
GSK-3β in Dendritic Cells Exerts Opposite Functions in Regulating Cross-Priming and Memory CD8 T Cell Responses Independent of β-Catenin
 , ,
, ,  , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
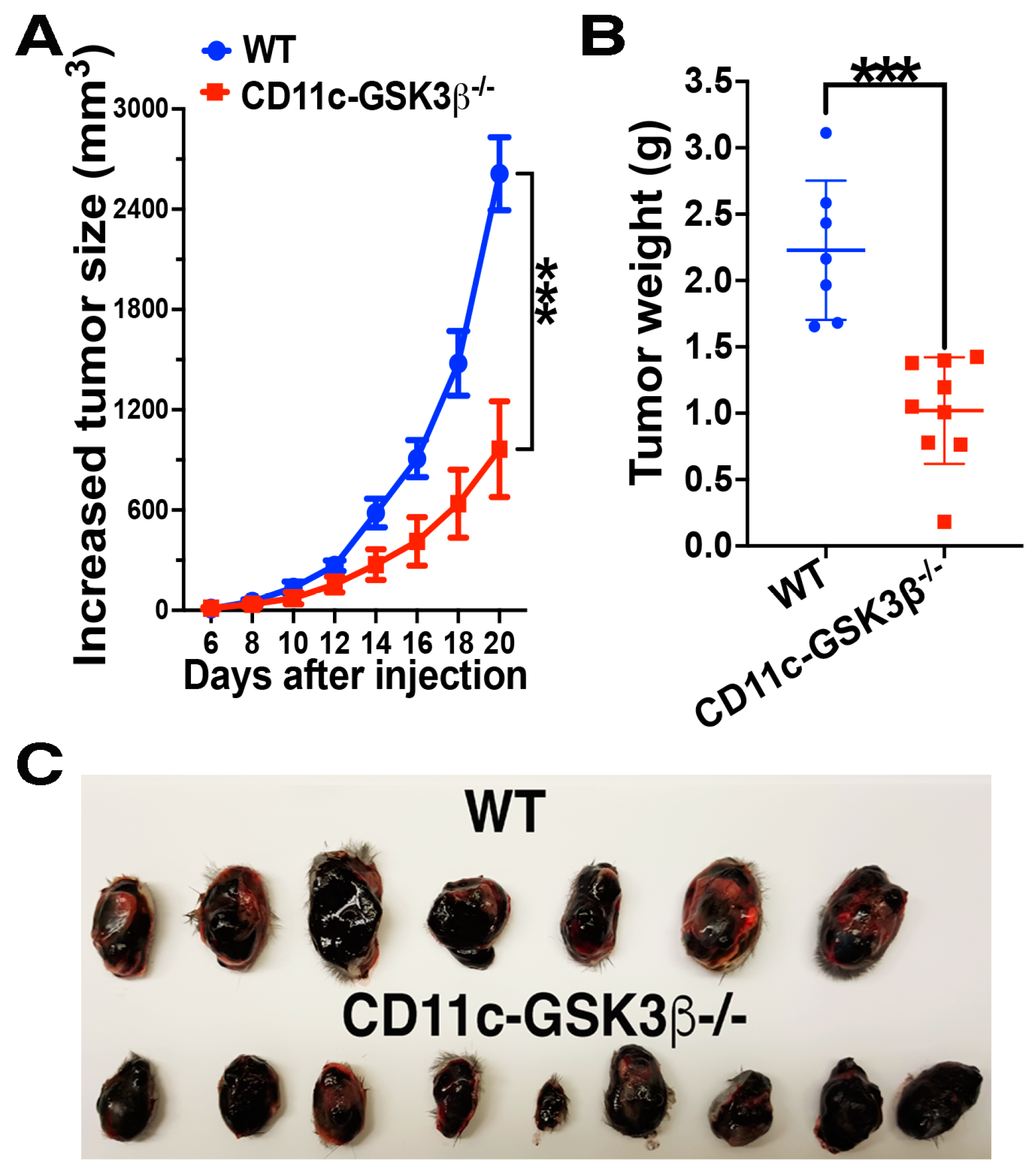
2.1. CD11c-GSK-3β−/− Mice Exhibit Augmented Antitumor Immunity Compared to GSK-3βFlox/Flox Mice
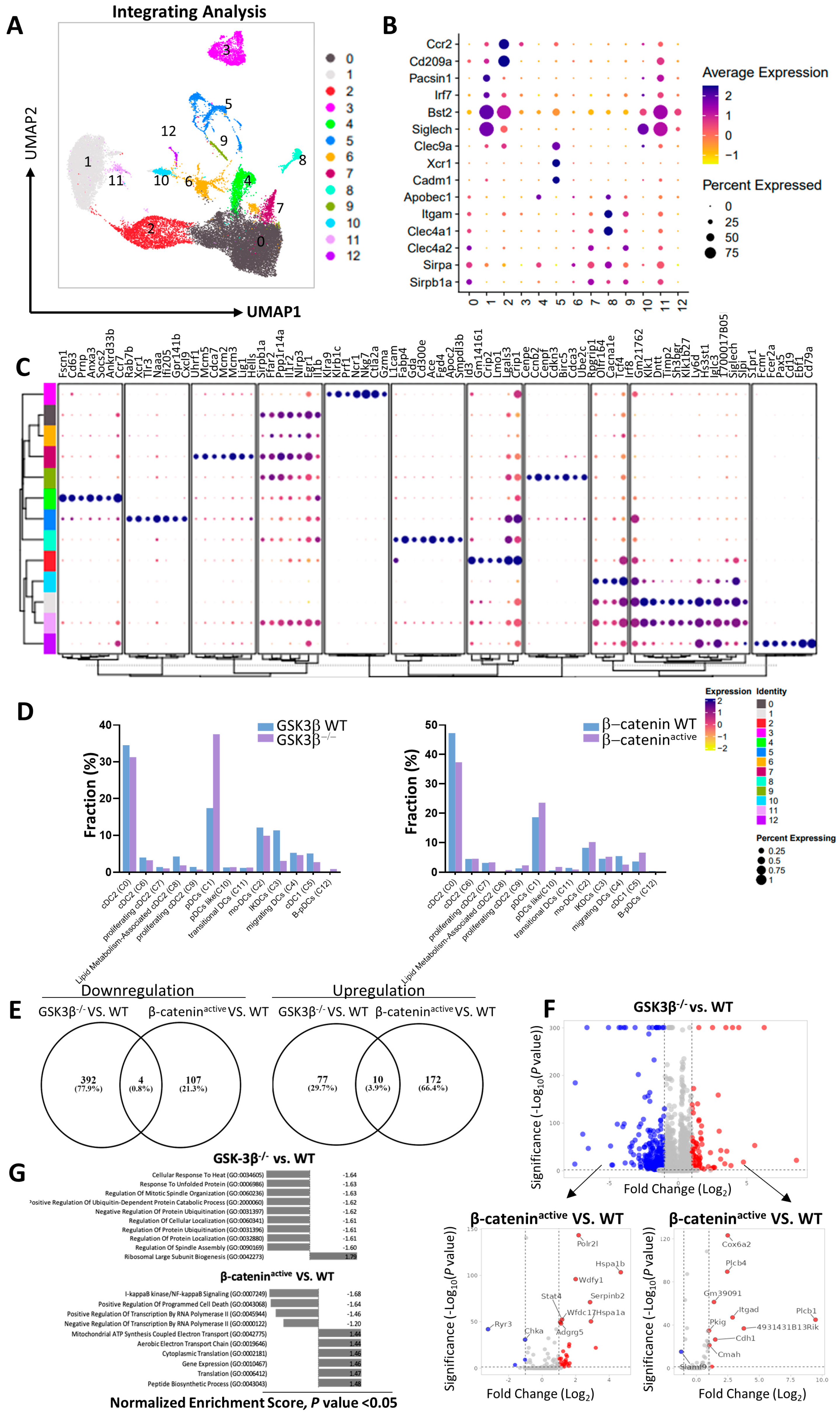
2.2. Distinct Gene Expression Profiles of DCs from CD11c-GSK-3β−/− and CD11c-β-cateninactive Mice
2.3. Lack of GSK-3β Does Not Lead to Accumulation of β-catenin in DCs and GSK-3β−/− DCs Exhibit Different Phenotypes than β-cateninactive DCs
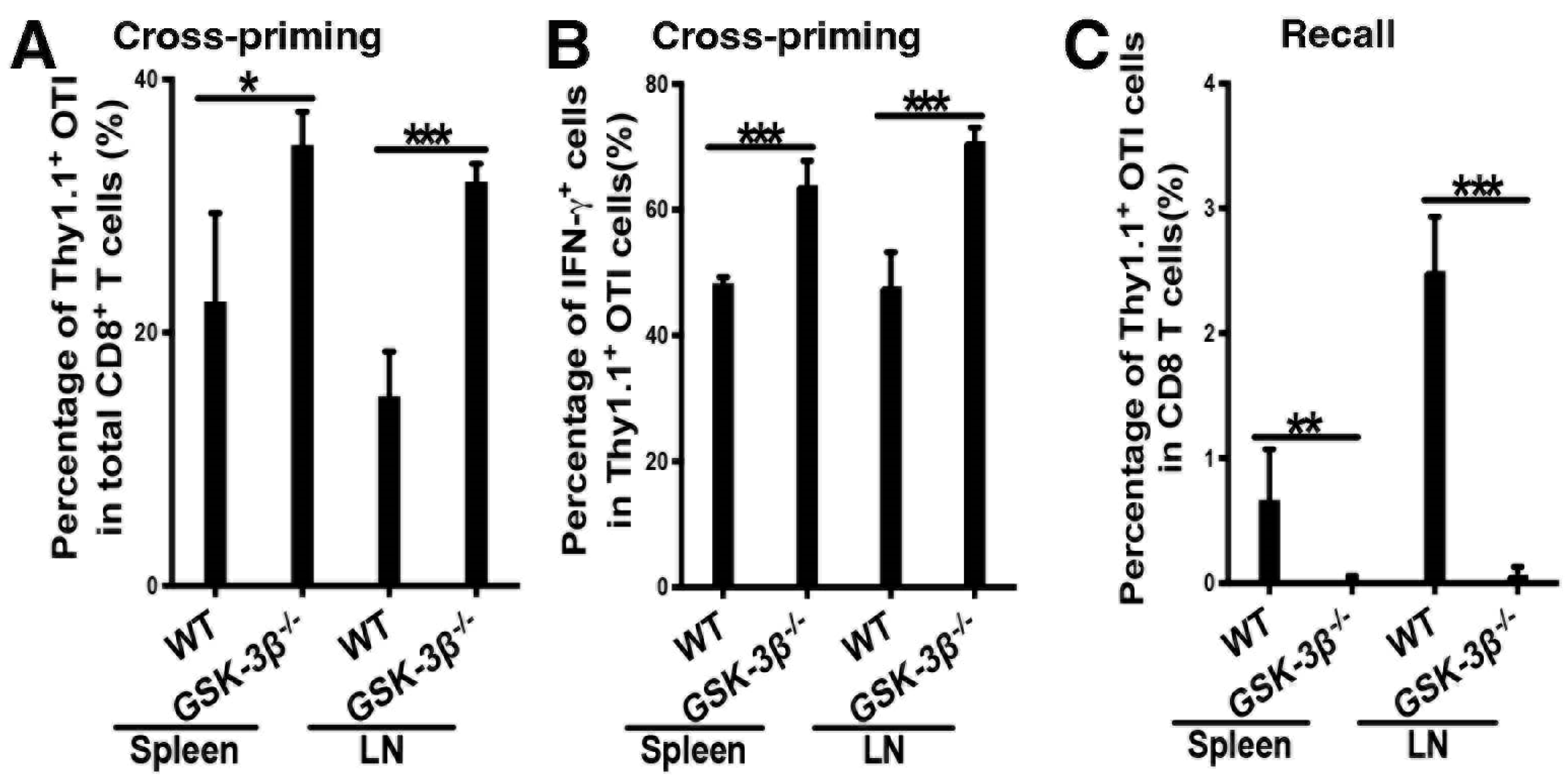
2.4. Deletion of GSK-3β in DCs Augments DC Vaccine-Induced Cross-Priming of Antigen-Specific CD8 T Cells
2.5. Deletion of GSK-3β in DCs Led to Diminished Memory Responses Despite Augmented Cross-Priming
2.6. scRNA-Seq Analysis of OTI CD8 T Cells Primed in WT and CD11c-GSK-3β −/− Mice
3. Discussion
4. Conclusions
5. Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, A.; Bloom, O.; Ono, S.; Cui, W.; Unternaehrer, J.; Jiang, S.; Whitney, J.A.; Connolly, J.; Banchereau, J.; Mellman, I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 2007, 27, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Fu, C.; Cui, W.; Ober-Blobaum, J.L.; Zahner, S.P.; Shrikant, P.A.; Clausen, B.E.; Flavell, R.A.; Mellman, I.; Jiang, A. beta-catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8(+) T cells. J. Leukoc. Biol. 2014, 95, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liang, X.; Cui, W.; Ober-Blobaum, J.L.; Vazzana, J.; Shrikant, P.A.; Lee, K.P.; Clausen, B.E.; Mellman, I.; Jiang, A. beta-Catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc. Natl. Acad. Sci. USA 2015, 112, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Majumdar, T.; Angus-Hill, M.L.; Koni, P.A.; Manicassamy, B.; Mellor, A.L.; Munn, D.H.; Manicassamy, S. beta-catenin promotes regulatory T-cell responses in tumors by inducing vitamin A metabolism in dendritic cells. Cancer Res. 2015, 75, 656–665. [Google Scholar] [CrossRef]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-beta-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160.e147. [Google Scholar] [CrossRef]
- Augello, G.; Emma, M.R.; Cusimano, A.; Azzolina, A.; Montalto, G.; McCubrey, J.A.; Cervello, M. The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells 2020, 9, 1427. [Google Scholar] [CrossRef]
- Borden, B.A.; Baca, Y.; Xiu, J.; Tavora, F.; Winer, I.; Weinberg, B.A.; Vanderwalde, A.M.; Darabi, S.; Korn, W.M.; Mazar, A.P.; et al. The Landscape of Glycogen Synthase Kinase-3 Beta Genomic Alterations in Cancer. Mol. Cancer Ther. 2021, 20, 183–190. [Google Scholar] [CrossRef]
- Liang, J.; Yu, M.; Li, Y.; Zhao, L.; Wei, Q. Glycogen synthase kinase-3: A potential immunotherapeutic target in tumor microenvironment. Biomed. Pharmacother. 2024, 173, 116377. [Google Scholar] [CrossRef]
- Thapa, R.; Gupta, G.; Bhat, A.A.; Almalki, W.H.; Alzarea, S.I.; Kazmi, I.; Saleem, S.; Khan, R.; Altwaijry, N.; Dureja, H.; et al. A review of Glycogen Synthase Kinase-3 (GSK3) inhibitors for cancers therapies. Int. J. Biol. Macromol. 2023, 253, 127375. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, A.; De Haseth, S.; Fray, M.; Miyajima, M.; Colvin, R.B.; Williams, W.W.; Benedict Cosimi, A.; Benichou, G. Dendritic cell maturation occurs through the inhibition of GSK-3beta. Cell Immunol. 2011, 270, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Escribano, C.; Delgado-Martin, C.; Rodriguez-Fernandez, J.L. CCR7-dependent stimulation of survival in dendritic cells involves inhibition of GSK3beta. J. Immunol. 2009, 183, 6282–6295. [Google Scholar] [CrossRef]
- Qian, C.; Qian, L.; Yu, Y.; An, H.; Guo, Z.; Han, Y.; Chen, Y.; Bai, Y.; Wang, Q.; Cao, X. Fas signal promotes the immunosuppressive function of regulatory dendritic cells via ERK/beta-catenin pathway. J. Biol. Chem. 2013, 288, 27825–27835. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.T.; Son, K.H.; Jung, I.D.; Kang, T.H.; Choi, C.H.; Park, Y.M. Glycogen Synthase Kinase-3beta (GSK-3beta) Inhibition Enhances Dendritic Cell-based Cancer Vaccine Potency via Suppression of Interferon-gamma-induced Indoleamine 2,3-Dioxygenase Expression. J. Biol. Chem. 2015, 290, 12394–12402. [Google Scholar] [CrossRef]
- Lopez Gonzalez, M.; Oosterhoff, D.; Lindenberg, J.J.; Milenova, I.; Lougheed, S.M.; Martianez, T.; Dekker, H.; Quixabeira, D.C.A.; Hangalapura, B.; Joore, J.; et al. Constitutively active GSK3beta as a means to bolster dendritic cell functionality in the face of tumour-mediated immune suppression. Oncoimmunology 2019, 8, e1631119. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Miao, C.; Xing, Q.; Wang, Z. High expression of CDCA7 predicts poor prognosis for clear cell renal cell carcinoma and explores its associations with immunity. Cancer Cell Int. 2021, 21, 140. [Google Scholar] [CrossRef]
- Chan, C.W.; Crafton, E.; Fan, H.N.; Flook, J.; Yoshimura, K.; Skarica, M.; Brockstedt, D.; Dubensky, T.W.; Stins, M.F.; Lanier, L.L.; et al. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat. Med. 2006, 12, 207–213. [Google Scholar] [CrossRef]
- Taieb, J.; Chaput, N.; Menard, C.; Apetoh, L.; Ullrich, E.; Bonmort, M.; Pequignot, M.; Casares, N.; Terme, M.; Flament, C.; et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nat. Med. 2006, 12, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallee, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e824. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, aah4573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gregorio, J.D.; Iwahori, T.; Zhang, X.; Choi, O.; Tolentino, L.L.; Prestwood, T.; Carmi, Y.; Engleman, E.G. A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.M.; Dekker, J.D.; Garrison, K.; Su, Z.; Rhee, C.; Hu, Z.; Lee, B.-K.; Hurtado, D.O.; Lee, J.; Iyer, V.R.; et al. Lymphoid origin of intrinsically activated plasmacytoid dendritic cells in mice eLife. eLife 2024, 13, RP96394. [Google Scholar] [CrossRef]
- Fu, C.; Wang, J.; Ma, T.; Yin, C.; Zhou, L.; Clausen, B.E.; Mi, Q.S.; Jiang, A. beta-Catenin in Dendritic Cells Negatively Regulates CD8 T Cell Immune Responses through the Immune Checkpoint Molecule Tim-3. Vaccines 2024, 12, 460. [Google Scholar] [CrossRef]
- Daniel, B.; Yost, K.E.; Hsiung, S.; Sandor, K.; Xia, Y.; Qi, Y.; Hiam-Galvez, K.J.; Black, M.; Raposo, C.J.; Shi, Q.; et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat. Immunol. 2022, 23, 1614–1627. [Google Scholar] [CrossRef]
- Rizzo, K.; Stetler-Stevenson, M.; Wilson, W.; Yuan, C.M. Novel CD19 expression in a peripheral T cell lymphoma: A flow cytometry case report with morphologic correlation. Cytom. B Clin. Cytom. 2009, 76, 142–149. [Google Scholar] [CrossRef]
- Vlaming, M.; Bilemjian, V.; Freile, J.A.; Lourens, H.J.; van Rooij, N.; Huls, G.; van Meerten, T.; de Bruyn, M.; Bremer, E. CD20 positive CD8 T cells are a unique and transcriptionally-distinct subset of T cells with distinct transmigration properties. Sci. Rep. 2021, 11, 20499. [Google Scholar] [CrossRef]
- Yi, Q.; Wang, J.; Liu, T.; Yao, Y.; Loveless, I.; Subedi, K.; Toor, J.; Adrianto, I.; Xiao, H.; Chen, B.; et al. scRNA-Seq and imaging mass cytometry analyses unveil iNKT cells-mediated anti-tumor immunity in pancreatic cancer liver metastasis. Cancer Lett. 2023, 561, 216149. [Google Scholar] [CrossRef]
- Ivanova, D.L.; Thompson, S.B.; Klarquist, J.; Harbell, M.G.; Kilgore, A.M.; Lasda, E.L.; Hesselberth, J.R.; Hunter, C.A.; Kedl, R.M. Vaccine adjuvant-elicited CD8(+) T cell immunity is co-dependent on T-bet and FOXO1. Cell Rep. 2023, 42, 112911. [Google Scholar] [CrossRef] [PubMed]
- Preiss, N.K.; Kamal, Y.; Wilkins, O.M.; Li, C.; Kolling, F.W.t.; Trask, H.W.; Usherwood, Y.K.; Cheng, C.; Frost, H.R.; Usherwood, E.J. Characterizing control of memory CD8 T cell differentiation by BTB-ZF transcription factor Zbtb20. Life Sci. Alliance 2023, 6, e202201683. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brown, J.; Martin, M. Glycogen synthase kinase 3: A point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef]
- Rodionova, E.; Conzelmann, M.; Maraskovsky, E.; Hess, M.; Kirsch, M.; Giese, T.; Ho, A.D.; Zoller, M.; Dreger, P.; Luft, T. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood 2007, 109, 1584–1592. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Garcia, C.A.; Tang, Y.; Benakanakere, M.R.; Greenway, T.; Alard, P.; Kinane, D.F.; Martin, M. The role of glycogen synthase kinase 3 in regulating IFN-beta-mediated IL-10 production. J. Immunol. 2011, 186, 675–684. [Google Scholar] [CrossRef]
- Liu, K.J.; Lee, Y.L.; Yang, Y.Y.; Shih, N.Y.; Ho, C.C.; Wu, Y.C.; Huang, T.S.; Huang, M.C.; Liu, H.C.; Shen, W.W.; et al. Modulation of the development of human monocyte-derived dendritic cells by lithium chloride. J. Cell Physiol. 2011, 226, 424–433. [Google Scholar] [CrossRef]
- Ono, T.; Yanagawa, Y.; Iwabuchi, K.; Nonomura, K.; Onoe, K. Glycogen synthase kinase 3 activity during development of bone marrow-derived dendritic cells (DCs) essential for the DC function to induce T helper 2 polarization. Immunology 2007, 122, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, M.; Nagai, S.; Kondo, S.; Mizuno, S.; Nakamura, K.; Tanabe, M.; Takeuchi, T.; Matsuda, S.; Koyasu, S. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood 2008, 112, 635–643. [Google Scholar] [CrossRef]
- Xu, W.D.; Wang, J.; Yuan, T.L.; Li, Y.H.; Yang, H.; Liu, Y.; Zhao, Y.; Herrmann, M. Interactions between canonical Wnt signaling pathway and MAPK pathway regulate differentiation, maturation and function of dendritic cells. Cell Immunol. 2016, 310, 170–177. [Google Scholar] [CrossRef]
- Issa, M.E.; Rudd, C.E. Glycogen Synthase Kinase-3 (GSK-3) Regulation of Inhibitory Coreceptor Expression in T-cell Immunity. J. Cell Immunol. 2021, 3, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Rao, R.R.; Araki, K.; Pollizzi, K.; Odunsi, K.; Powell, J.D.; Shrikant, P.A. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity 2011, 34, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Adrianto, I.; Wang, J.; Wu, X.; Datta, I.; Mi, Q.S. Single-Cell RNA-Seq Analysis Uncovers Distinct Functional Human NKT Cell Sub-Populations in Peripheral Blood. Front. Cell Dev. Biol. 2020, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Adrianto, I.; Subedi, K.; Liu, T.; Wu, X.; Yi, Q.; Loveless, I.; Yin, C.; Datta, I.; Sant’Angelo, D.B.; et al. Integrative scATAC-seq and scRNA-seq analyses map thymic iNKT cell development and identify Cbfbeta for its commitment. Cell Discov. 2023, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Nakao, A.; Yoshihama, M.; Kenmochi, N. RPG: The Ribosomal Protein Gene database. Nucleic Acids Res. 2004, 32, D168–D170. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, C.; Wang, J.; Ma, T.; Yin, C.; Zhou, L.; Clausen, B.E.; Mi, Q.-S.; Jiang, A. GSK-3β in Dendritic Cells Exerts Opposite Functions in Regulating Cross-Priming and Memory CD8 T Cell Responses Independent of β-Catenin. Vaccines 2024, 12, 1037. https://doi.org/10.3390/vaccines12091037
Fu C, Wang J, Ma T, Yin C, Zhou L, Clausen BE, Mi Q-S, Jiang A. GSK-3β in Dendritic Cells Exerts Opposite Functions in Regulating Cross-Priming and Memory CD8 T Cell Responses Independent of β-Catenin. Vaccines. 2024; 12(9):1037. https://doi.org/10.3390/vaccines12091037
Chicago/Turabian StyleFu, Chunmei, Jie Wang, Tianle Ma, Congcong Yin, Li Zhou, Björn E. Clausen, Qing-Sheng Mi, and Aimin Jiang. 2024. "GSK-3β in Dendritic Cells Exerts Opposite Functions in Regulating Cross-Priming and Memory CD8 T Cell Responses Independent of β-Catenin" Vaccines 12, no. 9: 1037. https://doi.org/10.3390/vaccines12091037
APA StyleFu, C., Wang, J., Ma, T., Yin, C., Zhou, L., Clausen, B. E., Mi, Q.-S., & Jiang, A. (2024). GSK-3β in Dendritic Cells Exerts Opposite Functions in Regulating Cross-Priming and Memory CD8 T Cell Responses Independent of β-Catenin. Vaccines, 12(9), 1037. https://doi.org/10.3390/vaccines12091037