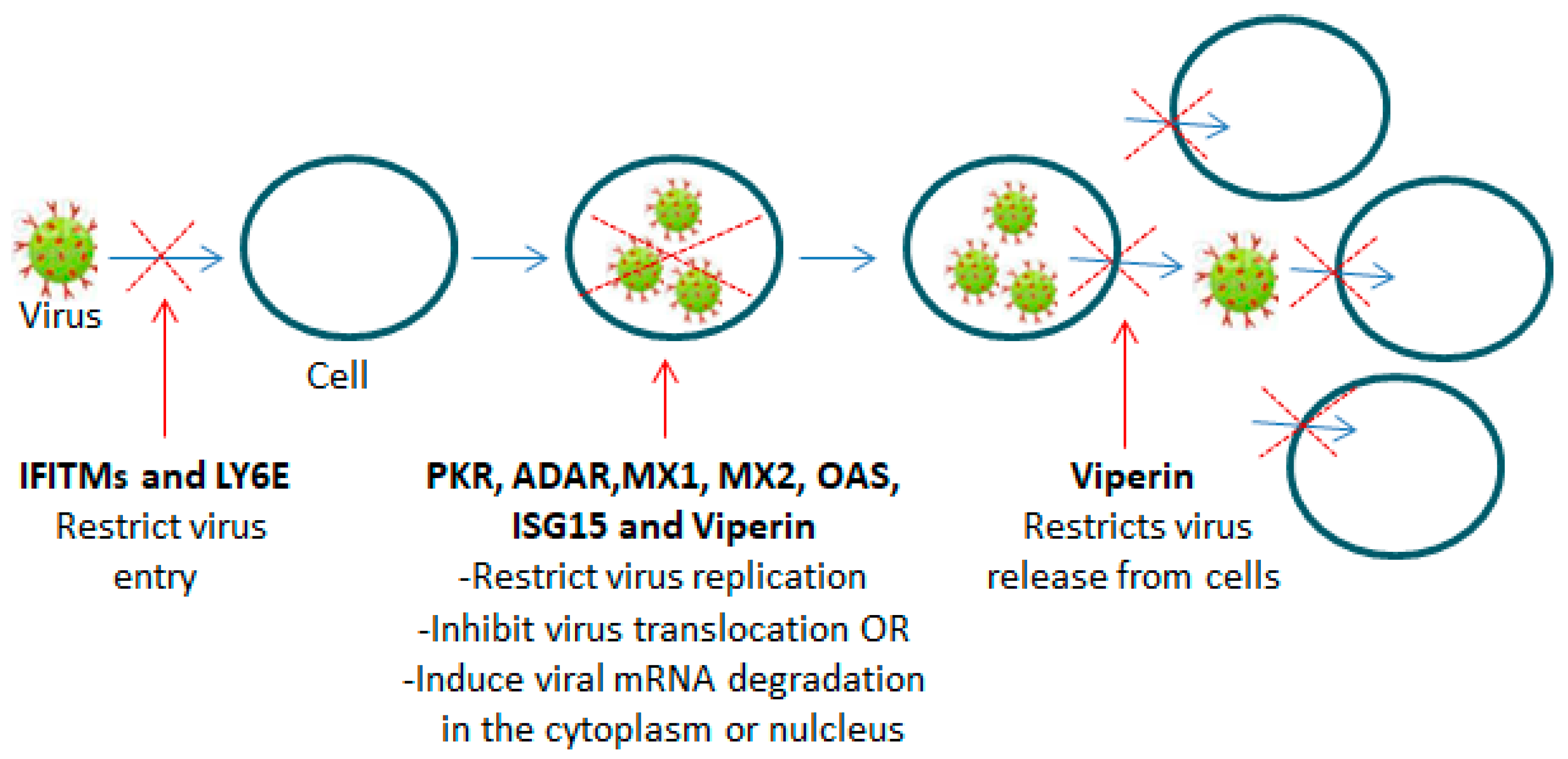
2.3. Interferon-Stimulated Genes (ISGs)
3. Potential Type 1 IFN Correlates of Vaccine Immune Response Efficacy and Safety That Might Be Used in the Future as Alternatives to Animal Models
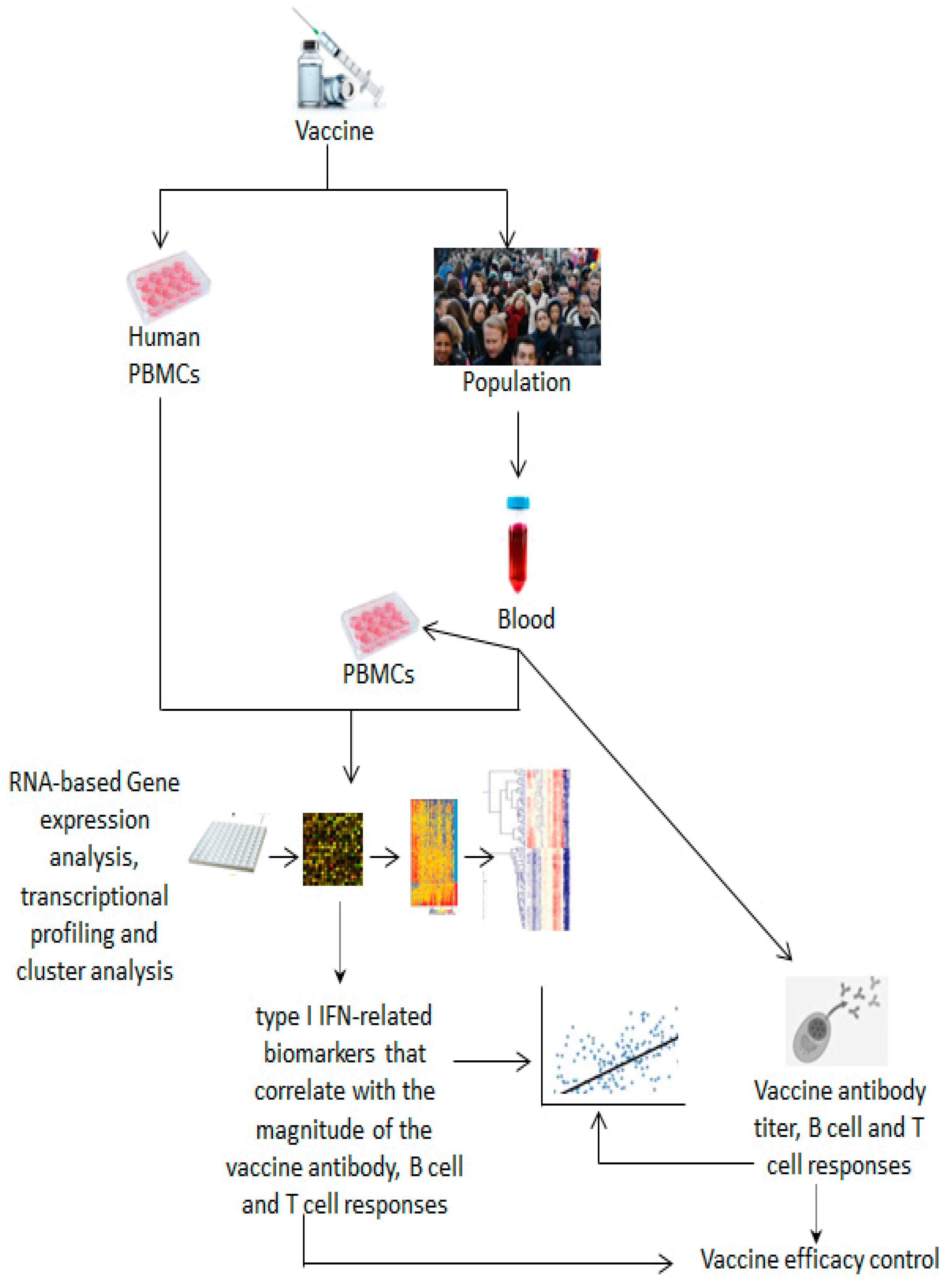
3.1. Methods, Assays and Systems Used to Identify Biomarkers for Vaccine Efficacy
3.2. Correlates of Vaccine Efficacy
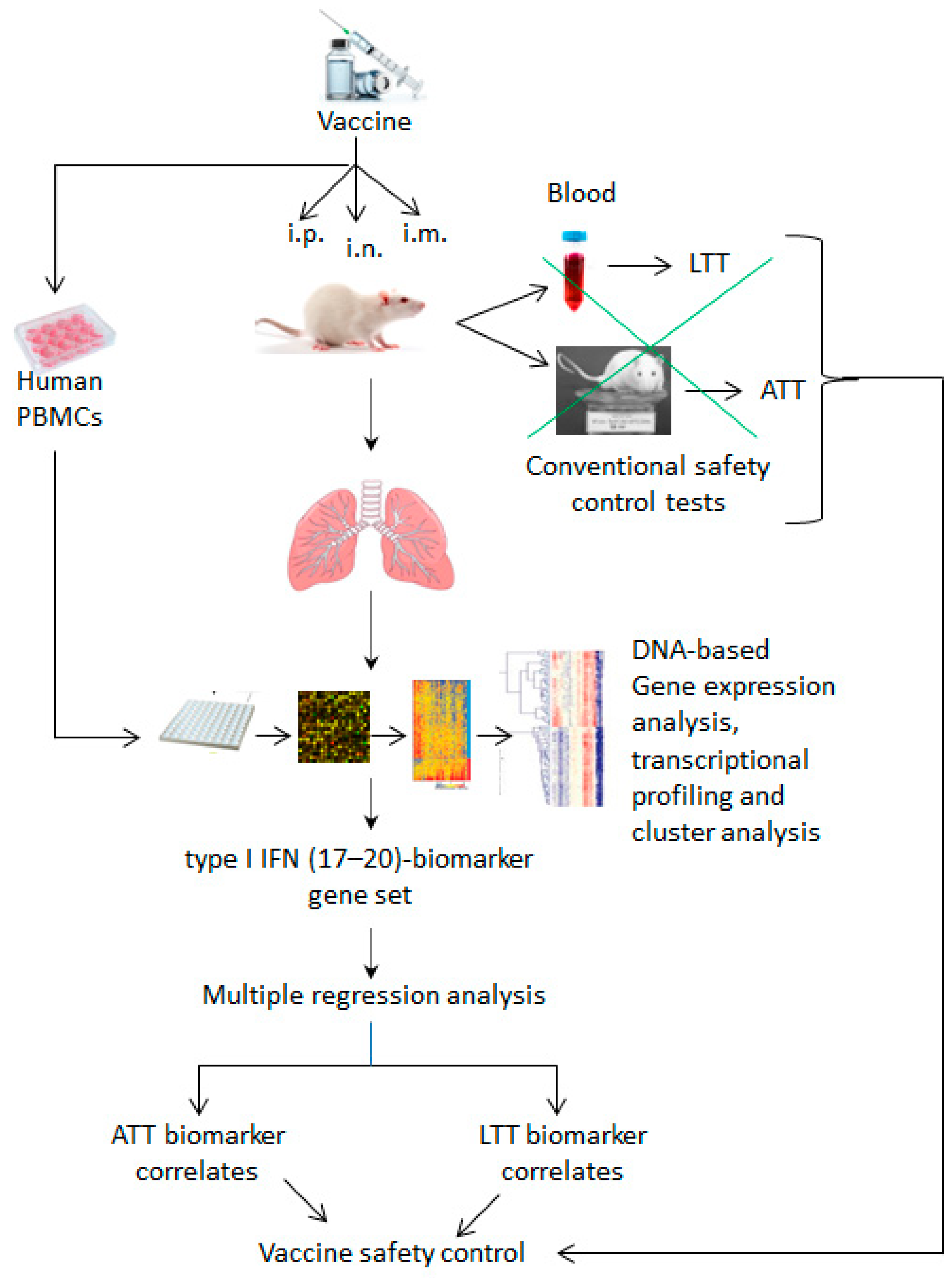
3.3. Methods, Assays and Systems Used to Identify Biomarkers for Vaccine Safety
3.4. Correlates of Vaccine Safety
4. Potential Limitations That May Hamper Vaccine Characterization Using Alternative Non-Animal Models
5. Discussion
- (1)
- Due to the dual effect of type I IFNs on the immune response, the reliability of the identified biomarkers in their ability to distinguish between vaccine efficacy and safety outcomes could be questionable.
- (2)
- Considering that type I IFN cellular sources, magnitude and timing of production, signaling and production of ISGs can differ in the host as well as in different infection models based on the infectious load and the route, type and stage of infection or vaccination, as well as host genetics and the immune-regulatory environmental background, type I IFN-related biomarkers associated with vaccine efficacy and safety may change as well.
- (3)
- Whereas the identified biomarkers may be applicable to vaccines belonging to the same vaccine type analyzed in this paper, whether these same biomarkers would be effective in predicting the immunogenicity and toxicity of vaccines belonging to other vaccine types, such as mRNA-based, vector-based or subunit-based vaccines, among others, remains unknown. The literature provides many examples of vaccines in which the production of type I IFNs is negligible or is exaggerated without benefit.
6. Conclusions and Perspectives
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; Del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed] [PubMed Central]
- Jameson, S.C.; Masopust, D. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? Reevaluating the Potential of Mouse Models for the Human Immune System. Cold Spring Harb. Perspect. Biol. 2018, 10, a029132. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lang, C.; Kolaj-Robin, O.; Cirefice, G.; Taconet, L.; Pel, E.; Jouette, S.; Buda, M.; Milne, C.; Charton, E. Replacement, Reduction, Refinement—Animal welfare progress in European Pharmacopoeia monographs: Activities of the European Pharmacopoeia Commission from 2007 to 2017. Pharmeur. Bio. Sci. Notes 2018, 2018, 12–36. [Google Scholar] [PubMed]
- Akkermans, A.; Chapsal, J.M.; Coccia, E.M.; Depraetere, H.; Dierick, J.F.; Duangkhae, P.; Goel, S.; Halder, M.; Hendriksen, C.; Levis, R.; et al. Animal testing for vaccines. Implementing replacement, reduction and refinement: Challenges and priorities. Biologicals 2020, 68, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Barrila, J.; Crabbé, A.; Yang, J.; Franco, K.; Nydam, S.D.; Forsyth, R.J.; Davis, R.R.; Gangaraju, S.; Ott, C.M.; Coyne, C.B.; et al. Modeling Host-Pathogen Interactions in the Context of the Microenvironment: Three-Dimensional Cell Culture Comes of Age. Infect. Immun. 2018, 86, e00282-18. [Google Scholar] [CrossRef] [PubMed]
- Varan, G.; Unal, S. Three-Dimensional Cell Culture Methods in Infectious Diseases and Vaccine Research. Future Pharmacol. 2023, 3, 48–60. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wagar, L.E.; DiFazio, R.M.; Davis, M.M. Advanced model systems and tools for basic and translational human immunology. Genome Med. 2018, 10, 73. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, I.M.; Bett, A.J.; Cristescu, R.; Loboda, A.; ter Meulen, J. Transcriptional profiling of vaccine-induced immune responses in humans and non-human primates. Microb. Biotechnol. 2012, 5, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Studholme, L.; Sutherland, J.; Desai, T.; Hockley, J.; Care, R.; Nordgren, I.K.; Vipond, C.; Collaborative Study Group. Evaluation of the monocyte activation test for the safety testing of meningococcal B vaccine Bexsero: A collaborative study. Vaccine 2019, 37, 3761–3769. [Google Scholar] [CrossRef] [PubMed]
- Shank-Retzlaff, M.; Wang, F.; Morley, T.; Anderson, C.; Hamm, M.; Brown, M.; Rowland, K.; Pancari, G.; Zorman, J.; Lowe, R.; et al. Correlation between mouse potency and in vitro relative potency for human papillomavirus Type 16 virus-like particles and Gardasil vaccine samples. Hum. Vaccines 2005, 1, 191–197. [Google Scholar] [CrossRef]
- Hamidi, A.; Kreeftenberg, H. Use of immuno assays during the development of a Hemophilus influenzae type b vaccine for technology transfer to emerging vaccine manufacturers. Hum. Vaccines Immunother. 2014, 10, 2697–2703. [Google Scholar] [CrossRef]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2009, 10, 116–125. [Google Scholar] [CrossRef]
- Nakaya, H.I.; Wrammert, J.; Lee, E.K.; Racioppi, L.; Marie-Kunze, S.; Haining, W.N.; Means, A.R.; Kasturi, S.P.; Khan, N.; Li, G.M.; et al. Systems biology of vaccination for seasonal influenza in humans. Nat. Immunol. 2011, 12, 786–795. [Google Scholar] [CrossRef]
- Gonçalves, E.; Bonduelle, O.; Soria, A.; Loulergue, P.; Rousseau, A.; Cachanado, M.; Bonnabau, H.; Thiebaut, R.; Tchitchek, N.; Behillil, S.; et al. Innate gene signature distinguishes humoral versus cytotoxic responses to influenza vaccination. J. Clin. Investig. 2019, 129, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Bucasas, K.L.; Franco, L.M.; Shaw, C.A.; Bray, M.S.; Wells, J.M.; Niño, D.; Arden, N.; Quarles, J.M.; Couch, R.B.; Belmont, J.W. Early patterns of gene expression correlate with the humoral immune response to influenza vaccination in humans. J. Infect. Dis. 2011, 203, 921–929. [Google Scholar] [CrossRef]
- Obermoser, G.; Presnell, S.; Domico, K.; Xu, H.; Wang, Y.; Anguiano, E.; Thompson-Snipes, L.; Ranganathan, R.; Zeitner, B.; Bjork, A.; et al. Systems scale interactive exploration reveals quantitative and qualitative differences in response to influenza and pneumococcal vaccines. Immunity 2013, 38, 831–844. [Google Scholar] [CrossRef]
- Li, S.; Sullivan, N.L.; Rouphael, N.; Yu, T.; Banton, S.; Maddur, M.S.; McCausland, M.; Chiu, C.; Canniff, J.; Dubey, S.; et al. Metabolic Phenotypes of Response to Vaccination in Humans. Cell 2017, 169, 862–877.e17. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, D.; Therrien, R.; Kettaf, N.; Angermann, B.R.; Boucher, G.; Filali-Mouhim, A.; Moser, J.M.; Mehta, R.S.; Drake, D.R., 3rd; Castro, E.; et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 2008, 205, 3119–3131. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zhang, J.; Wu, X.; Mao, Q.; Chen, P.; Zhu, F.; Xu, M.; Kong, W.; Liang, Z.; Wang, J. Comparing the Primary and Recall Immune Response Induced by a New EV71 Vaccine Using Systems Biology Approaches. PLoS ONE 2015, 10, e0140515. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, I.; Imai, J.; Momose, H.; Kawamura, M.; Mizukami, T.; Naito, S.; Maeyama, J.; Masumi, A.; Kuramitsu, M.; Takizawa, K.; et al. Application of quantitative gene expression analysis for pertussis vaccine safety control. Vaccine 2008, 26, 4686–4696. [Google Scholar] [CrossRef]
- Mizukami, T.; Imai, J.; Hamaguchi, I.; Kawamura, M.; Momose, H.; Naito, S.; Maeyama, J.; Masumi, A.; Kuramitsu, M.; Takizawa, K.; et al. Application of DNA microarray technology to influenza A/Vietnam/1194/2004 (H5N1) vaccine safety evaluation. Vaccine 2004, 26, 2270–2283. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, T.; Momose, H.; Kuramitsu, M.; Takizawa, K.; Araki, K.; Furuhata, K.; Ishii, K.J.; Hamaguchi, I.; Yamaguchi, K. System vaccinology for the evaluation of influenza vaccine safety by multiplex gene detection of novel biomarkers in a preclinical study and batch release test. PLoS ONE 2014, 9, e101835. [Google Scholar] [CrossRef] [PubMed]
- Momose, H.; Mizukami, T.; Kuramitsu, M.; Takizawa, K.; Masumi, A.; Araki, K.; Furuhata, K.; Yamaguchi, K.; Hamaguchi, I. Establishment of a new quality control and vaccine safety test for influenza vaccines and adjuvants using gene expression profiling. PLoS ONE 2015, 10, e0124392. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Momose, H.; Hiradate, Y.; Furuhata, K.; Takai, M.; Kamachi, K.; Asanuma, H.; Ishii, K.J.; Mizukami, T.; Hamaguchi, I. Evaluation of marker gene expression as a potential predictive marker of leukopenic toxicity for inactivated influenza vaccines. Biologicals 2017, 50, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Kuramitsu, M.; Momose, H.; Kobiyama, K.; Aoshi, T.; Yamada, H.; Ishii, K.J.; Mizukami, T.; Hamaguchi, I. A novel vaccinological evaluation of intranasal vaccine and adjuvant safety for preclinical tests. Vaccine 2017, 35, 821–830. [Google Scholar] [CrossRef]
- Sasaki, E.; Momose, H.; Hiradate, Y.; Furuhata, K.; Takai, M.; Asanuma, H.; Ishii, K.J.; Mizukami, T.; Hamaguchi, I. Modeling for influenza vaccines and adjuvants profile for safety prediction system using gene expression profiling and statistical tools. PLoS ONE 2018, 13, e0191896. [Google Scholar] [CrossRef]
- Cao, R.G.; Suarez, N.M.; Obermoser, G.; Lopez, S.M.; Flano, E.; Mertz, S.E.; Albrecht, R.A.; García-Sastre, A.; Mejias, A.; Xu, H.; et al. Differences in antibody responses between trivalent inactivated influenza vaccine and live attenuated influenza vaccine correlate with the kinetics and magnitude of interferon signaling in children. J. Infect. Dis. 2014, 210, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Chan, K.R.; Chua, C.J.; Nur Hazirah, S.; Ghosh, S.; Ooi, E.E.; Low, J.G. Early molecular correlates of adverse events following yellow fever vaccination. JCI Insight 2017, 2, e96031. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fratzke, A.P.; van Schaik, E.J.; Samuel, J.E. Immunogenicity and Reactogenicity in Q Fever Vaccine Development. Front. Immunol. 2022, 13, 886810. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ato, M.; Takahashi, Y.; Fujii, H.; Hashimoto, S.; Kaji, T.; Itamura, S.; Horiuchi, Y.; Arakawa, Y.; Tashiro, M.; Takemori, T. Influenza A whole virion vaccine induces a rapid reduction of peripheral blood leukocytes via interferon-α-dependent apoptosis. Vaccine 2013, 31, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stifter, S.A.; Feng, C.G. Interfering with immunity: Detrimental role of type I IFNs during infection. J. Immunol. 2015, 194, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Detournay, O.; Morrison, D.A.; Wagner, B.; Zarnegar, B.; Wattrang, E. Genomic analysis and mRNA expression of equine type I interferon genes. J. Interferon Cytokine Res. 2013, 33, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Gan, Z.; Hou, J.; Yang, Y.C.; Huang, L.; Huang, B.; Wang, S.; Nie, P. Identification and establishment of type IV interferon and the characterization of interferon-υ including its class II cytokine receptors IFN-υR1 and IL-10R2. Nat. Commun. 2022, 13, 999. [Google Scholar] [CrossRef] [PubMed]
- Björck, P.; Leong, H.X.; Engleman, E.G. Plasmacytoid dendritic cell dichotomy: Identification of IFN-α producing cells as a phenotypically and functionally distinct subset. J. Immunol. 2011, 186, 1477–1485. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mann-Nüttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Swiecki, M.; Colonna, M. Type I interferons: Diversity of sources, production pathways and effects on immune responses. Curr. Opin. Virol. 2011, 1, 463–475. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Melero, I.; Quetglas, J.I.; Reboredo, M.; Dubrot, J.; Rodriguez-Madoz, J.R.; Mancheño, U.; Casales, E.; Riezu-Boj, J.I.; Ruiz-Guillen, M.; Ochoa, M.C.; et al. Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res. 2015, 75, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Antonenko, S.; Lau, J.Y.; Liu, Y.J. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 2000, 192, 219–226. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F. Type I interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk. Lymphoma 2004, 45, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Kappler, J.; Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med. 1999, 189, 521–530. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chill, J.H.; Quadt, S.R.; Levy, R.; Schreiber, G.; Anglister, J. The human type I interferon receptor: NMR structure reveals the molecular basis of ligand binding. Structure 2003, 11, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Locke, M.C.; Fox, L.E.; Dunlap, B.F.; Young, A.R.; Monte, K.; Lenschow, D.J. Interferon Alpha, but Not Interferon Beta, Acts Early to Control Chronic Chikungunya Virus Pathogenesis. J. Virol. 2022, 96, e0114321. [Google Scholar] [CrossRef]
- Cook, L.E.; Locke, M.C.; Young, A.R.; Monte, K.; Hedberg, M.L.; Shimak, R.M.; Sheehan, K.C.F.; Veis, D.J.; Diamond, M.S.; Lenschow, D.J. Distinct Roles of Interferon Alpha and Beta in Controlling Chikungunya Virus Replication and Modulating Neutrophil-Mediated Inflammation. J. Virol. 2019, 94, e00841-19. [Google Scholar] [CrossRef]
- Fox, L.E.; Locke, M.C.; Lenschow, D.J. Context Is Key: Delineating the Unique Functions of IFNα and IFNβ in Disease. Front. Immunol. 2020, 11, 606874. [Google Scholar] [CrossRef]
- Wong, M.T.; Chen, S.S. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, L.; Sandbulte, M.R.; Thomas, P.G.; Webby, R.J.; Homayouni, R.; Pfeffer, L.M. NFkappaB negatively regulates interferon-induced gene expression and anti-influenza activity. J. Biol. Chem. 2006, 281, 11678–11684. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360, Erratum in Immunity 2006, 25, 849. [Google Scholar] [CrossRef] [PubMed]
- Ciancanelli, M.J.; Huang, S.X.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef]
- Thomsen, M.M.; Jørgensen, S.E.; Gad, H.H.; Storgaard, M.; Gjedsted, J.; Christiansen, M.; Hartmann, R.; Mogensen, T.H. Defective interferon priming and impaired antiviral responses in a patient with an IRF7 variant and severe influenza. Med. Microbiol. Immunol. 2019, 208, 869–876. [Google Scholar] [CrossRef]
- Tailor, P.; Tamura, T.; Ozato, K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res. 2006, 16, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Marié, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef]
- Johnson, K.E.; Sylvester, P.A.; Jondle, C.N.; Aurubin, C.A.; Tarakanova, V.L. Interferon Regulatory Factor 3 Supports the Establishment of Chronic Gammaherpesvirus Infection in a Route- and Dose-Dependent Manner. J. Virol. 2021, 95, e02208-20. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Aurubin, C.A.; Jondle, C.N.; Lange, P.T.; Tarakanova, V.L. Interferon Regulatory Factor 7 Attenuates Chronic Gammaherpesvirus Infection. J. Virol. 2020, 94, e01554-20. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Saleiro, D.; Platanias, L.C. Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin. Immunol. 2019, 43, 101299. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Tsurumi, A.; Zhao, C.; Li, W.X. Canonical and non-canonical JAK/STAT transcriptional targets may be involved in distinct and overlapping cellular processes. BMC Genom. 2017, 18, 718. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Carayol, N.; Woodard, J.; Konieczna, I.; Majchrzak-Kita, B.; Jordan, A.; Sassano, A.; Eklund, E.A.; Fish, E.N.; Platanias, L.C. Role of Schlafen 2 (SLFN2) in the generation of interferon alpha-induced growth inhibitory responses. J. Biol. Chem. 2009, 284, 25051–25064. [Google Scholar] [CrossRef]
- Mavrommatis, E.; Fish, E.N.; Platanias, L.C. The schlafen family of proteins and their regulation by interferons. J. Interferon Cytokine Res. 2013, 33, 206–210. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [PubMed]
- Ketkar, H.; Harrison, A.G.; Graziano, V.R.; Geng, T.; Yang, L.; Vella, A.T.; Wang, P. UBX Domain Protein 6 Positively Regulates JAK-STAT1/2 Signaling. J. Immunol. 2021, 206, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Piganis, R.A.; De Weerd, N.A.; Gould, J.A.; Schindler, C.W.; Mansell, A.; Nicholson, S.E.; Hertzog, P.J. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J. Biol. Chem. 2011, 286, 33811–33818. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Cao, W.; Chen, S.; Tian, R.; Wang, L.; Liu, Q.; Zhang, L.; Wang, Z.; Zhao, M.; Lu, Q.; et al. TRIM10 binds to IFN-α/β receptor 1 to negatively regulate type I IFN signal transduction. Eur. J. Immunol. 2021, 51, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- François-Newton, V.; Magno de Freitas Almeida, G.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uzé, G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE 2011, 6, e22200. [Google Scholar] [CrossRef] [PubMed]
- Dagenais-Lussier, X.; Loucif, H.; Cadorel, H.; Blumberger, J.; Isnard, S.; Bego, M.G.; Cohen, É.A.; Routy, J.P.; van Grevenynghe, J.; Montreal Primary Infection Study Group. USP18 is a significant driver of memory CD4 T-cell reduced viability caused by type I IFN signaling during primary HIV-1 infection. PLoS Pathog. 2019, 15, e1008060. [Google Scholar] [CrossRef]
- Arslan, A.D.; Sassano, A.; Saleiro, D.; Lisowski, P.; Kosciuczuk, E.M.; Fischietti, M.; Eckerdt, F.; Fish, E.N.; Platanias, L.C. Human SLFN5 is a transcriptional co-repressor of STAT1-mediated interferon responses and promotes the malignant phenotype in glioblastoma. Oncogene 2017, 36, 6006–6019. [Google Scholar] [CrossRef]
- Fischietti, M.; Arslan, A.D.; Sassano, A.; Saleiro, D.; Majchrzak-Kita, B.; Ebine, K.; Munshi, H.G.; Fish, E.N.; Platanias, L.C. Slfn2 Regulates Type I Interferon Responses by Modulating the NF-κB Pathway. Mol. Cell. Biol. 2018, 38, e00053-18. [Google Scholar] [CrossRef]
- Zhang, J.; Shao, J.; Wu, X.; Mao, Q.; Wang, Y.; Gao, F.; Kong, W.; Liang, Z. Type I interferon related genes are common genes on the early stage after vaccination by meta-analysis of microarray data. Hum. Vaccines Immunother. 2015, 11, 739–745. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pfaller, C.K.; Li, Z.; George, C.X.; Samuel, C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011, 23, 573–582. [Google Scholar] [CrossRef] [PubMed]
- McAllister, C.S.; Samuel, C.E. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 2009, 284, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246, Erratum in Proc. Natl. Acad. Sci. USA 2019, 116, 12573. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meischel, T.; Fritzlar, S.; Villalon-Letelier, F.; Tessema, M.B.; Brooks, A.G.; Reading, P.C.; Londrigan, S.L. IFITM Proteins That Restrict the Early Stages of Respiratory Virus Infection Do Not Influence Late-Stage Replication. J. Virol. 2021, 95, e0083721. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Nie, Y.; Hammond, G.L.; Yang, J.H. Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J. Virol. 2007, 81, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485, Erratum in Nature 2015, 525, 144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liao, G.R.; Tseng, Y.Y.; Tseng, C.Y.; Lin, F.Y.; Yamada, Y.; Liu, H.P.; Kuan, C.Y.; Hsu, W.L. Adenosine Deaminase Acting on RNA 1 Associates with Orf Virus OV20.0 and Enhances Viral Replication. J. Virol. 2019, 93, e01912-18. [Google Scholar] [CrossRef]
- Zhou, S.; Yang, C.; Zhao, F.; Huang, Y.; Lin, Y.; Huang, C.; Ma, X.; Du, J.; Wang, Y.; Long, G.; et al. Double-stranded RNA deaminase ADAR1 promotes the Zika virus replication by inhibiting the activation of protein kinase PKR. J. Biol. Chem. 2019, 294, 18168–18180. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Steiner, F.; Pavlovic, J. Subcellular Localization of MxB Determines Its Antiviral Potential against Influenza A Virus. J. Virol. 2020, 94, e00125-20. [Google Scholar] [CrossRef]
- Fitzgerald, K.A. The interferon inducible gene: Viperin. J. Interferon Cytokine Res. 2011, 31, 131–135. [Google Scholar] [CrossRef]
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Överby, A.K. Viperin Restricts Zika Virus and Tick-Borne Encephalitis Virus Replication by Targeting NS3 for Proteasomal Degradation. J. Virol. 2018, 92, e02054-17, Erratum in J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, R.; Kurhade, C.; Gilthorpe, J.D.; Överby, A.K. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J. Neuroinflamm. 2018, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Laurent-Rolle, M.; Pawlak, J.B.; Xia, H.; Kunte, A.; Hee, J.S.; Lim, J.; Harris, L.D.; Wood, J.M.; Evans, G.B.; et al. Viperin triggers ribosome collision-dependent translation inhibition to restrict viral replication. Mol. Cell. 2022, 82, 1631–1642.e6. [Google Scholar] [CrossRef]
- Wang, X.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef]
- Nasr, N.; Maddocks, S.; Turville, S.G.; Harman, A.N.; Woolger, N.; Helbig, K.J.; Wilkinson, J.; Bye, C.R.; Wright, T.K.; Rambukwelle, D.; et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 2012, 120, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Nandi, S.; Lo, M.; Gope, A.; Chawla-Sarkar, M. Viperin, an IFN-Stimulated Protein, Delays Rotavirus Release by Inhibiting Non-Structural Protein 4 (NSP4)-Induced Intrinsic Apoptosis. Viruses 2021, 13, 1324. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Okumura, F.; Okumura, A.J.; Uematsu, K.; Hatakeyama, S.; Zhang, D.E.; Kamura, T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013, 288, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.X.; Yang, K.; Liu, X.; Liu, X.Y.; Wei, B.; Shan, Y.F.; Zhu, L.H.; Wang, C. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bogunovic, D.; Payelle-Brogard, B.; Francois-Newton, V.; Speer, S.D.; Yuan, C.; Volpi, S.; Li, Z.; Sanal, O.; Mansouri, D.; et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 2015, 517, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Codias, E.K.; Malek, T.R. Regulation of B lymphocyte responses to IL-4 and IFN-gamma by activation through Ly-6A/E molecules. J. Immunol. 1990, 144, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiu, C.; Zhu, L.; Huang, J.; Li, L.; Fu, W.; Zhang, L.; Wei, J.; Wang, Y.; Geng, Y.; et al. IFN-stimulated gene LY6E in monocytes regulates the CD14/TLR4 pathway but inadequately restrains the hyperactivation of monocytes during chronic HIV-1 infection. J. Immunol. 2014, 193, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liang, C.; Liu, S.L. Interferon-inducible LY6E Protein Promotes HIV-1 Infection. J. Biol. Chem. 2017, 292, 4674–4685. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liang, C.; Liu, S.L. CD4-Dependent Modulation of HIV-1 Entry by LY6E. J. Virol. 2019, 93, e01866-18. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.; Cusimano, G.; Mege, N.; Woloszczuk, K.; Konopka, E.; Bell, M.; Joyner, D.; Marcy, J.; Tardif, V.; Kutzler, M.A.; et al. Using the power of innate immunoprofiling to understand vaccine design, infection, and immunity. Hum. Vaccines Immunother. 2023, 19, 2267295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakaya, H.I.; Clutterbuck, E.; Kazmin, D.; Wang, L.; Cortese, M.; Bosinger, S.E.; Patel, N.B.; Zak, D.E.; Aderem, A.; Dong, T.; et al. Systems biology of immunity to MF59-adjuvanted versus nonadjuvanted trivalent seasonal influenza vaccines in early childhood. Proc. Natl. Acad. Sci. USA 2016, 113, 1853–1858. [Google Scholar] [CrossRef]
- Zhu, W.; Higgs, B.W.; Morehouse, C.; Streicher, K.; Ambrose, C.S.; Woo, J.; Kemble, G.W.; Jallal, B.; Yao, Y. A whole genome transcriptional analysis of the early immune response induced by live attenuated and inactivated influenza vaccines in young children. Vaccine 2010, 28, 2865–2876. [Google Scholar] [CrossRef]
- Franco, L.M.; Bucasas, K.L.; Wells, J.M.; Niño, D.; Wang, X.; Zapata, G.E.; Arden, N.; Renwick, A.; Yu, P.; Quarles, J.M.; et al. Integrative genomic analysis of the human immune response to influenza vaccination. Elife 2013, 2, e00299, Erratum in Elife 2016, 5, e18898. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Bhattacharya, D. Basics of memory B-cell responses: Lessons from and for the real world. Immunology 2019, 156, 120–129. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Martinez-Sobrido, L.; Choi, C.; Petroff, N.; García-Sastre, A.; Niewiesk, S.; Carsillo, T. Induction of type I interferon secretion through recombinant Newcastle disease virus expressing measles virus hemagglutinin stimulates antibody secretion in the presence of maternal antibodies. J. Virol. 2011, 85, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, I.; Imai, J.; Momose, H.; Kawamura, M.; Mizukami, T.; Kato, H.; Naito, S.; Maeyama, J.; Masumi, A.; Kuramitsu, M.; et al. Two vaccine toxicity-related genes Agp and Hpx could prove useful for pertussis vaccine safety control. Vaccine 2007, 25, 3355–3364. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Momose, H.; Hiradate, Y.; Furuhata, K.; Mizukami, T.; Hamaguchi, I. Development of a preclinical humanized mouse model to evaluate acute toxicity of an influenza vaccine. Oncotarget 2018, 9, 25751–25763. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Momose, H.; Hiradate, Y.; Ishii, K.J.; Mizukami, T.; Hamaguchi, I. In vitro marker gene expression analyses in human peripheral blood mononuclear cells: A tool to assess safety of influenza vaccines in humans. J. Immunotoxicol. 2018, 15, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Reimann, K.A.; Chernoff, M.; Wilkening, C.L.; Nickerson, C.E.; Landay, A.L. Preservation of lymphocyte immunophenotype and proliferative responses in cryopreserved peripheral blood mononuclear cells from human immunodeficiency virus type 1-infected donors: Implications for multicenter clinical trials. The ACTG Immunology Advanced Technology Laboratories. Clin. Diagn. Lab. Immunol. 2000, 7, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, H.I.; Pulendran, B. Systems vaccinology: Its promise and challenge for HIV vaccine development. Curr. Opin. HIV AIDS 2012, 7, 24–31. [Google Scholar] [CrossRef]
- Maertzdorf, J.; Kaufmann, S.H.; Weiner, J., 3rd. Molecular signatures for vaccine development. Vaccine 2015, 33, 5256–5261. [Google Scholar] [CrossRef]
- Flanagan, K.L.; Noho-Konteh, F.; Ghazal, P.; Dickinson, P. Transcriptional profiling technology for studying vaccine responses: An untapped goldmine. Methods 2013, 60, 269–274. [Google Scholar] [CrossRef] [PubMed]
- García-Piñeres, A.J.; Hildesheim, A.; Dodd, L.; Kemp, T.J.; Yang, J.; Fullmer, B.; Harro, C.; Lowy, D.R.; Lempicki, R.A.; Pinto, L.A. Gene expression patterns induced by HPV-16 L1 virus-like particles in leukocytes from vaccine recipients. J. Immunol. 2009, 182, 1706–1729. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, Y.; Cheng, Y.; Yan, W.; Nardini, C. Exploring the molecular causes of hepatitis B virus vaccination response: An approach with epigenomic and transcriptomic data. BMC Med. Genom. 2014, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.T.; Oberg, A.L.; Grill, D.E.; Ovsyannikova, I.G.; Haralambieva, I.H.; Kennedy, R.B.; Poland, G.A. System-Wide Associations between DNA-Methylation, Gene Expression, and Humoral Immune Response to Influenza Vaccination. PLoS ONE 2016, 11, e0152034. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon ?? in acute influenza infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, H.; Goldsmith, C.; Thawatsupha, P.; Chittaganpitch, M.; Waicharoen, S.; Zaki, S.; Tumpey, T.M.; Katz, J.M. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type i interferon response in polarized human bronchial epithelial cells. J. Virol. 2007, 81, 12439–12449. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dittmann, J.; Stertz, S.; Grimm, D.; Steel, J.; García-Sastre, A.; Haller, O.; Kochs, G. Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J. Virol. 2008, 82, 3624–3631. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zimmermann, P.; Mänz, B.; Haller, O.; Schwemmle, M.; Kochs, G. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J. Virol. 2011, 85, 8133–8140. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cilloniz, C.; Pantin-Jackwood, M.J.; Ni, C.; Carter, V.S.; Korth, M.J.; Swayne, D.E.; Tumpey, T.M.; Katze, M.G. Molecular signatures associated with Mx1-mediated resistance to highly pathogenic influenza virus infection: Mechanisms of survival. J. Virol. 2012, 86, 2437–2446. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jaguva Vasudevan, A.A.; Bähr, A.; Grothmann, R.; Singer, A.; Häussinger, D.; Zimmermann, A.; Münk, C. MXB inhibits murine cytomegalovirus. Virology 2018, 522, 158–167. [Google Scholar] [CrossRef]
- Moser, M.; Leo, O. Key concepts in immunology. Vaccine 2010, 28 (Suppl. 3), C2–C13. [Google Scholar] [CrossRef] [PubMed]
- Galen, J.E.; Curtiss, R., 3rd. The delicate balance in genetically engineering live vaccines. Vaccine 2014, 32, 4376–4385. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prymula, R.; Siegrist, C.A.; Chlibek, R.; Zemlickova, H.; Vackova, M.; Smetana, J.; Lommel, P.; Kaliskova, E.; Borys, D.; Schuerman, L. Effect of prophylactic paracetamol administration at time of vaccination on febrile reactions and antibody responses in children: Two open-label, randomised controlled trials. Lancet 2009, 374, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Judson, R.S.; Casey, W.M.; Kleinstreuer, N.C.; Thomas, R.S. Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model. Environ. Sci. Technol. 2015, 49, 8804–8814, Erratum in Environ. Sci. Technol. 2017, 51, 9415. [Google Scholar] [CrossRef] [PubMed]
- Schiffelers, M.J.; Blaauboer, B.J.; Hendriksen, C.F.; Bakker, W.E. Regulatory acceptance and use of 3R models: A multilevel perspective. ALTEX 2012, 29, 287–300. [Google Scholar] [CrossRef] [PubMed]
- WHO Expert Committee on Biological Standardization. 30th Report. General Considerations for combined vaccine. WHO Tech. Rep. Ser. 1979, 638, 100. [Google Scholar]
- Minimal Requirements for Biological Products, National Institute of Infectious Diseases Japan. 2006. Available online: http://www.nih.go.jp/niid/MRBP/index-e.html (accessed on 1 September 2023).
- Wijesundara, D.K.; Xi, Y.; Ranasinghe, C. Unraveling the convoluted biological roles of type I interferons in infection and immunity: A way forward for therapeutics and vaccine design. Front. Immunol. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Temizoz, B.; Ishii, K.J. Type I and II interferons toward ideal vaccine and immunotherapy. Expert Rev. Vaccines 2021, 20, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; Chung, Y.R.; Eitzinger, S.; Palacio, N.; Gregory, S.; Bhattacharyya, M.; Penaloza-MacMaster, P. TLR4 signaling improves PD-1 blockade therapy during chronic viral infection. PLoS Pathog. 2019, 15, e1007583. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palacio, N.; Dangi, T.; Chung, Y.R.; Wang, Y.; Loredo-Varela, J.L.; Zhang, Z.; Penaloza-MacMaster, P. Early type I IFN blockade improves the efficacy of viral vaccines. J. Exp. Med. 2020, 217, e20191220. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated Intravenous Delivery of Antigen mRNA Results in Strong Antigen-specific T-cell Responses Despite the Induction of Type I Interferon. Mol. Ther. Nucleic Acids. 2016, 5, e326. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van Hoecke, L.; Roose, K.; Ballegeer, M.; Zhong, Z.; Sanders, N.N.; De Koker, S.; Saelens, X.; Van Lint, S. The Opposing Effect of Type I IFN on the T Cell Response by Non-modified mRNA-Lipoplex Vaccines Is Determined by the Route of Administration. Mol. Ther. Nucleic Acids. 2020, 22, 373–381. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cook, I.F. Subcutaneous vaccine administration—An outmoded practice. Hum. Vaccines Immunother. 2021, 17, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Syenina, A.; Gan, E.S.; Toh, J.Z.N.; de Alwis, R.; Lin, L.Z.; Tham, C.Y.L.; Yee, J.X.; Leong, Y.S.; Sam, H.; Cheong, C.; et al. Adverse effects following anti-COVID-19 vaccination with mRNA-based BNT162b2 are alleviated by altering the route of administration and correlate with baseline enrichment of T and NK cell genes. PLoS Biol. 2022, 20, e3001643. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jewell, N.A.; Vaghefi, N.; Mertz, S.E.; Akter, P.; Peebles, R.S., Jr.; Bakaletz, L.O.; Durbin, R.K.; Flaño, E.; Durbin, J.E. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J. Virol. 2007, 81, 9790–9800. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gautier, G.; Humbert, M.; Deauvieau, F.; Scuiller, M.; Hiscott, J.; Bates, E.E.; Trinchieri, G.; Caux, C.; Garrone, P. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J. Exp. Med. 2005, 201, 1435–1446. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Byrnes, A.A.; Ma, X.; Cuomo, P.; Park, K.; Wahl, L.; Wolf, S.F.; Zhou, H.; Trinchieri, G.; Karp, C.L. Type I interferons and IL-12: Convergence and cross-regulation among mediators of cellular immunity. Eur. J. Immunol. 2001, 31, 2026–2034. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Johnson, P.L.; Nakaya, H.I.; Edupuganti, S.; Mulligan, M.J.; Lawson, B.; Miller, J.D.; Pulendran, B.; Antia, R.; Ahmed, R. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc. Natl. Acad. Sci. USA 2015, 112, 3050–3055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilson, E.B.; Kidani, Y.; Elsaesser, H.; Barnard, J.; Raff, L.; Karp, C.L.; Bensinger, S.; Brooks, D.G. Emergence of distinct multiarmed immunoregulatory antigen-presenting cells during persistent viral infection. Cell Host Microbe 2012, 11, 481–491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, M.S.; Park, C.H.; Jeong, Y.H.; Kim, Y.J.; Ha, S.J. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8⁺ T-cell exhaustion. PLoS Pathog. 2013, 9, e1003478. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murira, A.; Lamarre, A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, W.; Teo, J.M.N.; Yau, S.W.; Wong, M.Y.; Lok, C.N.; Che, C.M.; Javed, A.; Huang, Y.; Ma, S.; Ling, G.S. Chronic type I interferon signaling promotes lipid-peroxidation-driven terminal CD8+ T cell exhaustion and curtails anti-PD-1 efficacy. Cell Rep. 2022, 41, 111647. [Google Scholar] [CrossRef] [PubMed]
- Knuschke, T.; Bayer, W.; Rotan, O.; Sokolova, V.; Wadwa, M.; Kirschning, C.J.; Hansen, W.; Dittmer, U.; Epple, M.; Buer, J.; et al. Prophylactic and therapeutic vaccination with a nanoparticle-based peptide vaccine induces efficient protective immunity during acute and chronic retroviral infection. Nanomedicine 2014, 10, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Knuschke, T.; Rotan, O.; Bayer, W.; Sokolova, V.; Hansen, W.; Sparwasser, T.; Dittmer, U.; Epple, M.; Buer, J.; Westendorf, A.M. Combination of nanoparticle-based therapeutic vaccination and transient ablation of regulatory T cells enhances anti-viral immunity during chronic retroviral infection. Retrovirology 2016, 13, 24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Knuschke, T.; Rotan, O.; Bayer, W.; Kollenda, S.; Dickow, J.; Sutter, K.; Hansen, W.; Dittmer, U.; Lang, K.S.; Epple, M.; et al. Induction of Type I Interferons by Therapeutic Nanoparticle-Based Vaccination Is Indispensable to Reinforce Cytotoxic CD8+ T Cell Responses During Chronic Retroviral Infection. Front. Immunol. 2018, 9, 614. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhong, C.; Liu, F.; Hajnik, R.J.; Yao, L.; Chen, K.; Wang, M.; Liang, Y.; Sun, J.; Soong, L.; Hou, W.; et al. Type I Interferon Promotes Humoral Immunity in Viral Vector Vaccination. J. Virol. 2021, 95, e0092521. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, F.; Niu, Q.; Fan, X.; Liu, C.; Zhang, J.; Wei, Z.; Hou, W.; Kanneganti, T.D.; Robb, M.L.; Kim, J.H.; et al. Priming and Activation of Inflammasome by Canarypox Virus Vector ALVAC via the cGAS/IFI16-STING-Type I IFN Pathway and AIM2 Sensor. J. Immunol. 2017, 199, 3293–3305. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carroll, E.C.; Jin, L.; Mori, A.; Muñoz-Wolf, N.; Oleszycka, E.; Moran, H.B.T.; Mansouri, S.; McEntee, C.P.; Lambe, E.; Agger, E.M.; et al. The Vaccine Adjuvant Chitosan Promotes Cellular Immunity via DNA Sensor cGAS-STING-Dependent Induction of Type I Interferons. Immunity 2016, 44, 597–608. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pennock, N.D.; Kedl, J.D.; Kedl, R.M. T Cell Vaccinology: Beyond the Reflection of Infectious Responses. Trends Immunol. 2016, 37, 170–180. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tomasello, E.; Pollet, E.; Vu Manh, T.P.; Uzé, G.; Dalod, M. Harnessing Mechanistic Knowledge on Beneficial Versus Deleterious IFN-I Effects to Design Innovative Immunotherapies Targeting Cytokine Activity to Specific Cell Types. Front. Immunol. 2014, 5, 526. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hagan, T.; Gerritsen, B.; Tomalin, L.E.; Fourati, S.; Mulè, M.P.; Chawla, D.G.; Rychkov, D.; Henrich, E.; Miller, H.E.R.; Diray-Arce, J.; et al. Transcriptional atlas of the human immune response to 13 vaccines reveals a common predictor of vaccine-induced antibody responses. Nat. Immunol. 2022, 23, 1788–1798. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khalaj-Hedayati, A.; Moosavi, S.; Manta, O.; Helal, M.H.; Ibrahim, M.M.; El-Bahy, Z.M.; Supriyanto, G. Identification and In Silico Characterization of a Conserved Peptide on Influenza Hemagglutinin Protein: A New Potential Antigen for Universal Influenza Vaccine Development. Nanomaterials 2023, 13, 2796. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cevirgel, A.; Shetty, S.A.; Vos, M.; Nanlohy, N.M.; Beckers, L.; Bijvank, E.; Rots, N.; van Beek, J.; Buisman, A.M.; van Baarle, D. Pre-vaccination immunotypes reveal weak and robust antibody responders to influenza vaccination. Aging Cell 2024, 23, e14048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Irimia, D.; Wang, X. Inflammation-on-a-Chip: Probing the Immune System Ex Vivo. Trends Biotechnol. 2018, 36, 923–937. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goodwin, T.J.; McCarthy, M.; Cohrs, R.J.; Kaufer, B.B. 3D tissue-like assemblies: A novel approach to investigate virus-cell interactions. Methods 2015, 90, 76–84. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, T.; Cao, Y.; Zhao, P.; Shen, S.; Xi, Y. Organoid: A powerful tool to study lung regeneration and disease. Cell Regen. 2021, 10, 21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wagar, L.E.; Salahudeen, A.; Constantz, C.M.; Wendel, B.S.; Lyons, M.M.; Mallajosyula, V.; Jatt, L.P.; Adamska, J.Z.; Blum, L.K.; Gupta, N.; et al. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 2021, 27, 125–135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hui, K.P.Y.; Ching, R.H.H.; Chan, S.K.H.; Nicholls, J.M.; Sachs, N.; Clevers, H.; Peiris, J.S.M.; Chan, M.C.W. Tropism, replication competence, and innate immune responses of influenza virus: An analysis of human airway organoids and ex-vivo bronchus cultures. Lancet Respir. Med. 2018, 6, 846–854. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Models | Non-Animal Models |
|---|---|
| Time-consuming, expensive and laborious | Rapid, cheap and easy in terms of vaccine testing and production time and cost |
| Issues of precision, accuracy and sensitivity | Accurate, precise and sensitive |
| Poorly predictive of human responses | Highly predictive of responses, selective and specific for the target |
| High individual variability and low translatability to humans | Provide reproducible and robust results that can be readily applicable to humans |
| Issues related to supplying and maintaining animals | Practice for routine use and flexible |
| Not sufficiently validated | Methods are sufficiently validated and cross-validated |
| Procedures and treatments can cause animal suffering | The used procedures can reduce or replace animal use |
| Present genetic and environmental differences with humans as well as limitations in reflecting some severe adverse effects (AEs) caused by cytokine-release syndrome | - |
| Model | Analytical Method | Vaccine | Main Investigated Issue | Identified Type I IFN-Related Biomarkers | Timing Used for the Expression Analysis & Kinetics | Reference |
|---|---|---|---|---|---|---|
| Whole blood from 40 subjects vaccinated with the YF17D vaccine | Microarray gene expression analysis and profiling using the open source Bioconductor platform (using the R software package) and CRAN package fastICA | Live attenuated yellow fever vaccine (YF17D) | Identification of immune response signatures to yellow fever vaccine | OAS1, OAS2, OAS3, MX1, MX2, IFI27, IFI30, IFI16, STAT1 and IRF7 | Gene expression analysis was performed on the day of vaccination and on days 3, 7, 10, 14, 28 and 60 after vaccination. Overexpression peaked on day 7 after vaccination. | [22] |
| -PBMCs isolated from the blood of 15 young adult vaccinees (18–45 years old) -Human PBMCs stimulated for 3 h or 12 h with the YF17D vaccine | Microarray gene expression and gene ontology analysis and profiling using hierarchical clustering, the DAVID bioinformatics database and the ingenuity pathway analysis software database | Live attenuated yellow fever vaccine (YF17D) | Identification of early gene signatures that predict adaptive immune responses after vaccination with yellow fever vaccine | OAS1, OAS2, OAS3, OASL, MX1, IFIH1, IRF7, ISG15 and STAT1 | Gene expression analysis was performed on the day of vaccination and on days 1, 3, 7 and 21 after vaccination. Overexpression peaked on day 7 after vaccination. | [16] |
| PBMCs isolated from the blood of 19 vaccinated children (6–35 months old) | Microarray gene expression analysis and profiling using the DAVID bioinformatics database and ingenuity pathway analysis | Inactivated enterovirus type 71 (EV71) whole-virus vaccine | Comparison between primary and recall immune responses | IRF7, ISG15, MX1, IFI27 and IFIH1 | Gene expression analysis was performed on the day of vaccination and on days 56 and 180 after vaccination. | [23] |
| Whole blood from healthy adult vaccinees (18 to 64 years old) | Microarray gene expression analysis and profiling using hierarchical clustering and ingenuity pathway analysis | Trivalent inactivated 2009–2010 seasonal influenza vaccine and pneumococcal vaccine (polysaccharide extracts from 23 serotypes) | Comparison between transcriptional signatures of a seasonal influenza vaccine and a 23-valent pneumococcal vaccine in blood | OAS1, OAS2, OAS3, OASL, IF135, IRF7, IFIH1, IFIT1, IFIT2, IFIT3, IFITM1, IFITM3, MX1, IRF9, IFI44L, ISG15, IFI16, IFI44l and STAT1 (only influenza) | Gene expression analysis was performed on the day of vaccination and on days 1, 3, 7, 10, 14, 21 and 28 after vaccination. Overexpression peaked at 24 h after vaccination for both vaccines and, more consistently, at day 10 after vaccination for the pneumococcal vaccine. | [20] |
| PBMCs from the blood of 119 healthy male adult vaccinees (18–40 years old) | Microarray gene expression and gene ontology analysis and profiling using hierarchical clustering, the DAVID bioinformatics database and the ingenuity pathway analysis software database | Trivalent inactivated influenza vaccine (TIV) | Analysis of the relationship between gene expression patterns and humoral immune response to vaccination | STAT1, IF135, IRF7, IFIT1, MX1 and IRF9 | Gene expression analysis was performed on the day of vaccination and on days 1, 3 and 14 after vaccination. Overexpression occurred within 24 h of vaccination. | [19] |
| -PBMCs isolated from the blood of 56 young adult vaccinees (18–50 years old) -Human PBMCs stimulated for 24 h with the vaccine | Microarray gene expression analysis and profiling using the ingenuity pathway analysis software database | Live attenuated influenza vaccine (LAIV) and trivalent inactivated influenza vaccine (TIV) | Characterization of innate and adaptive responses to different influenza vaccines, with the aim of identifying early molecular signatures that predict the later immune responses | OAS1, OAS2, OAS3, MX1, Mx2, IRF7 and STAT1 (only LAIV) | Gene expression analysis was performed before vaccination and on days 3 and 7 after vaccination. Overexpression peaked on day 3 after vaccination. | [17] |
| Blood from 90 children and adults (14–24 months old) vaccinated with the influenza vaccine | Microarray gene expression analysis and profiling using hierarchical clustering | Trivalent inactivated influenza vaccine (TIV) with and without the MF59 adjuvant | To show the potential utility of using systems approaches to delineate mechanisms of vaccine immunity and identify correlates of vaccine immunity in children | OAS1, OAS3, IFIT1, IRF7 and IFIH1 | Gene expression analysis was performed before vaccination and on days 1, 3, 7 and 28 after vaccination. | [109] |
| Whole blood from 85 infants (12–35 months old) vaccinated with influenza vaccine by intranasal (LAIV) or intramuscular (TIV) injection | Microarray gene expression analysis, ontology analysis and profiling by using hierarchical clustering (using the R package pvclust) and the ingenuity pathway analysis software database | Live attenuated influenza vaccine (LAIV) and trivalent inactivated influenza vaccine (TIV) | Identification of significantly differentially expressed genes after vaccination with LAIV and TIV influenza vaccines | IRF7, IFIT1, IFIT2, IFIT3, OAS1, OAS2, OAS3, MX1, MX2 and IFI44 (only LAIV) | Gene expression analysis was performed before vaccination and on days 7 to 10 after vaccination. Overexpression peaked on days 7–10 after vaccination. | [110] |
| Whole blood from 37 vaccinated infants and children (6 months to 14 years old) | Microarray gene expression analysis and profiling using GeneSpring GX 7.3 software and hierarchical clustering | Trivalent inactivated influenza vaccine (TIV) or live attenuated influenza vaccine (LAIV) | Comparing parameters of immune responses elicited by TIV and LAIV vaccines in children to determine whether the early changes in the expression of certain immune related genes correlates with the antibody responses | OASL, OAS3, IFIT1, IFIT3, IFI44L and ISG15 | Gene expression analysis was performed before vaccination and on days 1, 7 and 30 after vaccination. Overexpression occurred on day 1 after vaccination. | [31] |
| Whole blood and serum from 60 healthy adults (18–45 years old) enrolled in a randomized phase I/II clinical trial and vaccinated by transcutaneous, intradermal or intramuscular routes | Microarray gene expression analysis and profiling by hierarchical clustering, ingenuity pathway analysis and logistic regression analysis | Trivalent influenza vaccine (TIV, season 2012–2013, 1:1:1 ratio) | Comparison of the immunogenicity of a seasonal influenza vaccine after administration by different novel immunization routes to discover early biomarkers of the immune response and explore any possible influence of the administration routes | STAT1, IRF9, IFI35 and IRF7 | Gene expression analysis was performed before vaccination and on day 1 after vaccination. Overexpression peaked at 24 h after vaccination. | [18] |
| Blood from 119 healthy male adult vaccinees (18–40 years old) | Microarray gene expression analysis and integrative genomic analysis and profiling using the DAVID bioinformatics database and ingenuity pathway analysis | Trivalent influenza vaccine (TIV) | Identification of genetic factors that influence responsiveness and immunity to a seasonal influenza vaccine in healthy adults | OAS1 | Gene expression analysis was performed before vaccination and on days 1, 3 and 14 after vaccination | [111] |
| Model | Analytical Method | Vaccine | Main Investigated Issue | Conventional Animal Tests | Identified Type I IFN-Related Biomarkers | Reference |
|---|---|---|---|---|---|---|
| Lungs isolated from rats intraperitoneally injected with the pertussis vaccine | DNA microarray gene expression analysis, QuantiGene Plex (QGP) assay and profiling by two-dimensional hierarchical clustering | Pertussis vaccine and toxicity reference pertussis vaccine (whole-cell inactivated pertussis vaccine) | Identification of toxicity-related biomarkers of the pertussis vaccine | ATT | MX2, IRF7 and IFI27L | [24] |
| Lungs isolated from rats intraperitoneally injected with the influenza vaccine | DNA microarray gene expression analysis and profiling by hierarchical cluster analysis | Inactivated monovalent A/H5N1 whole-virion influenza vaccine adjuvanted with aluminum hydroxide (PDv), inactivated whole trivalent influenza vaccine (WPv) and trivalent HA influenza vaccine (HAv) | Comparison between quality of different influenza vaccines (PDv, WPv and HAv) | ATT and LTT | MX1, MX2, IRF7, IFI47, IFRD1 and FLN29 | [25] |
| Lungs isolated from rats intraperitoneally injected with the influenza vaccine | QuantiGene Plex (QGP) technology and hybridization with branched DNA (bDNA) amplifier combined with multi-analyte magnetic beads and profiling by hierarchical cluster analysis | Inactivated monovalent A/H5N1 whole-virion influenza vaccine adjuvanted with aluminum hydroxide (PDv), inactivated whole trivalent influenza vaccine (WPv) and trivalent HA influenza vaccine (HAV) | Comparison between the quality of influenza vaccine batches from different manufacturers | ATT | MX1, IRF7, IFI47, TRAFD1 and IFRD1 | [26] |
| Lungs isolated from rats intraperitoneally injected with the influenza vaccine | QuantiGene Plex (QGP) technology and hybridization with branched DNA (bDNA) amplifier combined with multi-analyte magnetic beads and profiling by hierarchical cluster analysis | Inactivated influenza virus vaccine used as a toxicity reference vaccine and concentrated bulk materials of influenza HA vaccine (HA-A to HA-D) | Comparison between biomarker evaluation and animal testing for dose-dependent behavior to show that animal testing does not produce dose-dependent results in serially diluted bulk materials of influenza HA vaccines | ATT and LTT | MX2, IRF7, IFI47 and TRAFD1 | [27] |
| Lungs isolated from mice injected with the influenza vaccine | QuantiGene Plex (QGP) assay and multiple regression analysis | Toxicity reference influenza vaccine (whole-virion inactivated influenza virus) and influenza hemagglutinin split vaccine (HAv) | Selection of potential biomarkers related to leukocyte reduction from the set of biomarkers | LTT | MX2, TRAFD1, IRF7, IFI47 and IFRD1 | [28] |
| Lungs isolated from mice after intranasal administration of the influenza vaccine | QuantiGene Plex (QGP) assay and multiple regression analysis | Toxicity reference influenza vaccine (whole-virion inactivated influenza virus) and influenza hemagglutinin split vaccine (HAv) | To demonstrate the utility of the biomarker gene set for the safety evaluation of the nasal route for influenza vaccine administration | ATT and LTT | MX2, IRF7, IFI47, TRAFD1 and IFRD1 | [29] |
| Lungs isolated from vaccine-treated mice after intramuscular, intraperitoneal and nasal administration | QuantiGene Plex (QGP) assay, logistic regression analyses and profiling by hierarchical clustering analyses | Subvirion influenza vaccine (HAv), poly I:C adjuvanted hemagglutinin split vaccine HAv, AddaVax™-adjuvanted HAv (squalene-based oil-in-water emulsion adjuvant similar to MF59, which is a safe and potent adjuvant for the use with human vaccines), non-adjuvanted hemagglutinin split vaccine and toxicity reference vaccine (whole-virion inactivated influenza vaccine) | To develop a practical system for the safety assessment of different vaccine and adjuvant inoculation routes | ATT and LTT | TRAFD1, IRF7, MX2 and IFI47 | [30] |
| Lungs isolated from humanized mice (NOG mice are intravenously engrafted with human PBMCs) injected with the influenza vaccine | QuantiGene Plex (QGP) assay, flow cytometric analysis and multiple regression analysis | Hemagglutinin split vaccine (HAv) and toxicity reference influenza vaccine (whole-virion inactivated influenza vaccine) | To develop a novel humanized mouse model retaining human innate immunity-related cells to assess the safety of influenza vaccines using the (17–20) biomarker gene set | LTT | MX2 and TRAFD1 (not significant) | [116] |
| Donor-derived human PBMCs stimulated for 16 h with different doses of influenza vaccine | QuantiGene Plex (QGP) assay, flow cytometric analysis and multiple regression analysis | Hemagglutinin split vaccine (HAv) and toxicity reference influenza vaccine (whole-virion inactivated influenza vaccine) | To confirm the utility of the (17–20)-biomarker gene set for the safety evaluation of vaccines in humans and to develop a novel in vitro system for the safety evaluation of vaccines using human samples | - | MX2, IRF7, TRAFD1 and TRAFD1 | [117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Haq, H. Feasibility of Using a Type I IFN-Based Non-Animal Approach to Predict Vaccine Efficacy and Safety Profiles. Vaccines 2024, 12, 583. https://doi.org/10.3390/vaccines12060583
Abdel-Haq H. Feasibility of Using a Type I IFN-Based Non-Animal Approach to Predict Vaccine Efficacy and Safety Profiles. Vaccines. 2024; 12(6):583. https://doi.org/10.3390/vaccines12060583
Chicago/Turabian StyleAbdel-Haq, Hanin. 2024. "Feasibility of Using a Type I IFN-Based Non-Animal Approach to Predict Vaccine Efficacy and Safety Profiles" Vaccines 12, no. 6: 583. https://doi.org/10.3390/vaccines12060583
APA StyleAbdel-Haq, H. (2024). Feasibility of Using a Type I IFN-Based Non-Animal Approach to Predict Vaccine Efficacy and Safety Profiles. Vaccines, 12(6), 583. https://doi.org/10.3390/vaccines12060583