

UL56 Is Essential for Herpes Simplex Virus-1 Virulence In Vivo but Is Dispensable for Induction of Host-Protective Immunity

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning of ΔUL56 Mutant and Revertant HSV-1 Strains
2.2. Cell Cultures and In Vitro Infections
2.3. Cell Staining, Imaging and Quantification
2.4. Mice and Genotyping
2.5. In Vivo Infections
2.6. Statistical Analyses
3. Results
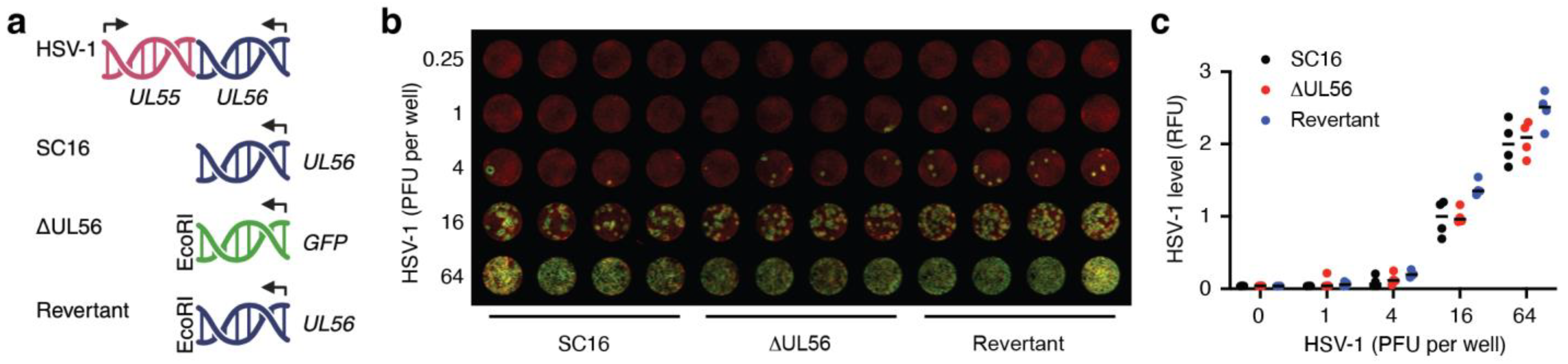
3.1. Generation of a UL56 Deletion Mutant, ΔUL56
3.2. UL56 Replicates in Skin Keratinocytes In Vitro
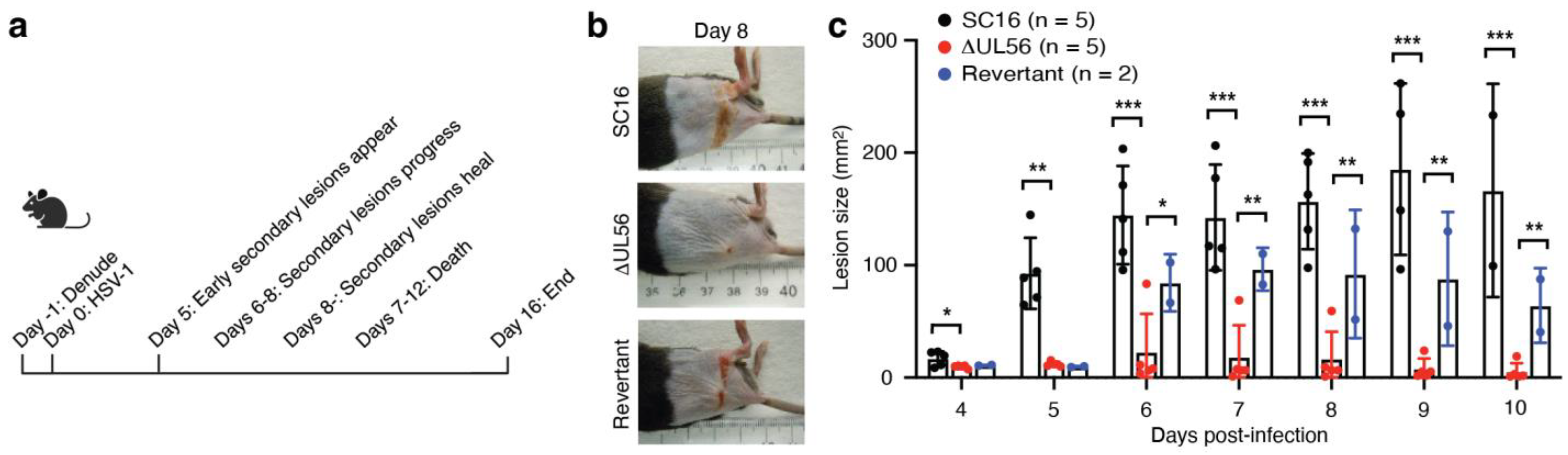
3.3. UL56 Has Diminished Capacity to Establish In Vivo Infections
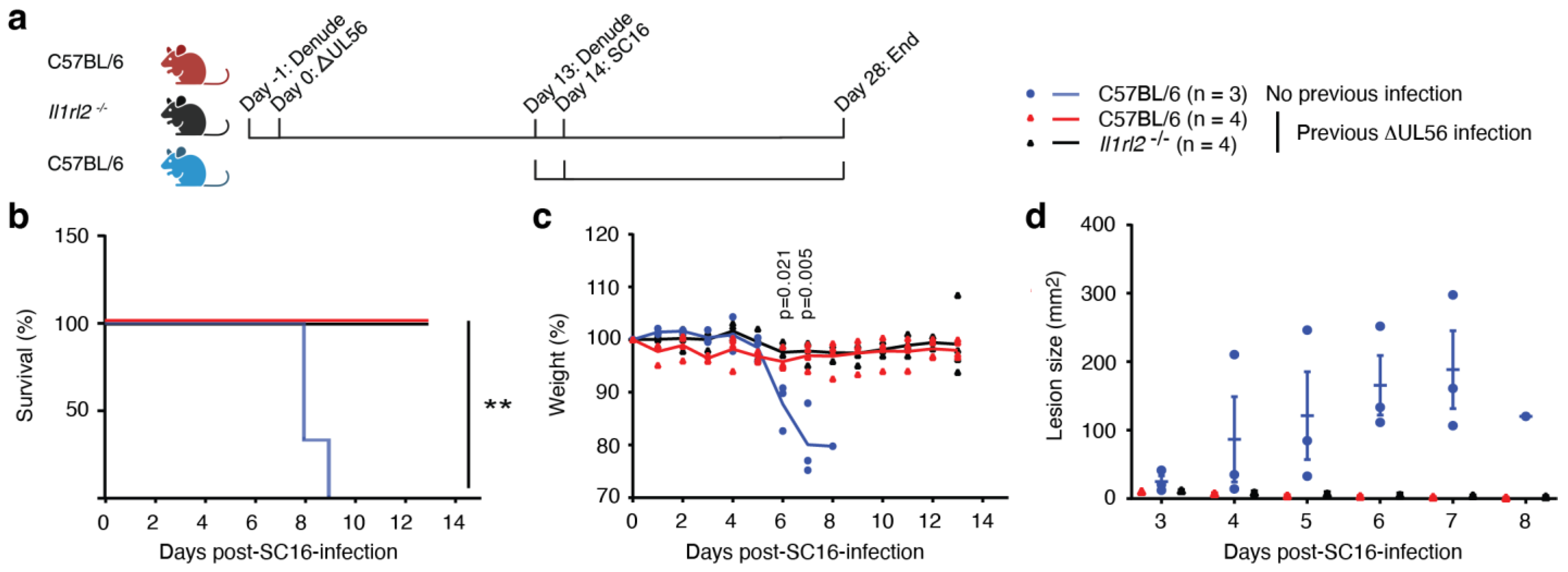
3.4. UL56 Induces Protective Immunity against a Lethal Challenge with Wild-Type Virus
3.5. Protective Immunity Induced by ΔUL56 is Independent of IL-36 Signaling
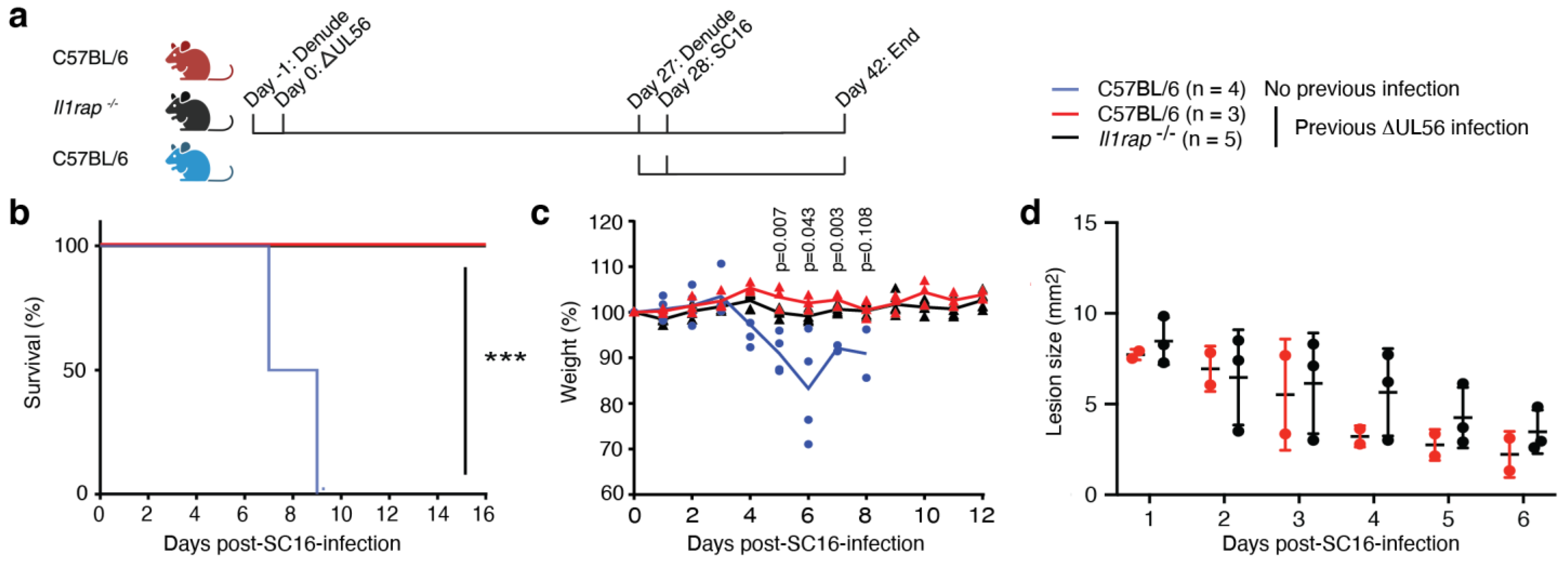
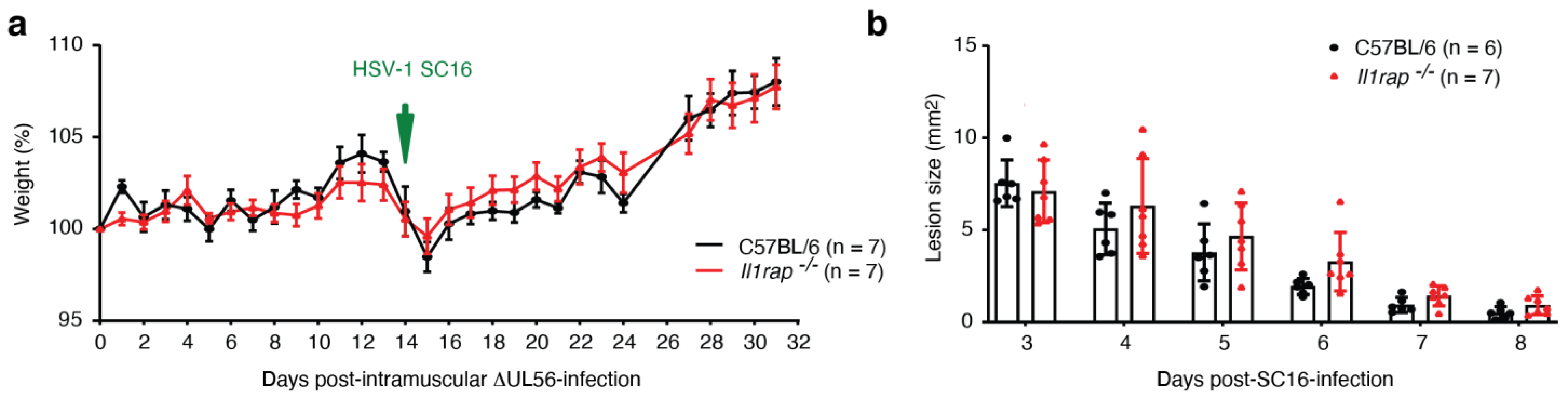
3.6. IL-1RAP Is Not Required for Protective Immunity Engaged by ΔUL56
3.7. Intramuscular Administration of ΔUL56 Provides Protective Immunity
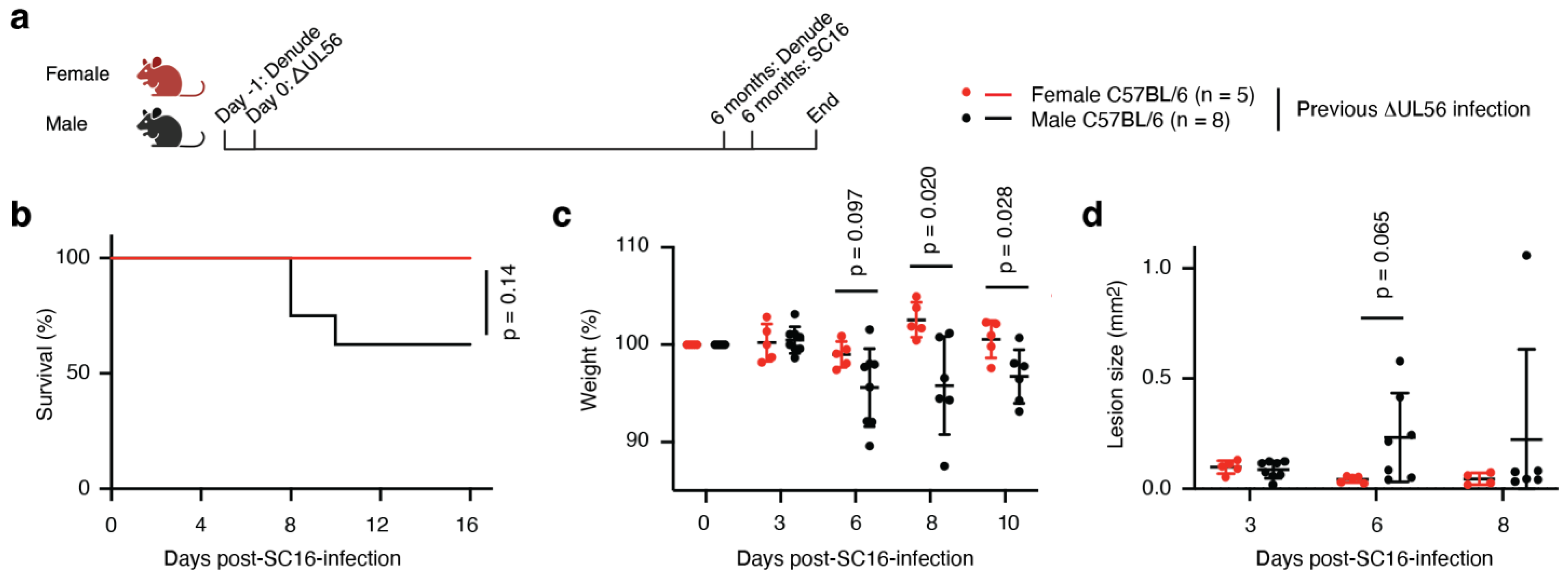
3.8. Protective Immunity Elicited by ΔUL56 Lasts Longer in Female Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soh, T.K.; Davies, C.T.R.; Muenzner, J.; Hunter, L.M.; Barrow, H.G.; Connor, V.; Bouton, C.R.; Smith, C.; Emmott, E.; Antrobus, R.; et al. Temporal Proteomic Analysis of Herpes Simplex Virus 1 Infection Reveals Cell-Surface Remodeling via pUL56-Mediated GOPC Degradation. Cell Rep. 2020, 33, 108235. [Google Scholar] [CrossRef]
- Zheng, Z.Q.; Fu, Y.Z.; Wang, S.Y.; Xu, Z.S.; Zou, H.M.; Wang, Y.Y. Herpes simplex virus protein UL56 inhibits cGAS-Mediated DNA sensing to evade antiviral immunity. Cell Insight 2022, 1, 100014. [Google Scholar] [CrossRef]
- Blest, H.T.W.; Redmond, A.; Avissar, J.; Barker, J.; Bridgeman, A.; Fowler, G.; Chauveau, L.; Hertzog, J.; Vendrell, I.; Fischer, R.; et al. HSV-1 employs UL56 to antagonize expression and function of cGAMP channels. Cell Rep. 2024, 43, 114122. [Google Scholar] [CrossRef]
- Koshizuka, T.; Kobayashi, T.; Ishioka, K.; Suzutani, T. Herpesviruses possess conserved proteins for interaction with Nedd4 family ubiquitin E3 ligases. Sci. Rep. 2018, 8, 4447. [Google Scholar] [CrossRef]
- Wang, J.; Wu, K.; Ni, L.; Li, C.; Peng, R.; Li, Y.; Fan, Z.; Yin, F.; Deng, F.; Shen, S.; et al. Effects of US7 and UL56 on Cell-to-Cell Spread of Human Herpes Simplex Virus 1. Viruses 2023, 15, 2256. [Google Scholar] [CrossRef]
- Nash, T.C.; Spivack, J.G. The UL55 and UL56 genes of herpes simplex virus type 1 are not required for viral replication, intraperitoneal virulence, or establishment of latency in mice. Virology 1994, 204, 794–798. [Google Scholar] [CrossRef]
- Berkowitz, C.; Moyal, M.; Rösen-Wolff, A.; Darai, G.; Becker, Y. Herpes simplex virus type 1 (HSV-1) UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch. Virol. 1994, 134, 73–83. [Google Scholar] [CrossRef]
- Rösen-Wolff, A.; Lamadé, W.; Berkowitz, C.; Becker, Y.; Darai, G. Elimination of UL56 gene by insertion of LacZ cassette between nucleotide position 116030 to 121753 of the herpes simplex virus type 1 genome abrogates intraperitoneal pathogenicity in tree shrews and mice. Virus Res. 1991, 20, 205–221. [Google Scholar] [CrossRef]
- Xiang, Z.; He, Y. Genome-wide prediction of vaccine targets for human herpes simplex viruses using Vaxign reverse vaccinology. BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef]
- Muñoz-Wolf, N.; Lavelle, E.C. A Guide to IL-1 family cytokines in adjuvanticity. Febs. J. 2018, 285, 2377–2401. [Google Scholar] [CrossRef]
- Reinke, S.; Thakur, A.; Gartlan, C.; Bezbradica, J.S.; Milicic, A. Inflammasome-Mediated Immunogenicity of Clinical and Experimental Vaccine Adjuvants. Vaccines 2020, 8, 554. [Google Scholar] [CrossRef]
- Van Den Eeckhout, B.; Tavernier, J.; Gerlo, S. Interleukin-1 as Innate Mediator of T Cell Immunity. Front. Immunol. 2020, 11, 621931. [Google Scholar] [CrossRef]
- Jensen, L.E. Interleukin-36 cytokines may overcome microbial immune evasion strategies that inhibit interleukin-1 family signaling. Sci. Signal. 2017, 10, eaan3589. [Google Scholar] [CrossRef]
- Milora, K.A.; Miller, S.L.; Sanmiguel, J.C.; Jensen, L.E. Interleukin-1α released from HSV-1 infected keratinocytes acts as a functional alarmin in the skin. Nat. Commun. 2014, 5, 5230. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Smith, A.; Jurado, K.A.; Iwasaki, A.; Garlick, J.A.; Kagan, J.C. An antiviral branch of the IL-1 signaling pathway restricts immune-evasive virus replication. Mol. Cell 2018, 71, 825–840.e826. [Google Scholar] [CrossRef]
- Gewaid, H.; Bowie, A.G. Regulation of type I and type III interferon induction in response to pathogen sensing. Curr. Opin. Immunol. 2024, 87, 102424. [Google Scholar] [CrossRef]
- Lukhele, S.; Boukhaled, G.M.; Brooks, D.G. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 2019, 43, 101277. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Cullinan, E.B.; Kwee, L.; Nunes, P.; Shuster, D.J.; Ju, G.; McIntyre, K.W.; Chizzonite, R.A.; Labow, M.A. IL-1 receptor accessory protein is an essential component of the IL-1 receptor. J. Immunol. 1998, 161, 5614–5620. [Google Scholar] [CrossRef]
- Wang, P.; Gamero, A.M.; Jensen, L.E. IL-36 promotes anti-viral immunity by boosting sensitivity to IFN-alpha/beta in IRF1 dependent and independent manners. Nat. Commun. 2019, 10, 4700. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Feng, C.; Weinstein, A.; Xia, R.; Wen, W.; Lv, Q.; Zuo, S.; Tang, P.; Yang, X.; et al. IL-36γ transforms the tumor microenvironment and promotes type 1 lymphocyte-mediated antitumor immune responses. Cancer Cell 2015, 28, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Louis, L.; Wise, M.C.; Choi, H.; Villarreal, D.O.; Muthumani, K.; Weiner, D.B. Designed DNA-Encoded IL-36 Gamma Acts as a Potent Molecular Adjuvant Enhancing Zika Synthetic DNA Vaccine-Induced Immunity and Protection in a Lethal Challenge Model. Vaccines 2019, 7, 42. [Google Scholar] [CrossRef]
- Stanfield, B.A.; Rider, P.J.F.; Caskey, J.; Del Piero, F.; Kousoulas, K.G. Intramuscular vaccination of guinea pigs with the live-attenuated human herpes simplex vaccine VC2 stimulates a transcriptional profile of vaginal Th17 and regulatory Tr1 responses. Vaccine 2018, 36, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Aoki, R.; Kawamura, T.; Goshima, F.; Ogawa, Y.; Nakae, S.; Nakao, A.; Moriishi, K.; Nishiyama, Y.; Shimada, S. Mast cells play a key role in host defense against herpes simplex virus infection through TNF-α and IL-6 production. J. Investig. Dermatol. 2013, 133, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Kim, B.C.; Chang, D.H.; Kwon, M.; Lee, S.Y.; Kang, D.; Kim, J.Y.; Hwang, I.; Yu, J.W.; Nakae, S.; et al. Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc. Natl. Acad. Sci. USA 2016, 113, E762–E771. [Google Scholar] [CrossRef] [PubMed]
- Aoki, R.; Kawamura, T.; Goshima, F.; Ogawa, Y.; Nakae, S.; Moriishi, K.; Nakao, A.; Shimada, S. The Alarmin IL-33 Derived from HSV-2-Infected Keratinocytes Triggers Mast Cell-Mediated Antiviral Innate Immunity. J. Investig. Dermatol. 2016, 136, 1290–1292. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Robert, C.; Kupper, T.S. Interleukin-1 and cutaneous inflammation: A crucial link between innate and acquired immunity. J. Investig. Dermatol. 2000, 114, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Straface, G.; Selmin, A.; Zanardo, V.; De Santis, M.; Ercoli, A.; Scambia, G. Herpes simplex virus infection in pregnancy. Infect. Dis. Obstet Gynecol. 2012, 2012, 385697. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Friedman, H.M. An mRNA vaccine to prevent genital herpes. Transl. Res. 2022, 242, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.A.; Pahar, B.; Chouljenko, V.N.; Veazey, R.; Kousoulas, K.G. Vaccination of rhesus macaques with the live-attenuated HSV-1 vaccine VC2 stimulates the proliferation of mucosal T cells and germinal center responses resulting in sustained production of highly neutralizing antibodies. Vaccine 2017, 35, 536–543. [Google Scholar] [CrossRef]
- Richards, A.L.; Sollars, P.J.; Pitts, J.D.; Stults, A.M.; Heldwein, E.E.; Pickard, G.E.; Smith, G.A. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog. 2017, 13, e1006741. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Therapeutic vaccines for herpesviruses. J. Clin. Investig. 2024, 134, e179483. [Google Scholar] [CrossRef] [PubMed]
- Mork, N.; Kofod-Olsen, E.; Sorensen, K.B.; Bach, E.; Orntoft, T.F.; Ostergaard, L.; Paludan, S.R.; Christiansen, M.; Mogensen, T.H. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. 2015, 16, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L.; Klein, S.L. The evolution of greater humoral immunity in females than males: Implications for vaccine efficacy. Curr. Opin. Physiol. 2018, 6, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, K.J.; Truong, N.R.; Smith, J.B.; Bertram, K.; Cunningham, A.L. Vaccines for Herpes Simplex: Recent Progress Driven by Viral and Adjuvant Immunology. Methods Mol. Biol. 2020, 2060, 31–56. [Google Scholar] [CrossRef]
- Benedetti, J.; Corey, L.; Ashley, R. Recurrence rates in genital herpes after symptomatic first-episode infection. Ann. Intern. Med. 1994, 121, 847–854. [Google Scholar] [CrossRef]
- Zhou, J.; Zhuang, Z.; Li, J.; Feng, Z. Significance of the cGAS-STING Pathway in Health and Disease. Int. J. Mol. Sci. 2023, 24, 13316. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5’–3’) | Target Direction |
|---|---|---|---|
| Il1rap | oIMR6916 | CTT GGG TGG AGA GGC TAT TC | Mutant Forward |
| oIMR6917 | AGG TGA GAT GAC AGG AGA TC | Mutant Reverse | |
| 9084 | ACT ACA GCA CTG CCC ATT CC | Wildtype Forward | |
| oIMR4232 | TGT AAT TGC CCG TGT CAT TG | Wildtype Reverse | |
| Il1rl2 | Il1rl2_comF | GGG CTA TTT TAT GGT CCA AAA CTA CCA G | Common Forward |
| Il1rl2-wtR | GCT CTG GTC AAT GTG AAA TTG CAT TTA | Wildtype Reverse | |
| Il1rl2_mutR | ACC TCT CTC TGT AAG CTG GTC TGG G | Mutant Reverse |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tongmuang, N.; Krishnan, M.; Connor, V.; Crump, C.; Jensen, L.E. UL56 Is Essential for Herpes Simplex Virus-1 Virulence In Vivo but Is Dispensable for Induction of Host-Protective Immunity. Vaccines 2024, 12, 837. https://doi.org/10.3390/vaccines12080837
Tongmuang N, Krishnan M, Connor V, Crump C, Jensen LE. UL56 Is Essential for Herpes Simplex Virus-1 Virulence In Vivo but Is Dispensable for Induction of Host-Protective Immunity. Vaccines. 2024; 12(8):837. https://doi.org/10.3390/vaccines12080837
Chicago/Turabian StyleTongmuang, Nopprarat, Meera Krishnan, Viv Connor, Colin Crump, and Liselotte E. Jensen. 2024. "UL56 Is Essential for Herpes Simplex Virus-1 Virulence In Vivo but Is Dispensable for Induction of Host-Protective Immunity" Vaccines 12, no. 8: 837. https://doi.org/10.3390/vaccines12080837
APA StyleTongmuang, N., Krishnan, M., Connor, V., Crump, C., & Jensen, L. E. (2024). UL56 Is Essential for Herpes Simplex Virus-1 Virulence In Vivo but Is Dispensable for Induction of Host-Protective Immunity. Vaccines, 12(8), 837. https://doi.org/10.3390/vaccines12080837