
Nano-Pulse Treatment Overcomes the Immunosuppressive Tumor Microenvironment to Elicit In Situ Vaccination Protection against Breast Cancer
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Cell Lines
2.2. In Vivo Nano-Pulse Treatment and the Secondary Live Tumor Challenge
2.3. Tissue Harvesting and Processing for the Analysis of Immune Cells
2.4. Preparation for Single-Cell Suspensions
2.5. Flow Cytometry
2.6. In Vitro Treg Suppression Assay
2.7. Statistical Analysis
3. Results
3.1. NPT Elicits Strong In Situ Vaccination (ISV) Protection
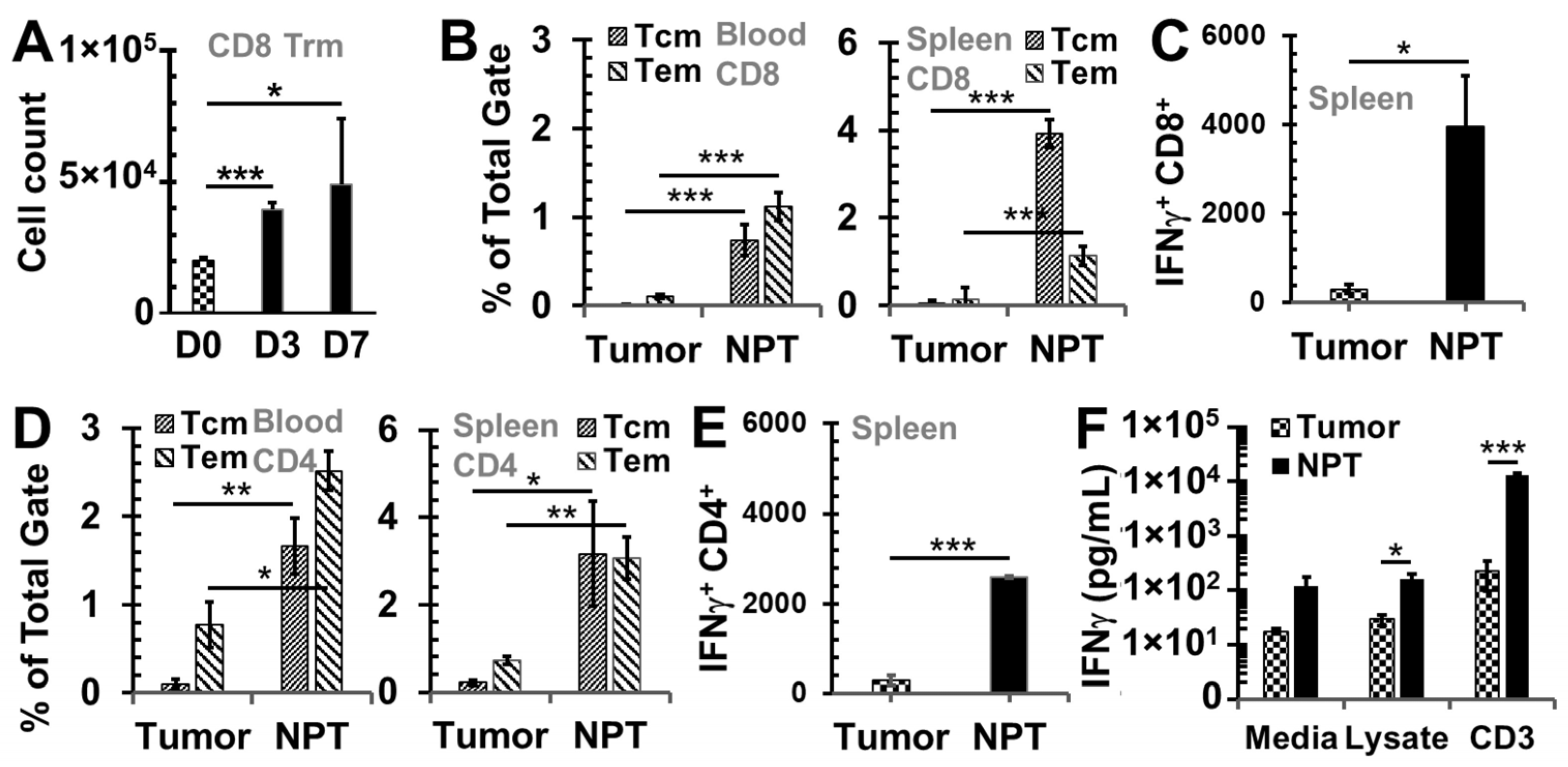
3.2. NPT Induces Antitumor Immune Memory Responses
3.3. NPT Overturns the Treg Dominance in the TME and Systemically
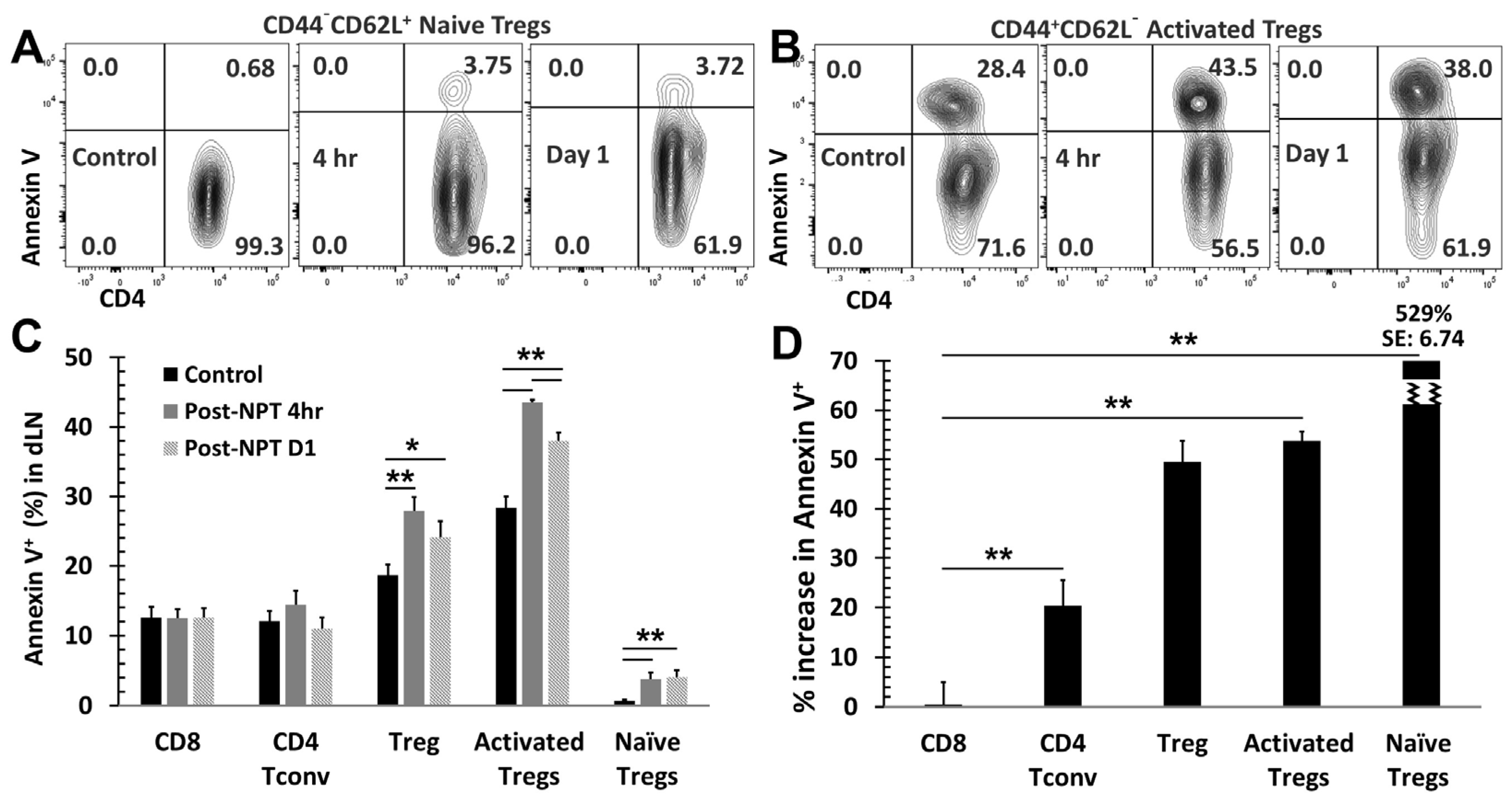
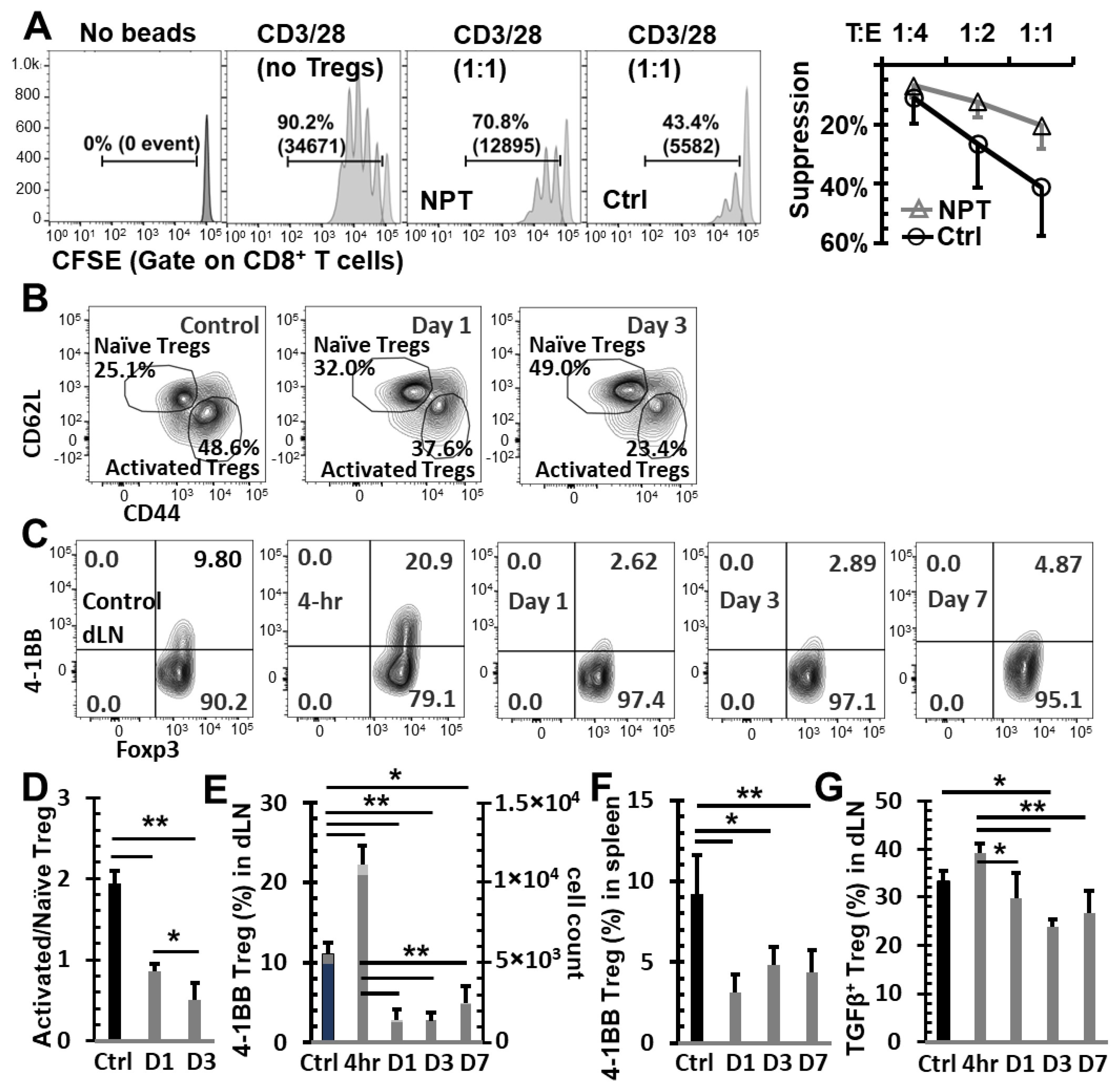
3.4. NPT Selectively Eradicates Tregs but Spares CD8 and CD4 Tconv Cells
3.5. NPT Reduces Treg Suppression Capacity
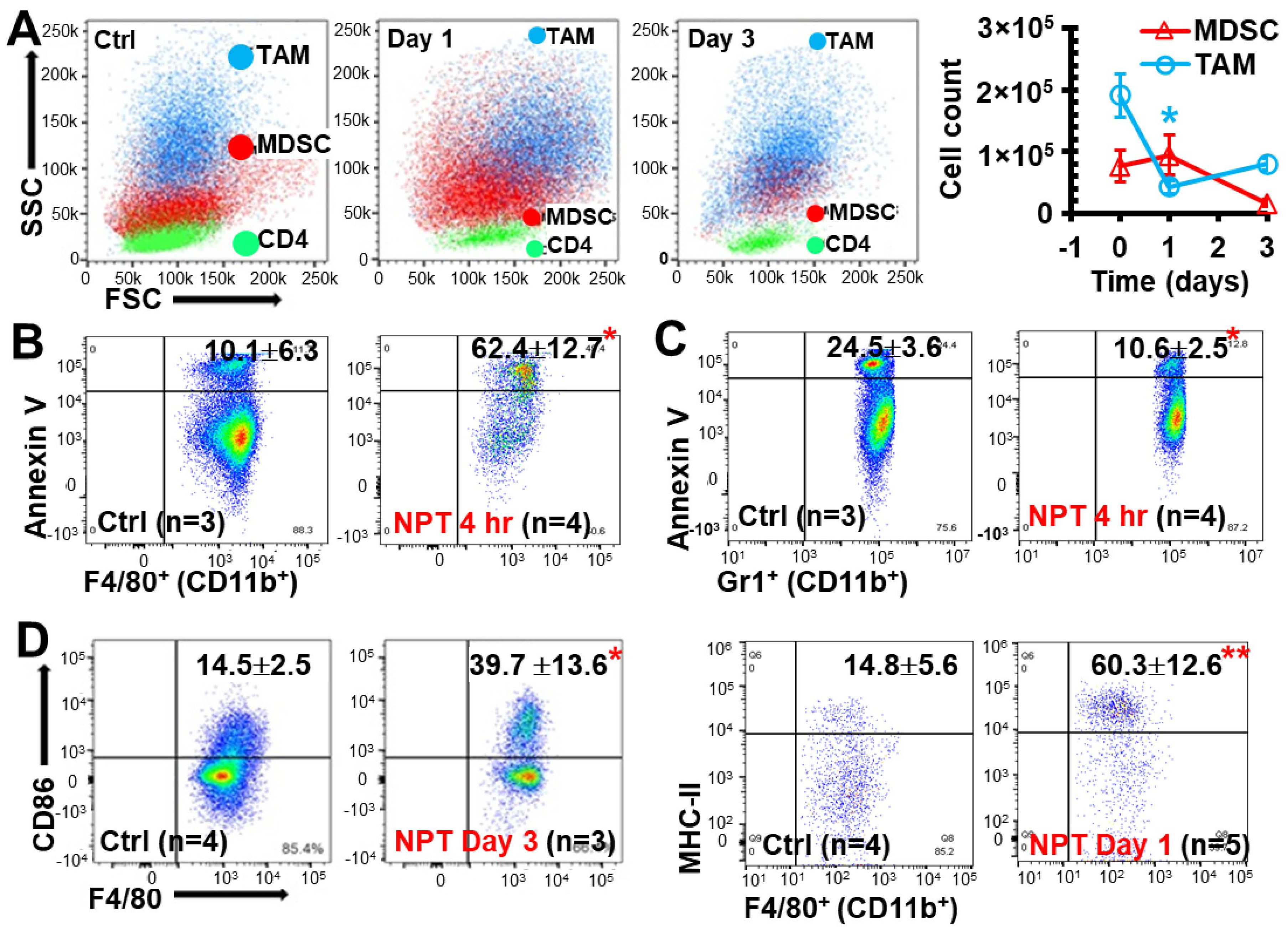
3.6. NPT Decreases MDSCs and TAMs with Distinctive Dynamics and Mechanisms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, C.A.; Nanda, R. Immune Checkpoint Inhibitor Therapy in Breast Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Luen, S.J.; Savas, P.; Salgado, R.; Loi, S. Checkpoint blockade in the treatment of breast cancer: Current status and future directions. Br. J. Cancer 2018, 119, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Liu, Y.; Lofland, D.; Lin, J. Breast Cancer Tumor Microenvironment and Molecular Aberrations Hijack Tumoricidal Immunity. Cancers 2022, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- Retecki, K.; Seweryn, M.; Graczyk-Jarzynka, A.; Bajor, M. The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance. Cancers 2021, 13, 6012. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Veena, M.S.; Shin, D.S. Key Players of the Immunosuppressive Tumor Microenvironment and Emerging Therapeutic Strategies. Front. Cell Dev. Biol. 2022, 10, 830208. [Google Scholar] [CrossRef] [PubMed]
- Barnestein, R.; Galland, L.; Kalfeist, L.; Ghiringhelli, F.; Ladoire, S.; Limagne, E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. Oncoimmunology 2022, 11, 2120676. [Google Scholar] [CrossRef] [PubMed]
- Prehn, R.T.; Main, J.M. Immunity to methylcholanthrene-induced sarcomas. J. Natl. Cancer Inst. 1957, 18, 769–778. [Google Scholar] [PubMed]
- Old, L.J.; Clarke, D.A.; Benacerraf, B. Effect of Bacillus Calmette-Guerin infection on transplanted tumours in the mouse. Nature 1959, 184 (Suppl. S5), 291–292. [Google Scholar] [CrossRef] [PubMed]
- Sheen, M.R.; Fiering, S. In situ vaccination: Harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1524. [Google Scholar] [CrossRef] [PubMed]
- Sersa, G.; Teissie, J.; Cemazar, M.; Signori, E.; Kamensek, U.; Marshall, G.; Miklavcic, D. Electrochemotherapy of tumors as in situ vaccination boosted by immunogene electrotransfer. Cancer Immunol. Immun. 2015, 64, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Bernat, C.; Al-Sakere, B.; Ghiringhelli, F.; Opolon, P.; Carpentier, A.F.; Zitvogel, L.; Mir, L.M.; Robert, C. Tumor destruction using electrochemotherapy followed by CpG oligodeoxynucleotide injection induces distant tumor responses. Cancer Immunol. Immunother. 2008, 57, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Marrero, B.; Shirley, S.; Heller, R. Delivery of interleukin-15 to B16 melanoma by electroporation leads to tumor regression and long-term survival. Technol. Cancer Res. Treat. 2014, 13, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.L.; Heller, R. IL-12 gene therapy using an electrically mediated nonviral approach reduces metastatic growth of melanoma. DNA Cell Biol. 2003, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Jing, Y.; Burcus, N.I.; Lassiter, B.P.; Tanaz, R.; Heller, R.; Beebe, S.J. Nano-pulse stimulation induces potent immune responses, eradicating local breast cancer while reducing distant metastases. Int. J. Cancer 2018, 142, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Sain, N.M.; Harlow, K.T.; Chen, Y.J.; Shires, P.K.; Heller, R.; Beebe, S.J. A protective effect after clearance of orthotopic rat hepatocellular carcinoma by nanosecond pulsed electric fields. Eur. J. Cancer 2014, 50, 2705–2713. [Google Scholar] [CrossRef]
- Guo, S.; Burcus, N.I.; Edelblute, C.M.; Hornef, J.; Jiang, C.; Schoenbach, K.; Heller, R.; Beebe, S.J. Enhanced electric pulse technology for the ablation of pancreatic cancer. In Advances in Pancreatic Cancer; Rodrigo, L., Ed.; Intech: Rijeka, Croatia, 2018. [Google Scholar]
- Toraya-Brown, S.; Sheen, M.R.; Zhang, P.; Chen, L.; Baird, J.R.; Demidenko, E.; Turk, M.J.; Hoopes, P.J.; Conejo-Garcia, J.R.; Fiering, S. Local hyperthermia treatment of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomedicine 2014, 10, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Castiello, L.; Arico, E.; D’Agostino, G.; Santodonato, L.; Belardelli, F. In situ Vaccination by Direct Dendritic Cell Inoculation: The Coming of Age of an Old Idea? Front. Immunol. 2019, 10, 2303. [Google Scholar] [CrossRef] [PubMed]
- Schoenbach, K.H.; Beebe, S.J.; Buescher, E.S. Intracellular effect of ultrashort electrical pulses. Bioelectromagnetics 2001, 22, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Kolb, J.F.; Kono, S.; Schoenbach, K.H. Nanosecond pulsed electric field generators for the study of subcellular effects. Bioelectromagnetics 2006, 27, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Burcus, N.I.; Hornef, J.; Jing, Y.; Jiang, C.; Heller, R.; Beebe, S.J. Nano-Pulse Stimulation for the Treatment of Pancreatic Cancer and the Changes in Immune Profile. Cancers 2018, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Edelblute, C.M.; Guo, S.; Hornef, J.; Yang, E.; Jiang, C.; Schoenbach, K.; Heller, R. Moderate Heat Application Enhances the Efficacy of Nanosecond Pulse Stimulation for the Treatment of Squamous Cell Carcinoma. Technol. Cancer Res. Treat. 2018, 17, 1533033818802305. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhuang, J.; Kolb, J.F.; Schoenbach, K.H.; Beebe, S.J. Long term survival of mice with hepatocellular carcinoma after pulse power ablation with nanosecond pulsed electric fields. Technol. Cancer Res. Treat. 2012, 11, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, R.; Pliquett, U.; Chen, X.; Ford, W.; James Swanson, R.; Beebe, S.J.; Kolb, J.F.; Schoenbach, K.H. Nanosecond pulsed electric fields cause melanomas to self-destruct. Biochem. Biophys. Res. Commun. 2006, 343, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Bao, X.; Dong, L.; Guo, Q.Q.; Guo, J.; Xie, Y.; Zhou, Y.; Yu, B.; Wu, H.; Wu, J.X.; et al. Doxorubicin pretreatment enhances FAPalpha/survivin co-targeting DNA vaccine anti-tumor activity primarily through decreasing peripheral MDSCs in the 4T1 murine breast cancer model. Oncoimmunology 2020, 9, 1747350. [Google Scholar] [CrossRef] [PubMed]
- Steenbrugge, J.; Vander Elst, N.; Demeyere, K.; De Wever, O.; Sanders, N.N.; Van Den Broeck, W.; Dirix, L.; Van Laere, S.; Meyer, E. Comparative Profiling of Metastatic 4T1- vs. Non-metastatic Py230-Based Mammary Tumors in an Intraductal Model for Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 2928. [Google Scholar] [CrossRef] [PubMed]
- Ledys, F.; Kalfeist, L.; Galland, L.; Limagne, E.; Ladoire, S. Therapeutic Associations Comprising Anti-PD-1/PD-L1 in Breast Cancer: Clinical Challenges and Perspectives. Cancers 2021, 13, 5999. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, M.M.; Tiainen, S.; Siiskonen, H.; Ahtiainen, M.; Kuopio, T.; Ronka, A.; Kettunen, T.; Hamalainen, K.; Rilla, K.; Harvima, I.; et al. The prognostic and predictive role of tumor-infiltrating lymphocytes (FoxP3+ and CD8+) and tumor-associated macrophages in early HER2 + breast cancer. Breast Cancer Res. Treat. 2023, 201, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Stenstrom, J.; Hedenfalk, I.; Hagerling, C. Regulatory T lymphocyte infiltration in metastatic breast cancer-an independent prognostic factor that changes with tumor progression. Breast Cancer Res. 2021, 23, 27. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic review and meta-analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Sirvent, C.; Schuster, M.; Neumann, Y.; Heise, U.; Pils, M.C.; Schulze-Osthoff, K.; Schmitz, I. c-FLIP Expression in Foxp3-Expressing Cells Is Essential for Survival of Regulatory T Cells and Prevention of Autoimmunity. Cell Rep. 2017, 18, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, P.; Ponti, C.; Gobbi, G.; Sponzilli, I.; Vaccarezza, M.; Cocco, L.; Zauli, G.; Secchiero, P.; Manzoli, F.A.; Vitale, M. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood 2004, 104, 2418–2424. [Google Scholar] [CrossRef] [PubMed]
- Overacre-Delgoffe, A.E.; Vignali, D.A.A. Treg Fragility: A Prerequisite for Effective Antitumor Immunity? Cancer Immunol. Res. 2018, 6, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, Y.; Nirschl, T.R.; Kochel, C.M.; Lopez-Bujanda, Z.; Theodros, D.; Mao, W.; Carrera-Haro, M.A.; Ghasemzadeh, A.; Marciscano, A.E.; Velarde, E.; et al. Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment. Cancer Immunol. Res. 2017, 5, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.; Hagekyriakou, J.; Chindris, I.; Albarakati, H.; Leong, T.; Schlenker, R.; Keam, S.P.; Williams, S.G.; Neeson, P.J.; Johnstone, R.W.; et al. Regulatory T Cells Shape the Differential Impact of Radiation Dose-Fractionation Schedules on Host Innate and Adaptive Antitumor Immune Defenses. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 502–514. [Google Scholar] [CrossRef]
- Anderson, B.E.; McNiff, J.M.; Matte, C.; Athanasiadis, I.; Shlomchik, W.D.; Shlomchik, M.J. Recipient CD4+ T cells that survive irradiation regulate chronic graft-versus-host disease. Blood 2004, 104, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing tumor immunity with fractionated radiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1306–1310. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, J.R.; Lax, B.M.; Peters, J.M.; Duhamel, L.; Stinson, J.A.; Santollani, L.; Lutz, E.A.; Pinney, W., 3rd; Bryson, B.D.; Dane Wittrup, K. CD8(+) T cell priming that is required for curative intratumorally anchored anti-4-1BB immunotherapy is constrained by Tregs. Nat. Commun. 2024, 15, 1900. [Google Scholar] [CrossRef] [PubMed]
- Freeman, Z.T.; Nirschl, T.R.; Hovelson, D.H.; Johnston, R.J.; Engelhardt, J.J.; Selby, M.J.; Kochel, C.M.; Lan, R.Y.; Zhai, J.; Ghasemzadeh, A.; et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J. Clin. Investig. 2020, 130, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pakhomova, O.N.; Pakhomov, A.G.; Weygandt, S.; Bulysheva, A.A.; Murray, L.E.; Mollica, P.A.; Muratori, C. Mechanisms and immunogenicity of nsPEF-induced cell death in B16F10 melanoma tumors. Sci. Rep. 2019, 9, 431. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanajian, A.; Scott, M.; Burcus, N.I.; Ruedlinger, B.L.; Oshin, E.A.; Beebe, S.J.; Guo, S. Nano-Pulse Treatment Overcomes the Immunosuppressive Tumor Microenvironment to Elicit In Situ Vaccination Protection against Breast Cancer. Vaccines 2024, 12, 633. https://doi.org/10.3390/vaccines12060633
Nanajian A, Scott M, Burcus NI, Ruedlinger BL, Oshin EA, Beebe SJ, Guo S. Nano-Pulse Treatment Overcomes the Immunosuppressive Tumor Microenvironment to Elicit In Situ Vaccination Protection against Breast Cancer. Vaccines. 2024; 12(6):633. https://doi.org/10.3390/vaccines12060633
Chicago/Turabian StyleNanajian, Anthony, Megan Scott, Niculina I. Burcus, Brittney L. Ruedlinger, Edwin A. Oshin, Stephen J. Beebe, and Siqi Guo. 2024. "Nano-Pulse Treatment Overcomes the Immunosuppressive Tumor Microenvironment to Elicit In Situ Vaccination Protection against Breast Cancer" Vaccines 12, no. 6: 633. https://doi.org/10.3390/vaccines12060633