
Navigating the Purification Process: Maintaining the Integrity of Replication-Competent Enveloped Viruses


Abstract
:1. Introduction
1.1. Challenges in the Purification of Enveloped VPs
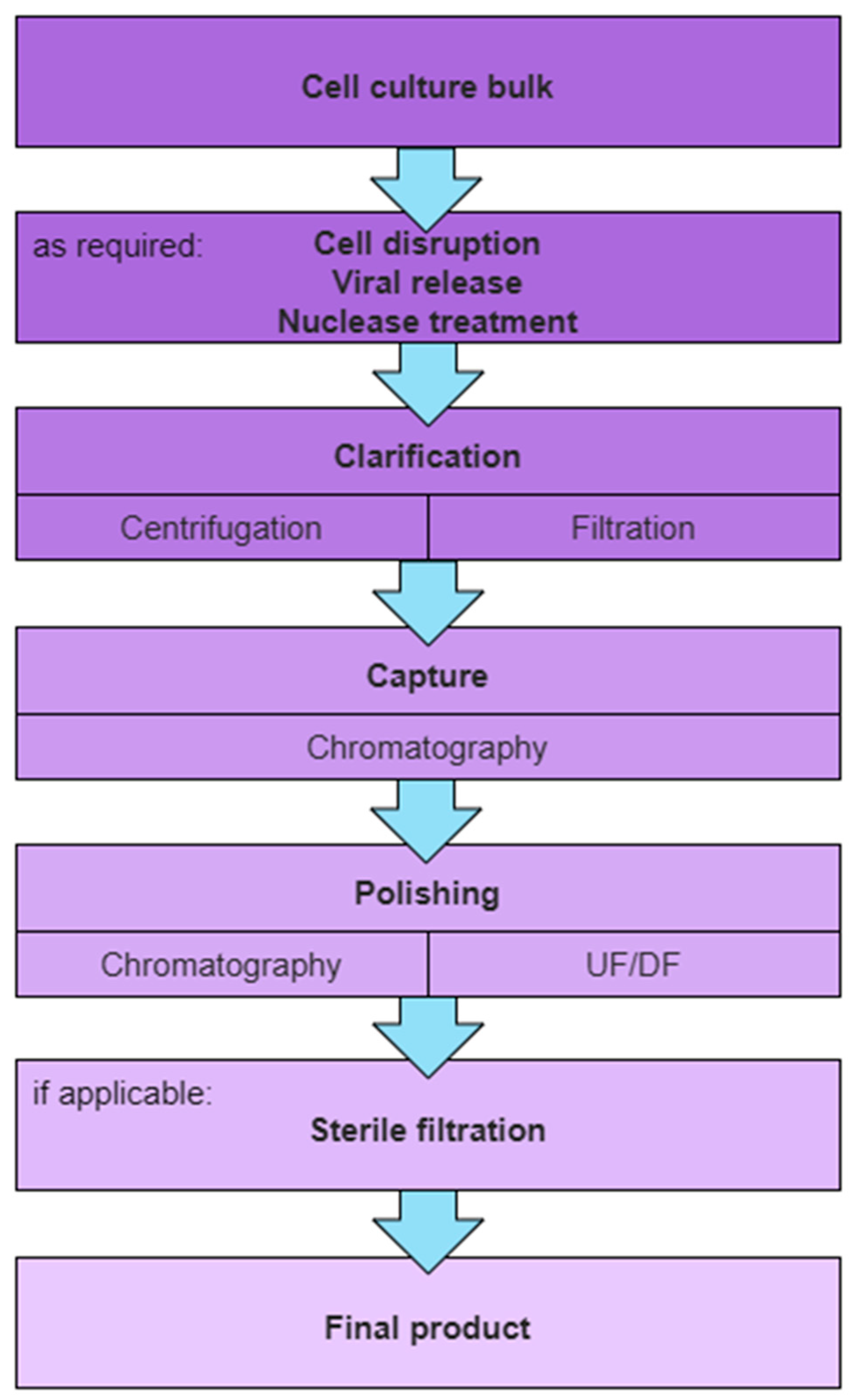
1.2. General Purification Scheme for Therapeutic Enveloped VPs
2. VP Integrity and Analytical Methods for Its Assessment
2.1. Quantification Methods
2.1.1. Infectious Titer Assays
2.1.2. Total Virus Particles
2.1.3. Total Particles by Light Scattering (LS)
2.2. Structure and Composition
2.2.1. Structure
2.2.2. Composition
2.2.3. Flow Virometry
2.2.4. Mass Balance for Virus Process Development
2.3. Further Commentary
3. Cell Culture and Infection
4. Harvest of Viral Particles
4.1. Continuous Harvest
4.2. Cell Disruption
4.3. Viral Release
4.4. Nuclease
5. Centrifugation Methods
5.1. Low-Speed Centrifugation: Clarification
5.2. Differential Centrifugation: Concentration and Partial Purification
5.3. Density Gradient Centrifugation: Concentration and Purification
5.4. Centrifugation Summary
6. Filtration Methods
6.1. Normal Flow Filtration (NFF)
6.1.1. NFF for Clarification
6.1.2. NFF for Sterile Filtration
6.2. Tangential Flow Filtration (TFF)
6.2.1. TFF for Clarification
6.2.2. TFF for Buffer Exchange and Concentration
6.3. Filtration Summary
7. Chromatography Methods
7.1. Chromatographic Modalities
7.1.1. Ion Exchange Chromatography (IEX)
7.1.2. Affinity Chromatography
7.1.3. Hydrophobic Interaction Chromatography (HIC)
7.1.4. Steric Exclusion Chromatography (SXC)
7.1.5. Surface Functionalization
7.1.6. Size Exclusion Chromatography (SEC)
7.2. Chromatographic Stationary Phases
7.2.1. Bead-Based Resins
7.2.2. Membrane Adsorber
7.2.3. Monolith Resins
7.2.4. Monolith-like Particles (MLP)
7.2.5. Cryogels
7.2.6. Fibers
7.2.7. Restricted-Access-Media (RAM)
7.3. Chromatography Summary
8. Further VP Purification Techniques
8.1. Flocculation
8.2. Aqueous Two-Phase (ATP) System
9. Formulation and Storage
9.1. Liquid Formulation
9.2. Lyophilized Formulation
10. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | adeno-associated viral vectors |
| AEX | anion-exchange chromatography |
| ATF | alternating TFF |
| ATMP | advanced therapy medicinal product |
| ATP | aqueous two-phase |
| B&E | bind-and-elute |
| BV | baculovirus |
| CA | cellulose acetate |
| CC700 | CaptoCore700 |
| CEX | cation-exchange chromatography |
| CIP | cleaning-in-place |
| DBC | dynamic binding capacity |
| DE | diatomaceous earth |
| DENV | dengue virus |
| DF | diafiltration |
| DIP | defective interfering particle |
| DLS | dynamic light scattering |
| DNA | deoxyribonucleic acid |
| ELISA | enzyme-linked immunosorbent assay |
| EM | electron microscopy |
| EU | European Union |
| EV | extracellular vesicle |
| FDA | food and drug administration |
| FFF | field flow fractionation |
| FMDV | foot-and-mouth-disease virus |
| FT | flowthrough |
| FT-cycle | freeze/thaw cycle |
| GF | glass fibers |
| HA | hemagglutination assay |
| HIC | hydrophobic interaction chromatography |
| HIV | human immunodeficiency viruses |
| HPLC | high-performance liquid chromatography |
| HPV | human papillomavirus |
| HSV | herpes simplex virus |
| IEP | isoelectric point |
| IEX | ion exchange chromatography |
| IU | infectious unit |
| LCMV | lymphocytic choriomeningitis |
| LS | light scattering |
| LV | lentivirus |
| MALDI-MS | matrix-assisted laser desorption/ionization MS |
| MALS | multi-angle LS |
| MLP | monolith-like particles |
| MOI | multiplicity of infection |
| MS | mass spectrometry |
| MVA | modified vaccinia ankara |
| NC | nitrocellulose |
| NDV | Newcastle disease virus |
| NFF | normal flow filtration |
| NGS | next-generation sequencing |
| NTA | nanoparticle tracking analysis |
| NYL | nylon |
| OV | oncolytic virus |
| P:IU | particles-to-infectious unit ratio |
| PAT | process analytical technology |
| PCC | periodic countercurrent chromatography |
| PCR | polymerase chain reaction |
| PEG | polyethylene glycol |
| PES | polyethersulfone |
| PFAS | polyfluoroalkyl substance |
| PP | polypropylene |
| PS | polysulfone |
| PVDF | polyvinylidene fluoride |
| RAM | restricted access media |
| RC | regenerated cellulose |
| RI | refractive index |
| RID | radial immunodiffusion |
| RNA | ribonucleic acid |
| RSV | respiratory syncytial virus |
| SBC | static binding capacity |
| SC | sucrose cushion |
| SEC | size exclusion chromatography |
| SLS | static LS |
| SMB | simulated moving bed |
| STR | stirred tank reactor |
| SXC | steric exclusion chromatography |
| T-VEC | Talimogene laherparepvec |
| TCID50 | 50% tissue culture infective dose assay |
| TEM | transmission electron microscopy |
| TFDF | tangential flow depth filtration |
| TFF | tangential flow filtration |
| TMP | transmembrane pressure |
| TOH | timepoints of harvest |
| TOI | timepoint of infection |
| UF | ultrafiltration |
| UV | ultraviolet |
| VLP | virus-like particle |
| VP | virus particle |
| VSV | vesicular stomatitis virus |
| aMPV | avian metapneumovirus |
| cGE | capillary gel electrophoresis |
| cyro-EM | cryogenic EM |
| dPCR | digital PCR |
| ddPCR | digital droplet PCR |
| env. VP | enveloped VP |
| hcDNA | host cell DNA |
| hcP | host cell protein |
| hpi | hours post-infection |
| inf. VP | infectious VP |
| non-env. VP | non-enveloped VP |
| non-inf. VP | non-infectious VP |
| nsEM | negative staining EM |
| pfu | plaque-forming units |
| qPCR | quantitative PCR |
References
- bin Umair, M.; Akusa, F.N.; Kashif, H.; Seerat-e-Fatima; Butt, F.; Azhar, M.; Munir, I.; Ahmed, M.; Khalil, W.; Sharyar, H.; et al. Viruses as Tools in Gene Therapy, Vaccine Development, and Cancer Treatment. Arch. Virol. 2022, 167, 1387–1404. [Google Scholar] [CrossRef]
- Montero, D.A.; Vidal, R.M.; Velasco, J.; Carreño, L.J.; Torres, J.P.; Benachi O., M.A.; Tovar-Rosero, Y.-Y.; Oñate, A.A.; O’Ryan, M. Two Centuries of Vaccination: Historical and Conceptual Approach and Future Perspectives. Front. Public. Health 2024, 11, 1326154. [Google Scholar] [CrossRef]
- Salmon, F.; Grosios, K.; Petry, H. Safety Profile of Recombinant Adeno-Associated Viral Vectors: Focus on Alipogene Tiparvovec (Glybera®). Expert Rev. Clin. Pharmacol. 2014, 7, 53–65. [Google Scholar] [CrossRef] [PubMed]
- White, M.; Whittaker, R.; Gándara, C.; Stoll, E.A. A Guide to Approaching Regulatory Considerations for Lentiviral-Mediated Gene Therapies. Hum. Gene Ther. Methods 2017, 28, 163–176. [Google Scholar] [CrossRef]
- Alwithenani, A.; Hengswat, P.; Chiocca, E.A. Oncolytic Viruses as Cancer Therapeutics: From Mechanistic Insights to Clinical Translation. Mol. Ther. 2025. [Google Scholar] [CrossRef] [PubMed]
- Schoeps, B.; Lauer, U.M.; Elbers, K. Deciphering Permissivity of Human Tumor Ecosystems to Oncolytic Viruses. Oncogene 2025, 44, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic Viruses as Therapeutic Cancer Vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef]
- Kohlhapp, F.J.; Kaufman, H.L. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. 2016, 22, 1048–1054. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. On Clinicaltrials.gov, the Search Term “Oncolytic Virus” was Used as a Keyword in the Field Other Terms. The Recruitment Status Was Filtered for not yet Recruiting; Recruiting; Enrolling by Invitation; Active, Not Recruiting. The search was Conducted on 21 February 2025 with 103 Results (67 using Enveloped, 33 non-Enveloped VPs). Available online: https://clinicaltrials.gov/search?term=oncolytic%20virus (accessed on 21 February 2025).
- He, B.; Wilson, B.; Chen, S.-H.; Sharma, K.; Scappini, E.; Cook, M.; Petrovich, R.; Martin, N.P. Molecular Engineering of Virus Tropism. Int. J. Mol. Sci. 2024, 25, 11094. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy: Where Do We Stand in 2023? Viruses 2023, 15, 698. [Google Scholar] [CrossRef]
- EMA (European Medicines Agency), Guideline on Quality, Non-Clinical and Clinical Requirements for Investigational Advanced Therapy Medicinal Products in Clinical Trials; Ref. No. EMA/CAT/22473/2025, 2025.
- EMA (European Medicines Agency), Guideline on the Quality, Non-Clinical and Clinical Aspects of Gene Therapy Medicinal Products; Ref. No. EMA/CAT/80183/2014, 2018.
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving Oncolytic Viruses into the Clinic: Clinical-Grade Production, Purification, and Characterization of Diverse Oncolytic Viruses. Mol. Ther. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [PubMed]
- Eilts, F.; Steger, M.; Pagallies, F.; Rziha, H.-J.; Hardt, M.; Amann, R.; Wolff, M.W. Comparison of Sample Preparation Techniques for the Physicochemical Characterization of Orf Virus Particles. J. Virol. Methods 2022, 310, 114614. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, Z.; Chiocca, E.A.; Caligiuri, M.A.; Yu, J. The Emerging Field of Oncolytic Virus-Based Cancer Immunotherapy. Trends Cancer 2023, 9, 122–139. [Google Scholar] [CrossRef]
- Rossini, E.; Bazzucchi, M.; Trocchi, V.; Merzoni, F.; Bertasio, C.; Knauf, S.; Lavazza, A.; Cavadini, P. Identification and Characterisation of a Myxoma Virus Detected in the Italian Hare (Lepus Corsicanus). Viruses 2024, 16, 437. [Google Scholar] [CrossRef]
- Ge, P.; Tsao, J.; Schein, S.; Green, T.J.; Luo, M.; Hong Zhou, Z. Cryo-EM Model of the Bullet-Shaped Vesicular Stomatitis Virus. Science 2010, 327, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, T.; Xi, G.; Ampey, A.; Borman, J.; Jaroudi, S.; Pappas, D.; Linke, T. Purification of a Recombinant Oncolytic Virus from Clarified Cell Culture Media by Anion Exchange Monolith Chromatography. Electrophoresis 2023, 44, 1923–1933. [Google Scholar] [CrossRef]
- Stachura, P.; Stencel, O.; Lu, Z.; Borkhardt, A.; Pandyra, A.A. Arenaviruses: Old Viruses Present New Solutions for Cancer Therapy. Front. Immunol. 2023, 14, 1110522. [Google Scholar] [CrossRef]
- Lee, K.J.; Novella, I.S.; Teng, M.N.; Oldstone, M.B.A.; de la Torre, J.C. NP and L Proteins of Lymphocytic Choriomeningitis Virus (LCMV) Are Sufficient for Efficient Transcription and Replication of LCMV Genomic RNA Analogs. J. Virol. 2000, 74, 3470–3477. [Google Scholar] [CrossRef]
- Neuman, B.W.; Adair, B.D.; Burns, J.W.; Milligan, R.A.; Buchmeier, M.J.; Yeager, M. Complementarity in the Supramolecular Design of Arenaviruses and Retroviruses Revealed by Electron Cryomicroscopy and Image Analysis. J. Virol. 2005, 79, 3822–3830. [Google Scholar] [CrossRef]
- Kabiljo, J.; Laengle, J.; Bergmann, M. From Threat to Cure: Understanding of Virus-Induced Cell Death Leads to Highly Immunogenic Oncolytic Influenza Viruses. Cell Death Discov. 2020, 6, 48. [Google Scholar] [CrossRef]
- Brown, E.G. Influenza Virus Genetics. Biomed. Pharmacother. 2000, 54, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Yamasaki, H.; Ikeda, K.; Ejima, D.; Naito, T.; Koyama, A. Antiviral and Virucidal Activities of Natural Products. Curr. Med. Chem. 2009, 16, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Salzig, D.; Gerstenberger, J.; Röder, Y.; Cichutek, K.; Pörtner, R.; Czermak, P.; Mühlebach, M.D. Influence of Process Conditions on Measles Virus Stability. Am. J. Biochem. Biotechnol. 2013, 9, 243–254. [Google Scholar] [CrossRef]
- Loewe, D.; Häussler, J.; Grein, T.A.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Forced Degradation Studies to Identify Critical Process Parameters for the Purification of Infectious Measles Virus. Viruses 2019, 11, 725. [Google Scholar] [CrossRef]
- Sviben, D.; Forčić, D.; Kurtović, T.; Halassy, B.; Brgles, M. Stability, Biophysical Properties and Effect of Ultracentrifugation and Diafiltration on Measles Virus and Mumps Virus. Arch. Virol. 2016, 161, 1455–1467. [Google Scholar] [CrossRef]
- Higashikawa, F.; Chang, L.-J. Kinetic Analyses of Stability of Simple and Complex Retroviral Vectors. Virology 2001, 280, 124–131. [Google Scholar] [CrossRef]
- Segura, M.d.l.M.; Kamen, A.; Trudel, P.; Garnier, A. A Novel Purification Strategy for Retrovirus Gene Therapy Vectors Using Heparin Affinity Chromatography. Biotechnol. Bioeng. 2005, 90, 391–404. [Google Scholar] [CrossRef]
- Huangfu, C.; Zhang, J.; Ma, Y.; Jia, J.; Li, J.; Lv, M.; Ma, X.; Zhao, X.; Zhang, J. Large-scale Purification of High Purity A1-antitrypsin from Cohn Fraction IV with Virus Inactivation by Solvent/Detergent and Dry-heat Treatment. Biotechnol. Appl. Biochem. 2018, 65, 446–454. [Google Scholar] [CrossRef]
- Roberts, P.L. Virus Inactivation by Solvent/Detergent Treatment Using Triton X-100 in a High Purity Factor VIII. Biologicals 2008, 36, 330–335. [Google Scholar] [CrossRef]
- Meingast, C.; Heldt, C.L. Arginine-enveloped Virus Inactivation and Potential Mechanisms. Biotechnol. Prog. 2020, 36, e2931. [Google Scholar] [CrossRef]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular Stomatitis Virus G Glycoprotein Pseudotyped Retroviral Vectors: Concentration to Very High Titer and Efficient Gene Transfer into Mammalian and Nonmammalian Cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Eisensmith, R.C.; Mohuiddin, R.; Pyron, M.; Woo, S.L.C. A Simple and Efficient Method for the Concentration and Purification of Recombinant Retrovirus for Increased Hepatocyte Transduction In Vivo. Hum. Gene Ther. 1996, 7, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Michalsky, R.; Pfromm, P.H.; Czermak, P.; Sorensen, C.M.; Passarelli, A.L. Effects of Temperature and Shear Force on Infectivity of the Baculovirus Autographa Californica M Nucleopolyhedrovirus. J. Virol. Methods 2008, 153, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Schimek, A.; Ng, J.K.M.; Basbas, I.; Martin, F.; Xin, D.; Saleh, D.; Hubbuch, J. An HPLC-SEC-Based Rapid Quantification Method for Vesicular Stomatitis Virus Particles to Facilitate Process Development. Mol. Ther. Methods Clin. Dev. 2024, 32, 101252. [Google Scholar] [CrossRef]
- Eilts, F.; Labisch, J.J.; Orbay, S.; Harsy, Y.M.J.; Steger, M.; Pagallies, F.; Amann, R.; Pflanz, K.; Wolff, M.W. Stability Studies for the Identification of Critical Process Parameters for a Pharmaceutical Production of the Orf Virus. Vaccine 2023, 41, 4731–4742. [Google Scholar] [CrossRef]
- Lesch, H.P.; Laitinen, A.; Peixoto, C.; Vicente, T.; Makkonen, K.-E.; Laitinen, L.; Pikkarainen, J.T.; Samaranayake, H.; Alves, P.M.; Carrondo, M.J.T.; et al. Production and Purification of Lentiviral Vectors Generated in 293T Suspension Cells with Baculoviral Vectors. Gene Ther. 2011, 18, 531–538. [Google Scholar] [CrossRef]
- Flint, S.J.; Racaniello, V.R.; Rall, G.F.; Hatziioannou, T.; Skalka, A.M. Principles of Virology; The Biomedical & Life Sciences Collection; ASM Press: Washington, DC, USA, 2020; ISBN 978-1-683-67360-6. [Google Scholar]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.N.; Scheel, T.K.H.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable Region 1 Differentially Impacts Viability of Hepatitis C Virus Strains of Genotypes 1 to 6 and Impairs Virus Neutralization. J. Virol. 2010, 85, 2224–2234. [Google Scholar] [CrossRef]
- Miller, C.A.; Raine, C.S. Heterogeneity of Virus Particles in Measles Virus. J. Gen. Virol. 1979, 45, 441–453. [Google Scholar] [CrossRef]
- Lodish, H.F.; Porter, M. Heterogeneity of Vesicular Stomatitis Virus Particles: Implications for Virion Assembly. J. Virol. 1980, 33, 52–58. [Google Scholar] [CrossRef]
- DeLong, J.P.; Al-Sammak, M.A.; Al-Ameeli, Z.T.; Dunigan, D.D.; Edwards, K.F.; Fuhrmann, J.J.; Gleghorn, J.P.; Li, H.; Haramoto, K.; Harrison, A.O.; et al. Towards an Integrative View of Virus Phenotypes. Nat. Rev. Microbiol. 2022, 20, 83–94. [Google Scholar] [CrossRef]
- Sviben, D.; Forcic, D.; Ivancic-Jelecki, J.; Halassy, B.; Brgles, M. Recovery of Infective Virus Particles in Ion-Exchange and Hydrophobic Interaction Monolith Chromatography Is Influenced by Particle Charge and Total-to-Infective Particle Ratio. J. Chromatogr. B 2017, 1054, 10–19. [Google Scholar] [CrossRef]
- Cantin, R.; Méthot, S.; Tremblay, M.J. Plunder and Stowaways: Incorporation of Cellular Proteins by Enveloped Viruses. J. Virol. 2005, 79, 6577–6587. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.d.l.M.; Garnier, A.; Kamen, A. Purification and Characterization of Retrovirus Vector Particles by Rate Zonal Ultracentrifugation. J. Virol. Methods 2006, 133, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Aitken, J.; Rixon, H.W.M.; Sugrue, R.J. Caveolin-1 Is Incorporated into Mature Respiratory Syncytial Virus Particles during Virus Assembly on the Surface of Virus-Infected Cells. J. Gen. Virol. 2002, 83, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Hammarstedt, M.; Ahlqvist, J.; Jacobson, S.; Garoff, H.; Fogdell-Hahn, A. Purification of Infectious Human Herpesvirus 6A Virions and Association of Host Cell Proteins. Virol. J. 2007, 4, 101. [Google Scholar] [CrossRef]
- Stegen, C.; Yakova, Y.; Henaff, D.; Nadjar, J.; Duron, J.; Lippé, R. Analysis of Virion-Incorporated Host Proteins Required for Herpes Simplex Virus Type 1 Infection through a RNA Interference Screen. PLoS ONE 2013, 8, e53276. [Google Scholar] [CrossRef]
- Minh, A.D.; Kamen, A.A. Critical Assessment of Purification and Analytical Technologies for Enveloped Viral Vector and Vaccine Processing and Their Current Limitations in Resolving Co-Expressed Extracellular Vesicles. Vaccines 2021, 9, 823. [Google Scholar] [CrossRef]
- Cantin, R.; Diou, J.; Bélanger, D.; Tremblay, A.M.; Gilbert, C. Discrimination between Exosomes and HIV-1: Purification of Both Vesicles from Cell-Free Supernatants. J. Immunol. Methods 2008, 338, 21–30. [Google Scholar] [CrossRef]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef]
- Cordelier, P. Unveiling the Nexus: Oncolytic Viruses, Extracellular Vesicles, and Immune Modulation. Mol. Ther. Oncol. 2025, 33, 200940. [Google Scholar] [CrossRef]
- McCormick, W.; Mermel, L.A. The Basic Reproductive Number and Particle-to-Plaque Ratio: Comparison of These Two Parameters of Viral Infectivity. Virol. J. 2021, 18, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.; Cao, A.; Yin, J. Virus-like Particles: Measures and Biological Functions. Viruses 2022, 14, 383. [Google Scholar] [CrossRef] [PubMed]
- Frensing, T.; Heldt, F.S.; Pflugmacher, A.; Behrendt, I.; Jordan, I.; Flockerzi, D.; Genzel, Y.; Reichl, U. Continuous Influenza Virus Production in Cell Culture Shows a Periodic Accumulation of Defective Interfering Particles. PLoS ONE 2013, 8, e72288. [Google Scholar] [CrossRef]
- Tan, E.; Chin, C.S.H.; Lim, Z.F.S.; Ng, S.K. HEK293 Cell Line as a Platform to Produce Recombinant Proteins and Viral Vectors. Front. Bioeng. Biotechnol. 2021, 9, 796991. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, Z.; Zhang, Y.; Huang, X.; Liu, Q. Efficacy and Safety of Oncolytic Viruses in Randomized Controlled Trials: A Systematic Review and Meta-Analysis. Cancers 2020, 12, 1416. [Google Scholar] [CrossRef]
- Minh, A.D.; Star, A.T.; Stupak, J.; Fulton, K.M.; Haqqani, A.S.; Gélinas, J.-F.; Li, J.; Twine, S.M.; Kamen, A.A. Characterization of Extracellular Vesicles Secreted in Lentiviral Producing HEK293SF Cell Cultures. Viruses 2021, 13, 797. [Google Scholar] [CrossRef]
- Ng, J.K.M. Product-Related Impurities in Therapeutic Virus Bioprocessing. In Bioprocess and Analytics Development for Virus-Based Advanced Therapeutics and Medicinal Products (ATMPs); Springer: Cham, Switzerland, 2023; pp. 277–294. ISBN 9783031284885. [Google Scholar]
- Russell, S.J.; Federspiel, M.J.; Peng, K.-W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of Disseminated Cancer After Systemic Oncolytic Virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef]
- Cook, J.; Peng, K.-W.; Ginos, B.F.; Dueck, A.C.; Giers, M.; Packiriswamy, N.; Brunton, B.; Patnaik, M.M.; Witzig, T.E.; Buadi, F.K.; et al. Phase I Trial of Systemic Administration of Vesicular Stomatitis Virus Genetically Engineered to Express NIS and Human Interferon Beta, in Patients with Relapsed or Refractory Multiple Myeloma (MM), Acute Myeloid Leukemia (AML), and T-Cell Neoplasms (TCL). Blood 2020, 136, 7–8. [Google Scholar] [CrossRef]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Virion Structure and Composition. In Fenner and White’s Medical Virology, 5th ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 27–37. [Google Scholar] [CrossRef]
- Lothert, K.; Eilts, F.; Wolff, M.W. Quantification Methods for Viruses and Virus-like Particles Applied in Biopharmaceutical Production Processes. Expert Rev. Vaccines 2022, 21, 1029–1044. [Google Scholar] [CrossRef]
- LaBarre, D.D.; Lowy, R.J. Improvements in Methods for Calculating Virus Titer Estimates from TCID50 and Plaque Assays. J. Virol. Methods 2001, 96, 107–126. [Google Scholar] [CrossRef]
- Steppert, P.; Mosor, M.; Stanek, L.; Burgstaller, D.; Palmberger, D.; Preinsperger, S.; Aguilar, P.P.; Müllner, M.; Csar, P.; Jungbauer, A. A Scalable, Integrated Downstream Process for Production of a Recombinant Measles Virus-Vectored Vaccine. Vaccine 2022, 40, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Hochdorfer, D.; Businger, R.; Hotter, D.; Seifried, C.; Solzin, J. Automated, Label-Free TCID50 Assay to Determine the Infectious Titer of Virus-Based Therapeutics. J. Virol. Methods 2022, 299, 114318. [Google Scholar] [CrossRef] [PubMed]
- Hotter, D.; Kunzelmann, M.; Kiefer, F.; Leukhardt, C.; Fackler, C.; Jäger, S.; Solzin, J. High-Throughput Determination of Infectious Virus Titers by Kinetic Measurement of Infection-Induced Changes in Cell Morphology. Int. J. Mol. Sci. 2024, 25, 8076. [Google Scholar] [CrossRef]
- Lothert, K.; Pagallies, F.; Eilts, F.; Sivanesapillai, A.; Hardt, M.; Moebus, A.; Feger, T.; Amann, R.; Wolff, M.W. A Scalable Downstream Process for the Purification of the Cell Culture-Derived Orf Virus for Human or Veterinary Applications. J. Biotechnol. 2020, 323, 221–230. [Google Scholar] [CrossRef]
- Strain, M.C.; Lada, S.M.; Luong, T.; Rought, S.E.; Gianella, S.; Terry, V.H.; Spina, C.A.; Woelk, C.H.; Richman, D.D. Highly Precise Measurement of HIV DNA by Droplet Digital PCR. PLoS ONE 2013, 8, e55943. [Google Scholar] [CrossRef] [PubMed]
- Furuta-Hanawa, B.; Yamaguchi, T.; Uchida, E. Two-Dimensional Droplet Digital PCR as a Tool for Titration and Integrity Evaluation of Recombinant Adeno-Associated Viral Vectors. Hum. Gene Ther. Methods 2019, 30, 127–136. [Google Scholar] [CrossRef]
- Wong, K.; Mukherjee, B.; Kahler, A.M.; Zepp, R.; Molina, M. Influence of Inorganic Ions on Aggregation and Adsorption Behaviors of Human Adenovirus. Environ. Sci. Technol. 2012, 46, 11145–11153. [Google Scholar] [CrossRef]
- Bohren, C.F.; Huffman, D.R. Absorption and Scattering of Light by Small Particles; Wiley-VCH: Weinheim, Germany, 2023. [Google Scholar] [CrossRef]
- Schimek, A.; Strebl, M.; Blech, M.; Garidel, P. Challenges at Submicron Particle Characterisation: A Case Study Using Nanoparticle Tracking Analysis (NTA). J. Pharm. Innov. 2024, 19, 29. [Google Scholar] [CrossRef]
- Kramberger, P.; Ciringer, M.; Štrancar, A.; Peterka, M. Evaluation of Nanoparticle Tracking Analysis for Total Virus Particle Determination. Virol. J. 2012, 9, 265. [Google Scholar] [CrossRef]
- Aguilar, P.P.; González-Domínguez, I.; Schneider, T.A.; Gòdia, F.; Cervera, L.; Jungbauer, A. At-line Multi-angle Light Scattering Detector for Faster Process Development in Enveloped Virus-like Particle Purification. J. Sep. Sci. 2019, 42, 2640–2649. [Google Scholar] [CrossRef]
- Bousse, T.; Shore, D.A.; Goldsmith, C.S.; Hossain, M.J.; Jang, Y.; Davis, C.T.; Donis, R.O.; Stevens, J. Quantitation of Influenza Virus Using Field Flow Fractionation and Multi-Angle Light Scattering for Quantifying Influenza A Particles. J. Virol. Methods 2013, 193, 589–596. [Google Scholar] [CrossRef]
- Zamora, J.L.R.; Aguilar, H.C. Flow Virometry as a Tool to Study Viruses. Methods 2018, 134, 87–97. [Google Scholar] [CrossRef]
- Kiss, G.; Chen, X.; Brindley, M.A.; Campbell, P.; Afonso, C.L.; Ke, Z.; Holl, J.M.; Guerrero-Ferreira, R.C.; Byrd-Leotis, L.A.; Steel, J.; et al. Capturing Enveloped Viruses on Affinity Grids for Downstream Cryo-Electron Microscopy Applications. Microsc. Microanal. 2014, 20, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Stass, R.; Ng, W.M.; Kim, Y.C.; Huiskonen, J.T. Chapter Two Structures of Enveloped Virions Determined by Cryogenic Electron Microscopy and Tomography. Adv. Virus Res. 2019, 105, 35–71. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.A.; Earnest, L.; Montoya, J.C.; Yap, A.H.Y.; Mumford, J.; Roberts, J.; Wong, C.Y.; Hans, D.; Grima, J.; Bisset, N.; et al. Development of Methods to Produce SARS CoV-2 Virus-Like Particles at Scale. Biotechnol. Bioeng. 2025, 122, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Xin, D.; Kurien, L.; Briggs, K.; Schimek, A.; Dambra, R.; Hochdorfer, D.; Arnouk, T.A.; Brgles, M.; Gautam, S.; Hotter, D.; et al. Characterization of VSV-GP Morphology by CryoEM Imaging and SEC-MALS. Mol. Ther. Methods Clin. Dev. 2025, 33, 101429. [Google Scholar] [CrossRef]
- Rziha, H.-J.; Büttner, M.; Müller, M.; Salomon, F.; Reguzova, A.; Laible, D.; Amann, R. Genomic Characterization of Orf Virus Strain D1701-V (Parapoxvirus) and Development of Novel Sites for Multiple Transgene Expression. Viruses 2019, 11, 127. [Google Scholar] [CrossRef]
- Willemsen, A.; Zwart, M.P. On the Stability of Sequences Inserted into Viral Genomes. Virus Evol. 2019, 5, vez045. [Google Scholar] [CrossRef]
- Basu, R.; Dambra, R.; Jiang, D.; Schätzlein, S.A.; Njiyang, S.; Ashour, J.; Chiramel, A.I.; Vigil, A.; Papov, V.V. Absolute Quantification of Viral Proteins from Pseudotyped VSV-GP Using UPLC-MRM. Microbiol. Spectr. 2024, 12, e03651-23. [Google Scholar] [CrossRef]
- Sviben, D.; Forcic, D.; Halassy, B.; Allmaier, G.; Marchetti-Deschmann, M.; Brgles, M. Mass Spectrometry-Based Investigation of Measles and Mumps Virus Proteome. Virol. J. 2018, 15, 160. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Hwang, S.-I.; Grdzelishvili, V.Z. Cellular Proteins Associated with the Interior and Exterior of Vesicular Stomatitis Virus Virions. PLoS ONE 2014, 9, e104688. [Google Scholar] [CrossRef]
- Sousa, I.P.; Carvalho, C.A.M.; Ferreira, D.F.; Weissmüller, G.; Rocha, G.M.; Silva, J.L.; Gomes, A.M.O. Envelope Lipid-Packing as a Critical Factor for the Biological Activity and Stability of Alphavirus Particles Isolated from Mammalian and Mosquito Cells*. J. Biol. Chem. 2011, 286, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Calisan, K.K.; Stitz, J.; Barbe, S. The Effects of High Shear Rates on the Average Hydrodynamic Diameter Measured in Biomimetic HIV Gag Virus-like Particle Dispersions. Front. Bioeng. Biotechnol. 2024, 12, 1367405. [Google Scholar] [CrossRef]
- Kim, S.; Lim, K. Stability of Retroviral Vectors Against Ultracentrifugation Is Determined by the Viral Internal Core and Envelope Proteins Used for Pseudotyping. Mol. Cells 2017, 40, 339–345. [Google Scholar] [CrossRef]
- Brgles, M.; Bonta, M.; Šantak, M.; Jagušić, M.; Forčić, D.; Halassy, B.; Allmaier, G.; Marchetti-Deschmann, M. Identification of Mumps Virus Protein and Lipid Composition by Mass Spectrometry. Virol. J. 2016, 13, 9. [Google Scholar] [CrossRef]
- Fernandes, R.P.; Escandell, J.M.; Guerreiro, A.C.L.; Moura, F.; Faria, T.Q.; Carvalho, S.B.; Silva, R.J.S.; Gomes-Alves, P.; Peixoto, C. Assessing Multi-Attribute Characterization of Enveloped and Non-Enveloped Viral Particles by Capillary Electrophoresis. Viruses 2022, 14, 2539. [Google Scholar] [CrossRef]
- Minsker, K.; Rustandi, R.R.; Ha, S.; Loughney, J.W. Characterization of RVSVΔG-ZEBOV-GP Glycoproteins Using Automated Capillary Western Blotting. Vaccine 2020, 38, 7166–7174. [Google Scholar] [CrossRef] [PubMed]
- Lippé, R. Flow Virometry: A Powerful Tool To Functionally Characterize Viruses. J. Virol. 2018, 92, e01765-17. [Google Scholar] [CrossRef] [PubMed]
- Drori, P.; Mouhadeb, O.; Muñoz, G.G.M.; Razvag, Y.; Alcalay, R.; Klocke, P.; Cordes, T.; Zahavy, E.; Lerner, E. Rapid and Specific Detection of Nanoparticles and Viruses One at a Time Using Microfluidic Laminar Flow and Confocal Fluorescence Microscopy. iScience 2024, 27, 110982. [Google Scholar] [CrossRef]
- Ricci, G.; Minsker, K.; Kapish, A.; Osborn, J.; Ha, S.; Davide, J.; Califano, J.P.; Sehlin, D.; Rustandi, R.R.; Dick, L.W.; et al. Flow Virometry for Process Monitoring of Live Virus Vaccines-Lessons Learned from ERVEBO. Sci. Rep. 2021, 11, 7432. [Google Scholar] [CrossRef]
- Vlasak, J.; Hoang, V.M.; Christanti, S.; Peluso, R.; Li, F.; Culp, T.D. Use of Flow Cytometry for Characterization of Human Cytomegalovirus Vaccine Particles. Vaccine 2016, 34, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Torelli, A.; Piu, P.; Bonifazi, C.; Ganfini, L.; Montomoli, E. Flow Cytometry as an Integrative Method for the Evaluation of Vaccine Immunogenicity: A Validation Approach. Biochem. Biophys. Rep. 2023, 34, 101472. [Google Scholar] [CrossRef]
- Deng, J.Z.; Rustandi, R.R.; Swartz, A.; Shieh, Y.; Baker, J.B.; Vlasak, J.; Wang, S.; Loughney, J.W. SEC Coupled with In-Line Multiple Detectors for the Characterization of an Oncolytic Coxsackievirus. Mol. Ther. Oncolytics 2022, 24, 139–147. [Google Scholar] [CrossRef]
- Yi, S.; McCracken, R.; Davide, J.; Salovich, D.R.; Whitmer, T.; Bhat, A.; Vlasak, J.; Ha, S.; Sehlin, D.; Califano, J.; et al. Development of Process Analytical Tools for Rapid Monitoring of Live Virus Vaccines in Manufacturing. Sci. Rep. 2022, 12, 15494. [Google Scholar] [CrossRef] [PubMed]
- Vlecken, D.H.W.; Pelgrim, R.P.M.; Ruminski, S.; Bakker, W.A.M.; Pol, L.A. van der Comparison of Initial Feasibility of Host Cell Lines for Viral Vaccine Production. J. Virol. Methods 2013, 193, 28–41. [Google Scholar] [CrossRef]
- Sharma, R.; Harrison, S.T.L.; Tai, S.L. Advances in Bioreactor Systems for the Production of Biologicals in Mammalian Cells. ChemBioEng Rev. 2022, 9, 42–62. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Aeration and Shear Stress Are Critical Process Parameters for the Production of Oncolytic Measles Virus. Front. Bioeng. Biotechnol. 2019, 7, 78. [Google Scholar] [CrossRef]
- YekrangSafakar, A.; Acun, A.; Choi, J.; Song, E.; Zorlutuna, P.; Park, K. Hollow Microcarriers for Large-scale Expansion of Anchorage-dependent Cells in a Stirred Bioreactor. Biotechnol. Bioeng. 2018, 115, 1717–1728. [Google Scholar] [CrossRef]
- Kaiser, S.C.; Decaria, P.N.; Seidel, S.; Eibl, D. Scaling-up of an Insect Cell-based Virus Production Process in a Novel Single-use Bioreactor with Flexible Agitation. Chem. Ing. Tech. 2022, 94, 1950–1961. [Google Scholar] [CrossRef]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell Cycle Perturbations Induced by Infection with the Coronavirus Infectious Bronchitis Virus and Their Effect on Virus Replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef]
- Zhu, Y.; Yongky, A.; Yin, J. Growth of an RNA Virus in Single Cells Reveals a Broad Fitness Distribution. Virology 2009, 385, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Salzig, D.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Key Parameters of Measles Virus Production for Oncolytic Virotherapy. Am. J. Biochem. Biotechnol. 2012, 8, 81–98. [Google Scholar] [CrossRef]
- Eckhardt, D.; Mueller, J.; Friedrich, J.; Klee, J.-P.; Sardlishvili, I.; Walter, L.E.; Fey, S.; Czermak, P.; Salzig, D. Production of Oncolytic Measles Virus in Vero Cells: Impact of Culture Medium and Multiplicity of Infection. Viruses 2024, 16, 1740. [Google Scholar] [CrossRef]
- Frensing, T. Defective Interfering Viruses and Their Impact on Vaccines and Viral Vectors. Biotechnol. J. 2015, 10, 681–689. [Google Scholar] [CrossRef]
- Thompson, K.A.S.; Yin, J. Population Dynamics of an RNA Virus and Its Defective Interfering Particles in Passage Cultures. Virol. J. 2010, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Paillet, C.; Forno, G.; Kratje, R.; Etcheverrigaray, M. Suspension-Vero Cell Cultures as a Platform for Viral Vaccine Production. Vaccine 2009, 27, 6464–6467. [Google Scholar] [CrossRef]
- Elahi, S.M.; Shen, C.F.; Gilbert, R. Optimization of Production of Vesicular Stomatitis Virus (VSV) in Suspension Serum-Free Culture Medium at High Cell Density. J. Biotechnol. 2019, 289, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Göbel, S.; Pelz, L.; Silva, C.A.T.; Brühlmann, B.; Hill, C.; Altomonte, J.; Kamen, A.; Reichl, U.; Genzel, Y. Production of Recombinant Vesicular Stomatitis Virus-Based Vectors by Tangential Flow Depth Filtration. Appl. Microbiol. Biotechnol. 2024, 108, 240. [Google Scholar] [CrossRef]
- Gutiérrez-Granados, S.; Gòdia, F.; Cervera, L. Continuous Manufacturing of Viral Particles. Curr. Opin. Chem. Eng. 2018, 22, 107–114. [Google Scholar] [CrossRef]
- Gadaleta, P.; Vacotto, M.; Coulombié, F. Vesicular Stomatitis Virus Induces Apoptosis at Early Stages in the Viral Cycle and Does Not Depend on Virus Replication. Virus Res. 2002, 86, 87–92. [Google Scholar] [CrossRef]
- Eymieux, S.; Rouillé, Y.; Terrier, O.; Seron, K.; Blanchard, E.; Rosa-Calatrava, M.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Ultrastructural Modifications Induced by SARS-CoV-2 in Vero Cells: A Kinetic Analysis of Viral Factory Formation, Viral Particle Morphogenesis and Virion Release. Cell. Mol. Life Sci. 2021, 78, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.M. Membrane Uncoating of Intact Enveloped Viruses. J. Virol. 2010, 84, 10946–10955. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. High Titer Oncolytic Measles Virus Production Process by Integration of Dielectric Spectroscopy as Online Monitoring System. Biotechnol. Bioeng. 2018, 115, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Petiot, E.; Jacob, D.; Lanthier, S.; Lohr, V.; Ansorge, S.; Kamen, A.A. Metabolic and Kinetic Analyses of Influenza Production in Perfusion HEK293 Cell Culture. BMC Biotechnol. 2011, 11, 84. [Google Scholar] [CrossRef]
- Gränicher, G.; Coronel, J.; Trampler, F.; Jordan, I.; Genzel, Y.; Reichl, U. Performance of an Acoustic Settler versus a Hollow Fiber–Based ATF Technology for Influenza Virus Production in Perfusion. Appl. Microbiol. Biotechnol. 2020, 104, 4877–4888. [Google Scholar] [CrossRef]
- Weiss, K.; Gerstenberger, J.; Salzig, D.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Oncolytic Measles Viruses Produced at Different Scales under Serum-free Conditions. Eng. Life Sci. 2015, 15, 425–436. [Google Scholar] [CrossRef]
- Habisch, R.; Neubauer, P.; Soza-Ried, J.; Puschmann, E. Repeated Harvest Enables Efficient Production of VSV-GP. Front. Bioeng. Biotechnol. 2024, 12, 1505338. [Google Scholar] [CrossRef]
- Shen, C.F.; Burney, E.; Gilbert, R.; Elahi, S.M.; Parato, K.; Loignon, M. Development, Optimization, and Scale-up of Suspension Vero Cell Culture Process for High Titer Production of Oncolytic Herpes Simplex Virus-1. Biotechnol. J. 2024, 19, e2300244. [Google Scholar] [CrossRef]
- Nikolay, A.; de Grooth, J.; Genzel, Y.; Wood, J.A.; Reichl, U. Virus Harvesting in Perfusion Culture: Choosing the Right Type of Hollow Fiber Membrane. Biotechnol. Bioeng. 2020, 117, 3040–3052. [Google Scholar] [CrossRef]
- Vázquez-Ramírez, D.; Jordan, I.; Sandig, V.; Genzel, Y.; Reichl, U. High Titer MVA and Influenza A Virus Production Using a Hybrid Fed-Batch/Perfusion Strategy with an ATF System. Appl. Microbiol. Biotechnol. 2019, 103, 3025–3035. [Google Scholar] [CrossRef]
- Göbel, S.; Jaén, K.E.; Dorn, M.; Neumeyer, V.; Jordan, I.; Sandig, V.; Reichl, U.; Altomonte, J.; Genzel, Y. Process Intensification Strategies toward Cell Culture-based High-yield Production of a Fusogenic Oncolytic Virus. Biotechnol. Bioeng. 2023, 120, 2639–2657. [Google Scholar] [CrossRef]
- Gränicher, G.; Babakhani, M.; Göbel, S.; Jordan, I.; Marichal-Gallardo, P.; Genzel, Y.; Reichl, U. A High Cell Density Perfusion Process for Modified Vaccinia Virus Ankara Production: Process Integration with Inline DNA Digestion and Cost Analysis. Biotechnol. Bioeng. 2021, 118, 4720–4734. [Google Scholar] [CrossRef] [PubMed]
- Gränicher, G.; Tapia, F.; Behrendt, I.; Jordan, I.; Genzel, Y.; Reichl, U. Production of Modified Vaccinia Ankara Virus by Intensified Cell Cultures: A Comparison of Platform Technologies for Viral Vector Production. Biotechnol. J. 2021, 16, 2000024. [Google Scholar] [CrossRef] [PubMed]
- Schad, M.; Gautam, S.; Grein, T.A.; Käß, F. Process Analytical Technologies (PAT) and Quality by Design (QbD) for Bioprocessing of Virus-Based Therapeutics. In Bioprocess and Analytics Development for Virus-Based Advanced Therapeutics and Medicinal Products (ATMPs); Springer: Cham, Switzerland, 2023; pp. 295–328. [Google Scholar] [CrossRef]
- Rheinemann, L.; Sundquist, W.I. Virus Budding. In Encyclopedia of Virology, 4th ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 519–528. [Google Scholar] [CrossRef]
- Aggarwal, T.; Kondabagil, K. Poxviruses. Adv. Exp. Med. Biol. 2024, 1451, 35–54. [Google Scholar] [CrossRef]
- Roller, R.J.; Johnson, D.C. Herpesvirus Nuclear Egress across the Outer Nuclear Membrane. Viruses 2021, 13, 2356. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.G. The Existence of an Envelope on Extracellular Cowpox Virus and Its Antigenic Relationship to the Vaccinia Envelope. Arch. Virol. 1986, 90, 125–133. [Google Scholar] [CrossRef]
- Roberts, K.L.; Smith, G.L. Vaccinia Virus Morphogenesis and Dissemination. Trends Microbiol. 2008, 16, 472–479. [Google Scholar] [CrossRef]
- Appleyard, G.; Hapel, A.J.; Boulter, E.A. An Antigenic Difference between Intracellular and Extracellular Rabbitpox Virus. J. Gen. Virol. 1971, 13, 9–17. [Google Scholar] [CrossRef]
- Nguyen, H.-M.; Sah, N.; Humphrey, M.R.M.; Rabkin, S.D.; Saha, D. Growth, Purification, and Titration of Oncolytic Herpes Simplex Virus. J. Vis. Exp. 2021, 171, 10–3791. [Google Scholar] [CrossRef]
- van Vloten, J.P.; Minott, J.A.; McAusland, T.M.; Ingrao, J.C.; Santry, L.A.; McFadden, G.; Petrik, J.J.; Bridle, B.W.; Wootton, S.K. Production and Purification of High-Titer OrfV for Preclinical Studies in Vaccinology and Cancer Therapy. Mol. Ther. Methods Clin. Dev. 2021, 23, 434–447. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, X.; Zhang, Z.; Huang, R.; Peng, Q.; Zhao, Y.; Zhong, L.; Gan, L. Efficient Production of Evolutionary Nanoparticle Newcastle Disease Virus. J. Biomed. Nanotechnol. 2023, 19, 139–145. [Google Scholar] [CrossRef]
- Wallis, C.; Melnick, J.L. Stabilization of Enveloped Viruses by Dimethyl Sulfoxide. J. Virol. 1968, 2, 953–954. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S. 2D PAGE: Sample Preparation and Fractionation. Methods Mol. Biol. 2008, 424, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Pförringer, D.; Braun, K.F.; Mühlhofer, H.; Schneider, J.; Stemberger, A.; Seifried, E.; Pohlscheidt, E.; Seidel, M.; Edenharter, G.; Duscher, D.; et al. Novel Method for Reduction of Virus Load in Blood Plasma by Sonication. Eur. J. Méd. Res. 2020, 25, 12. [Google Scholar] [CrossRef]
- Mundle, S.T.; Kishko, M.; Groppo, R.; DiNapoli, J.; Hamberger, J.; McNeil, B.; Kleanthous, H.; Parrington, M.; Zhang, L.; Anderson, S.F. Core Bead Chromatography for Preparation of Highly Pure, Infectious Respiratory Syncytial Virus in the Negative Purification Mode. Vaccine 2016, 34, 3690–3696. [Google Scholar] [CrossRef] [PubMed]
- Laposova, K.; Oveckova, I.; Tomaskova, J. A Simple Method for Isolation of Cell-Associated Viral Particles from Cell Culture. J. Virol. Methods 2017, 249, 194–196. [Google Scholar] [CrossRef]
- Kong, B.-W.; Foster, L.K.; Foster, D.N. A Method for the Rapid Isolation of Virus from Cultured Cells. BioTechniques 2008, 44, 97–99. [Google Scholar] [CrossRef]
- Choi, H.-J.; Ebersbacher, C.F.; Kim, M.-C.; Kang, S.-M.; Montemagno, C.D. A Mechanistic Study on the Destabilization of Whole Inactivated Influenza Virus Vaccine in Gastric Environment. PLoS ONE 2013, 8, e66316. [Google Scholar] [CrossRef]
- O’Keeffe, R.; Johnston, M.D.; Slater, N.K.H. The Primary Production of an Infectious Recombinant Herpes Simplex Virus Vaccine. Biotechnol. Bioeng. 1998, 57, 262–271. [Google Scholar] [CrossRef]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane Proteins, Lipids and Detergents: Not Just a Soap Opera. Biochim. Biophys. Acta (BBA) Biomembr. 2004, 1666, 105–117. [Google Scholar] [CrossRef]
- Srivastava, A.; Mallela, K.M.G.; Deorkar, N.; Brophy, G. Manufacturing Challenges and Rational Formulation Development for AAV Viral Vectors. J. Pharm. Sci. 2021, 110, 2609–2624. [Google Scholar] [CrossRef]
- Mueller, D.; Pardo Garcia, A.; Grein, T.A.; Kaess, F.; Ng, J.; Pesta, K.; Schneider, S.; Turnbull, J. Purification of Recombinant Vesicular Stomatitis Virus-Based HIV Vaccine Candidate. US20220010286A1. Filed 9 September 2021, Patent Pending. Available online: https://patents.google.com/patent/US20220010286A1/en (accessed on 17 April 2025).
- Pagallies, F.; Labisch, J.J.; Wronska, M.; Pflanz, K.; Amann, R. Efficient and Scalable Clarification of Orf Virus from HEK Suspension for Vaccine Development. Vaccine X 2024, 18, 100474. [Google Scholar] [CrossRef]
- Gashti, A.B.; Chahal, P.S.; Gaillet, B.; Garnier, A. Purification of Recombinant Vesicular Stomatitis Virus-Based HIV Vaccine Candidate. Vaccine 2023, 41, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.P.; Göbel, S.; Reiter, M.; Bryan, A.; Altomonte, J.; Genzel, Y.; Peixoto, C. Streamlining the Purification of a Clinical-Grade Oncolytic Virus for Therapeutic Applications. Sep. Purif. Technol. 2025, 354, 128769. [Google Scholar] [CrossRef]
- Mayer, V.; Steiner, F.; Jungbauer, A.; Aguilar, P.P. Highly Pure Measles Virus Generated by Combination of Salt-Active Nuclease Treatment and Heparin Affinity Chromatography. J. Chromatogr. A 2024, 1738, 465470. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.C.; Maeshima, K.; Hendzel, M.J. The Solid and Liquid States of Chromatin. Epigenetics Chromatin 2021, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Mayer, V.; Frank, A.; Preinsperger, S.; Csar, P.; Steppert, P.; Jungbauer, A.; Aguilar, P.P. Removal of Chromatin by Salt-tolerant Endonucleases for Production of Recombinant Measles Virus. Biotechnol. Prog. 2023, 39, e3342. [Google Scholar] [CrossRef]
- Vincent, D.; Kramberger, P.; Hudej, R.; Štrancar, A.; Wang, Y.; Zhou, Y.; Velayudhan, A. The Development of a Monolith-Based Purification Process for Orthopoxvirus Vaccinia Virus Lister Strain. J. Chromatogr. A 2017, 1524, 87–100. [Google Scholar] [CrossRef]
- Besnard, L.; Fabre, V.; Fettig, M.; Gousseinov, E.; Kawakami, Y.; Laroudie, N.; Scanlan, C.; Pattnaik, P. Clarification of Vaccines: An Overview of Filter Based Technology Trends and Best Practices. Biotechnol. Adv. 2016, 34, 1–13. [Google Scholar] [CrossRef]
- Barbieri, E.; Mollica, G.N.; Moore, B.D.; Sripada, S.A.; Shastry, S.; Kilgore, R.E.; Loudermilk, C.M.; Whitacre, Z.H.; Kilgour, K.M.; Wuestenhagen, E.; et al. Peptide Ligands Targeting the Vesicular Stomatitis Virus G (VSV-G) Protein for the Affinity Purification of Lentivirus Particles. Biotechnol. Bioeng. 2024, 121, 618–639. [Google Scholar] [CrossRef]
- Lothert, K.; Harsy, Y.M.J.; Endres, P.; Müller, E.; Wolff, M.W. Evaluation of Restricted Access Media for the Purification of Cell Culture-derived Orf Viruses. Eng. Life Sci. 2023, 23, e2300009. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Xin, D.; Garcia, A.P.; Spiesschaert, B. Single-Step Rapid Chromatographic Purification and Characterization of Clinical Stage Oncolytic VSV-GP. Front. Bioeng. Biotechnol. 2022, 10, 992069. [Google Scholar] [CrossRef] [PubMed]
- Gerster, P.; Kopecky, E.-M.; Hammerschmidt, N.; Klausberger, M.; Krammer, F.; Grabherr, R.; Mersich, C.; Urbas, L.; Kramberger, P.; Paril, T.; et al. Purification of Infective Baculoviruses by Monoliths. J. Chromatogr. A 2013, 1290, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Luo, J.; Teng, M.; Xing, G.; Guo, J.; Zhang, Y. Purification of Cell-derived Japanese Encephalitis Virus by Dual-mode Chromatography. Biotechnol. Appl. Biochem. 2021, 68, 547–553. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Kennedy, A.; Gregory, B.; Brennan, F.M. Simplified Production and Concentration of Lentiviral Vectors to Achieve High Transduction in Primary Human T Cells. BMC Biotechnol. 2013, 13, 98. [Google Scholar] [CrossRef]
- Lotfian, P.; Levy, M.S.; Coffin, R.S.; Fearn, T.; Ayazi-Shamlou, P. Impact of Process Conditions on the Centrifugal Recovery of a Disabled Herpes Simplex Virus. Biotechnol. Prog. 2003, 19, 209–215. [Google Scholar] [CrossRef]
- Kuroda, S.; Miyagawa, Y.; Sukegawa, M.; Tomono, T.; Yamamoto, M.; Adachi, K.; Verlengia, G.; Goins, W.F.; Cohen, J.B.; Glorioso, J.C.; et al. Evaluation of Parameters for Efficient Purification and Long-Term Storage of Herpes Simplex Virus-Based Vectors. Mol. Ther. Methods Clin. Dev. 2022, 26, 132–143. [Google Scholar] [CrossRef]
- Baekelandt, V.; Eggermont, K.; Michiels, M.; Nuttin, B.; Debyser, Z. Optimized Lentiviral Vector Production and Purification Procedure Prevents Immune Response after Transduction of Mouse Brain. Gene Ther. 2003, 10, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Boroujeni, M.E.; Gardaneh, M. The Superiority of Sucrose Cushion Centrifugation to Ultrafiltration and PEGylation in Generating High-Titer Lentivirus Particles and Transducing Stem Cells with Enhanced Efficiency. Mol. Biotechnol. 2018, 60, 185–193. [Google Scholar] [CrossRef]
- Transfiguracion, J.; Jaalouk, D.E.; Ghani, K.; Galipeau, J.; Kamen, A. Size-Exclusion Chromatography Purification of High-Titer Vesicular Stomatitis Virus G Glycoprotein-Pseudotyped Retrovectors for Cell and Gene Therapy Applications. Hum. Gene Ther. 2003, 14, 1139–1153. [Google Scholar] [CrossRef]
- Wang, J.; Ma, J.; Wen, X. Basic Concepts of Density Gradient Ultracentrifugation. In Nanoseparation Using Density Gradient Ultracentrifugation; SpringerBriefs in Molecular Science; Springer: Singapore, 2018; pp. 21–36. ISBN 9789811051890. [Google Scholar]
- Nagano, H.; Yagyu, K.; Ohta, S. Purification of Infectious Bronchitis Coronavirus by Sephacryl S-1000 Gel Chromatography. Vet. Microbiol. 1989, 21, 115–123. [Google Scholar] [CrossRef]
- Saha, K.; Lin, Y.-C.; Wong, P.K.Y. A Simple Method for Obtaining Highly Viable Virus from Culture Supernatant. J. Virol. Methods 1994, 46, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-H.; Murthy, H.N.; Wu, C.-H.; Paek, K.-Y. Sucrose-Induced Osmotic Stress Affects Biomass, Metabolite, and Antioxidant Levels in Root Suspension Cultures of Hypericum Perforatum L. Plant Cell, Tissue Organ Cult. (PCTOC) 2010, 103, 7–14. [Google Scholar] [CrossRef]
- Helmholz, H.F.; Bollman, J.L. The Intravenous Administration of Sucrose Solutions as a Means of Producing Intense Diuresis. J. Lab. Clin. Med. 1939, 25, 1180–1187. [Google Scholar]
- Sandig, V.; Hofmann, C.; Steinert, S.; Jennings, G.; Schlag, P.; Strauss, M. Gene Transfer into Hepatocytes and Human Liver Tissue by Baculovirus Vectors. Hum. Gene Ther. 1996, 7, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Santry, L.A.; McAusland, T.M.; Susta, L.; Wood, G.A.; Major, P.P.; Petrik, J.J.; Bridle, B.W.; Wootton, S.K. Production and Purification of High-Titer Newcastle Disease Virus for Use in Preclinical Mouse Models of Cancer. Mol. Ther. Methods Clin. Dev. 2018, 9, 181–191. [Google Scholar] [CrossRef]
- Granier, C.; Toesca, J.; Mialon, C.; Ritter, M.; Freitas, N.; Boson, B.; Pécheur, E.-I.; Cosset, F.-L.; Denolly, S. Low-Density Hepatitis C Virus Infectious Particles Are Protected from Oxidation by Secreted Cellular Proteins. mBio 2023, 14, e01549-23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Feng, Y.; Tu, Z.; Lou, Z.; Tu, C. Proteomic Profiling of Purified Rabies Virus Particles. Virol. Sin. 2020, 35, 143–155. [Google Scholar] [CrossRef]
- Segura, M.M.; Kamen, A.A.; Garnier, A. Viral Vectors for Gene Therapy, Methods and Protocols. Methods Mol. Biol. 2011, 737, 89–116. [Google Scholar] [CrossRef]
- Loewe, D.; Dieken, H.; Grein, T.A.; Salzig, D.; Czermak, P. A Combined Ultrafiltration/Diafiltration Process for the Purification of Oncolytic Measles Virus. Membranes 2022, 12, 105. [Google Scholar] [CrossRef]
- Bandeira, V.; Peixoto, C.; Rodrigues, A.F.; Cruz, P.E.; Alves, P.M.; Coroadinha, A.S.; Carrondo, M.J.T. Downstream Processing of Lentiviral Vectors: Releasing Bottlenecks. Hum. Gene Ther. Part. B Methods 2012, 23, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Labisch, J.J.; Bollmann, F.; Wolff, M.W.; Pflanz, K. A New Simplified Clarification Approach for Lentiviral Vectors Using Diatomaceous Earth Improves Throughput and Safe Handling. J. Biotechnol. 2021, 326, 11–20. [Google Scholar] [CrossRef]
- Shoaebargh, S.; Gough, I.; Medina, M.F.; Smith, A.; van der Heijden, J.; Lichty, B.D.; Bell, J.C.; Latulippe, D.R. Sterile Filtration of Oncolytic Viruses: An Analysis of Effects of Membrane Morphology on Fouling and Product Recovery. J. Membr. Sci. 2018, 548, 239–246. [Google Scholar] [CrossRef]
- Labisch, J.J.; Evangelopoulou, M.; Schleuß, T.; Pickl, A. Investigating Ultrafiltration Membranes and Operation Modes for Improved Lentiviral Vector Processing. Eng. Life Sci. 2025, 25, e202400057. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.B.; Silva, R.J.S.; Moreira, A.S.; Cunha, B.; Clemente, J.J.; Alves, P.M.; Carrondo, M.J.T.; Xenopoulos, A.; Peixoto, C. Efficient Filtration Strategies for the Clarification of Influenza Virus-like Particles Derived from Insect Cells. Sep. Purif. Technol. 2019, 218, 81–88. [Google Scholar] [CrossRef]
- Loewe, D.; Grein, T.A.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Tangential Flow Filtration for the Concentration of Oncolytic Measles Virus: The Influence of Filter Properties and the Cell Culture Medium. Membranes 2019, 9, 160. [Google Scholar] [CrossRef]
- Jones, T.H.; Brassard, J.; Johns, M.W.; Gagné, M.-J. The Effect of Pre-Treatment and Sonication of Centrifugal Ultrafiltration Devices on Virus Recovery. J. Virol. Methods 2009, 161, 199–204. [Google Scholar] [CrossRef]
- Wright, E.; Kawka, K.; Medina, M.F.C.; Latulippe, D.R. Evaluation of Host Cell Impurity Effects on the Performance of Sterile Filtration Processes for Therapeutic Viruses. Membranes 2022, 12, 359. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.; Bons, J.; Kumar, A.; Kabir, M.H.; Liang, H. Forever Chemicals, Per-and Polyfluoroalkyl Substances (PFAS), in Lubrication. Lubricants 2024, 12, 114. [Google Scholar] [CrossRef]
- Mark, T.; Janice, W.; Akanksha, N. Methods for Purification of Viruses. US8785173B2, 22 July 2014. [Google Scholar]
- Rene, A.B.; Champluvier, B.P.S. Method for Producing Virus from Cell Culture Involving Homogenization. US20120058145A1, 6 May 2010. [Google Scholar]
- Transfiguracion, J.; Jorio, H.; Meghrous, J.; Jacob, D.; Kamen, A. High Yield Purification of Functional Baculovirus Vectors by Size Exclusion Chromatography. J. Virol. Methods 2007, 142, 21–28. [Google Scholar] [CrossRef]
- Taylor, N.; Ma, W.J.; Kristopeit, A.; Wang, S.-C.; Zydney, A.L. Enhancing the Performance of Sterile Filtration for Viral Vaccines and Model Nanoparticles Using an Appropriate Prefilter. J. Membr. Sci. 2022, 647, 120264. [Google Scholar] [CrossRef]
- EMA (European Medicines Agency). Guideline on the Sterilisation of the Medicinal Product, Active Substance, Excipient and Primary Container; Ref. No. EMA/CHMP/CVMP/QWP/850374/2015, 2019.
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Godfrey, S.; Ravikrishnan, J.; Lin, H.; Vogel, J.; Coffman, J. Shear Contributions to Cell Culture Performance and Product Recovery in ATF and TFF Perfusion Systems. J. Biotechnol. 2017, 246, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.; Rayat, A.C.M.E. Lentiviral Vector Bioprocessing. Viruses 2021, 13, 268. [Google Scholar] [CrossRef]
- Negrete, A.; Pai, A.; Shiloach, J. Use of Hollow Fiber Tangential Flow Filtration for the Recovery and Concentration of HIV Virus-like Particles Produced in Insect Cells. J. Virol. Methods 2014, 195, 240–246. [Google Scholar] [CrossRef]
- van Reis, R.; Zydney, A. Bioprocess Membrane Technology. J. Membr. Sci. 2007, 297, 16–50. [Google Scholar] [CrossRef]
- Heffron, J.; Mayer, B.K. Virus Isoelectric Point Estimation: Theories and Methods. Appl. Environ. Microb. 2020, 87, e02319-20. [Google Scholar] [CrossRef]
- Banjac, M.; Roethl, E.; Gelhart, F.; Kramberger, P.; Jarc, B.L.; Jarc, M.; Štrancar, A.; Muster, T.; Peterka, M. Purification of Vero Cell Derived Live Replication Deficient Influenza A and B Virus by Ion Exchange Monolith Chromatography. Vaccine 2014, 32, 2487–2492. [Google Scholar] [CrossRef]
- Eckhardt, D.; Dieken, H.; Loewe, D.; Grein, T.A.; Salzig, D.; Czermak, P. Purification of Oncolytic Measles Virus by Cation-Exchange Chromatography Using Resin-Based Stationary Phases. Sep. Sci. Technol. 2022, 57, 886–896. [Google Scholar] [CrossRef]
- Santry, L.A.; Jacquemart, R.; Vandersluis, M.; Zhao, M.; Domm, J.M.; McAusland, T.M.; Shang, X.; Major, P.M.; Stout, J.G.; Wootton, S.K. Interference Chromatography: A Novel Approach to Optimizing Chromatographic Selectivity and Separation Performance for Virus Purification. BMC Biotechnol. 2020, 20, 32. [Google Scholar] [CrossRef]
- Lothert, K.; Wolff, M.W. Affinity and Pseudo-Affinity Membrane Chromatography for Viral Vector and Vaccine Purifications: A Review. Membranes 2023, 13, 770. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Lynn, D.; van der Loo, J.C.; Malik, P. Purification of Baculovirus Vectors Using Heparin Affinity Chromatography. Mol. Ther. Methods Clin. Dev. 2016, 3, 16071. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.W.; Siewert, C.; Lehmann, S.; Hansen, S.P.; Djurup, R.; Faber, R.; Reichl, U. Capturing of Cell Culture-derived Modified Vaccinia Ankara Virus by Ion Exchange and Pseudo-affinity Membrane Adsorbers. Biotechnol. Bioeng. 2010, 105, 761–769. [Google Scholar] [CrossRef]
- Fortuna, A.R.; van Teeffelen, S.; Ley, A.; Fischer, L.M.; Taft, F.; Genzel, Y.; Villain, L.; Wolff, M.W.; Reichl, U. Use of Sulfated Cellulose Membrane Adsorbers for Chromatographic Purification of Cell Cultured-Derived Influenza A and B Viruses. Sep. Purif. Technol. 2019, 226, 350–358. [Google Scholar] [CrossRef]
- Mekkaoui, L.; Parekh, F.; Kotsopoulou, E.; Darling, D.; Dickson, G.; Cheung, G.W.; Chan, L.; MacLellan-Gibson, K.; Mattiuzzo, G.; Farzaneh, F.; et al. Lentiviral Vector Purification Using Genetically Encoded Biotin Mimic in Packaging Cell. Mol. Ther. Methods Clin. Dev. 2018, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Vandersluis, M.; Stout, J.; Haupts, U.; Sanders, M.; Jacquemart, R. Affinity Chromatography for Vaccines Manufacturing: Finally Ready for Prime Time? Vaccine 2019, 37, 5491–5503. [Google Scholar] [CrossRef]
- Cheung, R.C.F.; Wong, J.H.; Ng, T.B. Immobilized Metal Ion Affinity Chromatography: A Review on Its Applications. Appl. Microbiol. Biotechnol. 2012, 96, 1411–1420. [Google Scholar] [CrossRef]
- Moreira, A.S.; Bezemer, S.; Faria, T.Q.; Detmers, F.; Hermans, P.; Sierkstra, L.; Coroadinha, A.S.; Peixoto, C. Implementation of Novel Affinity Ligand for Lentiviral Vector Purification. Int. J. Mol. Sci. 2023, 24, 3354. [Google Scholar] [CrossRef]
- Ma, J.; Huang, Y.; Jia, G.; Dong, X.; Shi, Q.; Sun, Y. Discovery of Broad-Spectrum High-Affinity Peptide Ligands of Spike Protein for the Vaccine Purification of SARS-CoV-2 and Omicron Variants. Int. J. Biol. Macromol. 2024, 283, 137059. [Google Scholar] [CrossRef]
- Wolff, M.W.; Siewert, C.; Hansen, S.P.; Faber, R.; Reichl, U. Purification of Cell Culture-derived Modified Vaccinia Ankara Virus by Pseudo-affinity Membrane Adsorbers and Hydrophobic Interaction Chromatography. Biotechnol. Bioeng. 2010, 107, 312–320. [Google Scholar] [CrossRef]
- Diogo, M.M.; Queiroz, J.A.; Prazeres, D.M.F. Chromatography of Plasmid DNA. J. Chromatogr. A 2005, 1069, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Loa, C.C.; Lin, T.L.; Wu, C.C.; Bryan, T.A.; Thacker, H.L.; Hooper, T.; Schrader, D. Purification of Turkey Coronavirus by Sephacryl Size-Exclusion Chromatography. J. Virol. Methods 2002, 104, 187–194. [Google Scholar] [CrossRef]
- Eilts, F.; Steger, M.; Lothert, K.; Wolff, M.W. The Suitability of Latex Particles to Evaluate Critical Process Parameters in Steric Exclusion Chromatography. Membranes 2022, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Labisch, J.J.; Wiese, G.P.; Pflanz, K. Steric Exclusion Chromatography for Purification of Biomolecules—A Review. Separations 2023, 10, 183. [Google Scholar] [CrossRef]
- Labisch, J.J.; Kassar, M.; Bollmann, F.; Valentic, A.; Hubbuch, J.; Pflanz, K. Steric Exclusion Chromatography of Lentiviral Vectors Using Hydrophilic Cellulose Membranes. J. Chromatogr. A 2022, 1674, 463148. [Google Scholar] [CrossRef]
- Lothert, K.; Sprick, G.; Beyer, F.; Lauria, G.; Czermak, P.; Wolff, M.W. Membrane-Based Steric Exclusion Chromatography for the Purification of a Recombinant Baculovirus and Its Application for Cell Therapy. J. Virol. Methods 2020, 275, 113756. [Google Scholar] [CrossRef]
- Marichal-Gallardo, P.; Pieler, M.M.; Wolff, M.W.; Reichl, U. Steric Exclusion Chromatography for Purification of Cell Culture-Derived Influenza A Virus Using Regenerated Cellulose Membranes and Polyethylene Glycol. J. Chromatogr. A 2017, 1483, 110–119. [Google Scholar] [CrossRef]
- Lothert, K.; Offersgaard, A.F.; Pihl, A.F.; Mathiesen, C.K.; Jensen, T.B.; Alzua, G.P.; Fahnøe, U.; Bukh, J.; Gottwein, J.M.; Wolff, M.W. Development of a Downstream Process for the Production of an Inactivated Whole Hepatitis C Virus Vaccine. Sci. Rep. 2020, 10, 16261. [Google Scholar] [CrossRef] [PubMed]
- Labisch, J.; Paul, R.; Wiese, G.; Pflanz, K. Scaling Up of Steric Exclusion Membrane Chromatography for Lentiviral Vector Purification. Membranes 2023, 13, 149. [Google Scholar] [CrossRef]
- Nestola, P.; Villain, L.; Peixoto, C.; Martins, D.L.; Alves, P.M.; Carrondo, M.J.T.; Mota, J.P.B. Impact of Grafting on the Design of New Membrane Adsorbers for Adenovirus Purification. J. Biotechnol. 2014, 181, 1–11. [Google Scholar] [CrossRef]
- Aguilar, P.P.; Schneider, T.A.; Wetter, V.; Maresch, D.; Ling, W.L.; Tover, A.; Steppert, P.; Jungbauer, A. Polymer-Grafted Chromatography Media for the Purification of Enveloped Virus-like Particles, Exemplified with HIV-1 Gag VLP. Vaccine 2019, 37, 7070–7080. [Google Scholar] [CrossRef]
- Connell-Crowley, L.; Nguyen, T.; Bach, J.; Chinniah, S.; Bashiri, H.; Gillespie, R.; Moscariello, J.; Hinckley, P.; Dehghani, H.; Vunnum, S.; et al. Cation Exchange Chromatography Provides Effective Retrovirus Clearance for Antibody Purification Processes. Biotechnol. Bioeng. 2012, 109, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Wright, B.; Green, N.K.; Tarrant, R.; Roberts, I.; Hardick, O.; Bracewell, D.G. Adenovirus 5 Recovery Using Nanofiber Ion-exchange Adsorbents. Biotechnol. Bioeng. 2019, 116, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Vicente, T.; Fáber, R.; Alves, P.M.; Carrondo, M.J.T.; Mota, J.P.B. Impact of Ligand Density on the Optimization of Ion-exchange Membrane Chromatography for Viral Vector Purification. Biotechnol. Bioeng. 2011, 108, 1347–1359. [Google Scholar] [CrossRef]
- Pamenter, G.; Davies, L.; Knevelman, C.; Miskin, J.; Mitrophanous, K.; Dikicioglu, D.; Bracewell, D.G. Time-dependent Sorption Behavior of Lentiviral Vectors during Anion-exchange Chromatography. Biotechnol. Bioeng. 2023, 120, 2269–2282. [Google Scholar] [CrossRef]
- Boudeffa, D.; Bertin, B.; Biek, A.; Mormin, M.; Leseigneur, F.; Galy, A.; Merten, O.-W. Toward a Scalable Purification Protocol of GaLV-TR-Pseudotyped Lentiviral Vectors. Hum. Gene Ther. Methods 2019, 30, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Kröber, T.; Wolff, M.W.; Hundt, B.; Seidel-Morgenstern, A.; Reichl, U. Continuous Purification of Influenza Virus Using Simulated Moving Bed Chromatography. J. Chromatogr. A 2013, 1307, 99–110. [Google Scholar] [CrossRef]
- Aust, N.; Parth, M.; Lederer, K. SEC of Ultra-High Molar Mass Polymers: Optimization of Experimental Conditions to Avoid Molecular Degradation in the Case of Narrow Polystyrene Standards. Int. J. Polym. Anal. Charact. 2001, 6, 245–260. [Google Scholar] [CrossRef]
- Song, Y.; Yang, Y.; Lin, X.; Zhao, Q.; Su, Z.; Ma, G.; Zhang, S. Size Exclusion Chromatography Using Large Pore Size Media Induces Adverse Conformational Changes of Inactivated Foot-and-Mouth Disease Virus Particles. J. Chromatogr. A 2022, 1677, 463301. [Google Scholar] [CrossRef]
- D’Atri, V.; Imiołek, M.; Quinn, C.; Finny, A.; Lauber, M.; Fekete, S.; Guillarme, D. Size Exclusion Chromatography of Biopharmaceutical Products: From Current Practices for Proteins to Emerging Trends for Viral Vectors, Nucleic Acids and Lipid Nanoparticles. J. Chromatogr. A 2024, 1722, 464862. [Google Scholar] [CrossRef]
- Iyer, G.; Ramaswamy, S.; Asher, D.; Mehta, U.; Leahy, A.; Chung, F.; Cheng, K.-S. Reduced Surface Area Chromatography for Flow-through Purification of Viruses and Virus like Particles. J. Chromatogr. A 2011, 1218, 3973–3981. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Simons, J.; Hooson, S.; Abraham, D.; Carta, G. Protein and Virus-like Particle Adsorption on Perfusion Chromatography Media. J. Chromatogr. A 2013, 1297, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Abraham, D.; Carta, G. Particle Size Effects on Protein and Virus-like Particle Adsorption on Perfusion Chromatography Media. J. Chromatogr. A 2015, 1375, 92–100. [Google Scholar] [CrossRef]
- Pabst, T.M.; Thai, J.; Hunter, A.K. Evaluation of Recent Protein A Stationary Phase Innovations for Capture of Biotherapeutics. J. Chromatogr. A 2018, 1554, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Ta, D.T.; Chu, K.L.; Soonaan, N.I.B.; Chin, C.; Ng, S.K.; Zhang, W. A New and Simplified Anion Exchange Chromatographic Process for the Purification of Cell-Grown Influenza A H1N1 Virus. Sep. Purif. Technol. 2021, 263, 118412. [Google Scholar] [CrossRef]
- Hagemann, F.; Wypysek, D.; Baitalow, K.; Adametz, P.; Thom, V.; Wessling, M. Why Device Design Is Crucial for Membrane Adsorbers. J. Chromatogr. Open 2022, 2, 100029. [Google Scholar] [CrossRef]
- Roshankhah, R.; Pelton, R.; Ghosh, R. Optimization of Fluid Flow in Membrane Chromatography Devices Using Computational Fluid Dynamic Simulations. J. Chromatogr. A 2023, 1699, 464030. [Google Scholar] [CrossRef]
- Picard, G.; Ajdari, A.; Bocquet, L.; Lequeux, F. Simple Model for Heterogeneous Flows of Yield Stress Fluids. Phys. Rev. E 2002, 66, 051501. [Google Scholar] [CrossRef]
- Velali, E.; Stute, B.; Leuthold, M.; von Lieres, E. Model-Based Performance Analysis and Scale-up of Membrane Adsorbers with a Cassettes Format Designed for Parallel Operation. Chem. Eng. Sci. 2018, 192, 103–113. [Google Scholar] [CrossRef]
- Teepakorn, C.; Fiaty, K.; Charcosset, C. Effect of Geometry and Scale for Axial and Radial Flow Membrane Chromatography—Experimental Study of Bovin Serum Albumin Adsorption. J. Chromatogr. A 2015, 1403, 45–53. [Google Scholar] [CrossRef]
- Matos, T.; Hoying, D.; Kristopeit, A.; Wenger, M.; Joyce, J. Continuous Multi-Membrane Chromatography of Large Viral Particles. J. Chromatogr. A 2023, 1705, 464194. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, A.R.; Taft, F.; Villain, L.; Wolff, M.W.; Reichl, U. Continuous Purification of Influenza A Virus Particles Using Pseudo-Affinity Membrane Chromatography. J. Biotechnol. 2021, 342, 139–148. [Google Scholar] [CrossRef]
- Mendes, J.P.; Bergman, M.; Solbrand, A.; Peixoto, C.; Carrondo, M.J.T.; Silva, R.J.S. Continuous Affinity Purification of Adeno-Associated Virus Using Periodic Counter-Current Chromatography. Pharmaceutics 2022, 14, 1346. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.P.; Silva, R.J.S.; Berg, M.; Mathiasson, L.; Peixoto, C.; Alves, P.M.; Carrondo, M.J.T. Oncolytic Virus Purification with Periodic Counter-current Chromatography. Biotechnol. Bioeng. 2021, 118, 3522–3532. [Google Scholar] [CrossRef]
- Orr, V.; Zhong, L.; Moo-Young, M.; Chou, C.P. Recent Advances in Bioprocessing Application of Membrane Chromatography. Biotechnol. Adv. 2013, 31, 450–465. [Google Scholar] [CrossRef]
- Leinweber, F.C.; Tallarek, U. Chromatographic Performance of Monolithic and Particulate Stationary Phases Hydrodynamics and Adsorption Capacity. J. Chromatogr. A 2003, 1006, 207–228. [Google Scholar] [CrossRef]
- Vicente, T.; Peixoto, C.; Carrondo, M.J.T.; Alves, P.M. Gene Therapy of Cancer, Methods and Protocols. Methods Mol. Biol. 2009, 542, 447–470. [Google Scholar] [CrossRef] [PubMed]
- Koku, H.; Maier, R.S.; Czymmek, K.J.; Schure, M.R.; Lenhoff, A.M. Modeling of Flow in a Polymeric Chromatographic Monolith. J. Chromatogr. A 2011, 1218, 3466–3475. [Google Scholar] [CrossRef]
- Podgornik, A.; Jančar, J.; Mihelič, I.; Barut, M.; Strancar, A. Large Volume Monolithic Stationary Phases: Preparation, Properties, and Applications. Acta Chim. Slov. 2010, 57, 1–8. [Google Scholar]
- Hahn, R.; Tscheliessnig, A.; Bauerhansl, P.; Jungbauer, A. Dispersion Effects in Preparative Polymethacrylate Monoliths Operated in Radial-Flow Columns. J. Biochem. Biophys. Meth 2007, 70, 87–94. [Google Scholar] [CrossRef]
- Brgles, M.; Sviben, D.; Forčić, D.; Halassy, B. Nonspecific Native Elution of Proteins and Mumps Virus in Immunoaffinity Chromatography. J. Chromatogr. A 2016, 1447, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Trilisky, E.I.; Lenhoff, A.M. Flow-dependent Entrapment of Large Bioparticles in Porous Process Media. Biotechnol. Bioeng. 2009, 104, 127–133. [Google Scholar] [CrossRef]
- Kralj, Š.; Kodermac, Š.M.; Bergoč, I.; Kostelec, T.; Podgornik, A.; Štrancar, A.; Černigoj, U. Effect of Plasmid DNA Isoforms on Preparative Anion Exchange Chromatography. Electrophoresis 2023, 44, 1953–1966. [Google Scholar] [CrossRef]
- Pavlin, N.; Černigoj, U.; Bavčar, M.; Plesničar, T.; Mavri, J.; Zidar, M.; Bone, M.; Savič, U.K.; Sever, T.; Štrancar, A. Analytical Separation of Plasmid DNA Isoforms Using Anion Exchanging Chromatographic Monoliths with 6 Μm Channels. Electrophoresis 2023, 44, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Schimek, A.; Ng, J.; Will, F.; Hubbuch, J. Mechanistic Modeling of the Elution Behavior and Convective Entrapment of Vesicular Stomatitis Virus on an Ion Exchange Chromatography Monolith. J. Chromatogr. A 2025, 1748, 465832. [Google Scholar] [CrossRef] [PubMed]
- Jungreuthmayer, C.; Steppert, P.; Sekot, G.; Zankel, A.; Reingruber, H.; Zanghellini, J.; Jungbauer, A. The 3D Pore Structure and Fluid Dynamics Simulation of Macroporous Monoliths: High Permeability Due to Alternating Channel Width. J. Chromatogr. A 2015, 1425, 141–149. [Google Scholar] [CrossRef]
- Burden, C.S.; Jin, J.; Podgornik, A.; Bracewell, D.G. A Monolith Purification Process for Virus-like Particles from Yeast Homogenate. J. Chromatogr. B 2012, 880, 82–89. [Google Scholar] [CrossRef]
- Kadoi, K.; Iwamoto, E.; Nakama, T. Fabrication and Characterization of a Cellulose Monolith-like Particle for Virus Purification. Biochem. Eng. J. 2023, 192, 108849. [Google Scholar] [CrossRef]
- Babanejad, N.; Mfoafo, K.; Zhang, E.; Omidi, Y.; Razeghifard, R.; Omidian, H. Applications of Cryostructures in the Chromatographic Separation of Biomacromolecules. J. Chromatogr. A 2022, 1683, 463546. [Google Scholar] [CrossRef]
- Arvidsson, P.; Plieva, F.M.; Lozinsky, V.I.; Galaev, I.Y.; Mattiasson, B. Direct Chromatographic Capture of Enzyme from Crude Homogenate Using Immobilized Metal Affinity Chromatography on a Continuous Supermacroporous Adsorbent. J. Chromatogr. A 2003, 986, 275–290. [Google Scholar] [CrossRef]
- Cheeks, M.C.; Kamal, N.; Sorrell, A.; Darling, D.; Farzaneh, F.; Slater, N.K.H. Immobilized Metal Affinity Chromatography of Histidine-Tagged Lentiviral Vectors Using Monolithic Adsorbents. J. Chromatogr. A 2009, 1216, 2705–2711. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Eccleston, M.E.; Slater, N.K.H. Affinity Capture of a Biotinylated Retrovirus on Macroporous Monolithic Adsorbents: Towards a Rapid Single-step Purification Process. Biotechnol. Bioeng. 2005, 89, 783–787. [Google Scholar] [CrossRef]
- Savina, I.N.; Galaev, I.Y.; Mattiasson, B. Ion-exchange Macroporous Hydrophilic Gel Monolith with Grafted Polymer Brushes. J. Mol. Recognit. 2006, 19, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.; Fan, J.; Pourdeyhimi, B.; Boi, C.; Carbonell, R.G. Advances in High-throughput, High-capacity Nonwoven Membranes for Chromatography in Downstream Processing: A Review. Biotechnol. Bioeng. 2024, 121, 2300–2317. [Google Scholar] [CrossRef]
- Ruscic, J.; Perry, C.; Mukhopadhyay, T.; Takeuchi, Y.; Bracewell, D.G. Lentiviral Vector Purification Using Nanofiber Ion-Exchange Chromatography. Mol. Ther. Methods Clin. Dev. 2019, 15, 52–62. [Google Scholar] [CrossRef]
- Sánchez-Trasviña, C.; Fuks, P.; Mushagasha, C.; Kimerer, L.; Mayolo-Deloisa, K.; Rito-Palomares, M.; Carta, G. Structure and Functional Properties of CaptoTM Core 700 Core-Shell Particles. J. Chromatogr. A 2020, 1621, 461079. [Google Scholar] [CrossRef]
- Lothert, K.; Pagallies, F.; Feger, T.; Amann, R.; Wolff, M.W. Selection of Chromatographic Methods for the Purification of Cell Culture-Derived Orf Virus for Its Application as a Vaccine or Viral Vector. J. Biotechnol. 2020, 323, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Gencoglu, M.F.; Heldt, C.L. Enveloped Virus Flocculation and Removal in Osmolyte Solutions. J. Biotechnol. 2015, 206, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Heldt, C.L.; Saksule, A.; Joshi, P.U.; Ghafarian, M. A Generalized Purification Step for Viral Particles Using Mannitol Flocculation. Biotechnol. Prog. 2018, 34, 1027–1035. [Google Scholar] [CrossRef]
- Hasan, T.; Kumari, K.; Devi, S.C.; Handa, J.; Rehman, T.; Ansari, N.A.; Singh, L.R. Osmolytes in Vaccine Production, Flocculation and Storage: A Critical Review. Hum. Vaccines Immunother. 2019, 15, 514–525. [Google Scholar] [CrossRef]
- Rocha, J.M. Aqueous Two-phase Systems and Monolithic Chromatography as Alternative Technological Platforms for Virus and Virus-like Particle Purification. J. Chem. Technol. Biotechnol. 2021, 96, 309–317. [Google Scholar] [CrossRef]
- Turpeinen, D.G.; Joshi, P.U.; Kriz, S.A.; Kaur, S.; Nold, N.M.; O’Hagan, D.; Nikam, S.; Masoud, H.; Heldt, C.L. Continuous Purification of an Enveloped and Non-Enveloped Viral Particle Using an Aqueous Two-Phase System. Sep. Purif. Technol. 2021, 269, 118753. [Google Scholar] [CrossRef]
- Du, P.; Sun, P.; Sun, S.; Dong, J.; Dong, H.; Liu, R.; Guo, H.; Mu, K.; Liu, Z. Separation and Purification of Foot-and-Mouth Disease Virus by Multiple-Stage Aqueous Two-Phase Extraction System. Process Biochem. 2019, 77, 143–150. [Google Scholar] [CrossRef]
- Joshi, P.U.; Turpeinen, D.G.; Schroeder, M.; Jones, B.; Lyons, A.; Kriz, S.; Khaksari, M.; O’Hagan, D.; Nikam, S.; Heldt, C.L. Osmolyte Enhanced Aqueous Two-phase System for Virus Purification. Biotechnol. Bioeng. 2021, 118, 3251–3262. [Google Scholar] [CrossRef]
- Kim, H.; Yi, J.; Yu, J.; Park, J.; Jang, S.K. A Simple and Effective Method to Concentrate Hepatitis C Virus: Aqueous Two-Phase System Allows Highly Efficient Enrichment of Enveloped Viruses. Viruses 2022, 14, 1987. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liu, X.; Fan, D.; Qian, Z.; Sun, X.; Wu, P.; Zhong, L. Study of Oncolytic Virus Preservation and Formulation. Pharmaceuticals 2023, 16, 843. [Google Scholar] [CrossRef]
- Toniolo, S.P.; Afkhami, S.; Mahmood, A.; Fradin, C.; Lichty, B.D.; Miller, M.S.; Xing, Z.; Cranston, E.D.; Thompson, M.R. Excipient Selection for Thermally Stable Enveloped and Non-Enveloped Viral Vaccine Platforms in Dry Powders. Int. J. Pharm. 2019, 561, 66–73. [Google Scholar] [CrossRef]
- Kumru, O.S.; Saleh-Birdjandi, S.; Antunez, L.R.; Sayeed, E.; Robinson, D.; van den Worm, S.; Diemer, G.S.; Perez, W.; Caposio, P.; Früh, K.; et al. Stabilization and Formulation of a Recombinant Human Cytomegalovirus Vector for Use as a Candidate HIV-1 Vaccine. Vaccine 2019, 37, 6696–6706. [Google Scholar] [CrossRef]
- Homan, Y.; Rosenbloom, D.; Wong, S.; Lucchese, J.; Li, A.; Dubey, S.; Thomas, J.; Salituro, G.; Helmy, R.; Verch, T. Prediction of Frozen Virus Stability Based on Degradation Mechanisms, Real-Time Data and Modeling. Bioanalysis 2022, 14, 1177–1190. [Google Scholar] [CrossRef]
- Shi, L.; Sanyal, G.; Ni, A.; Luo, Z.; Doshna, S.; Wang, B.; Graham, T.L.; Wang, N.; Volkin, D.B. Stabilization of Human Papillomavirus Virus-like Particles by Non-Ionic Surfactants. J. Pharm. Sci. 2005, 94, 1538–1551. [Google Scholar] [CrossRef]
- Felix, M.N.; Waerner, T.; Lakatos, D.; Reisinger, B.; Fischer, S.; Garidel, P. Polysorbates Degrading Enzymes in Biotherapeutics—A Current Status and Future Perspectives. Front. Bioeng. Biotechnol. 2025, 12, 1490276. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Luo, W.; Hoffman, J.; Huang, L.; Sandefur, S.; Hall, T.; Murphy, M.; O’Donnell, S. Insights into Virus Inactivation by Polysorbate 80 in the Absence of Solvent. Biotechnol. Prog. 2020, 36, e2953. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.J.J.; Daoussi, R.; Vervaet, C.; Remon, J.-P.; Beer, T.R.M.D. Freeze-Drying of Live Virus Vaccines: A Review. Vaccine 2015, 33, 5507–5519. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Hansen, R.K.; Taylor, R.; Skepper, J.N.; Sanches, R.; Slater, N.K.H. Effect of Freezing Rates and Excipients on the Infectivity of a Live Viral Vaccine during Lyophilization. Biotechnol. Prog. 2004, 20, 1113–1120. [Google Scholar] [CrossRef]
- Coleman, H.J.; Schwartz, D.K.; Kaar, J.L.; Garcea, R.L.; Randolph, T.W. Stabilization of an Infectious Enveloped Virus by Spray-Drying and Lyophilization. J. Pharm. Sci. 2024, 113, 2072–2080. [Google Scholar] [CrossRef]
- Mundle, S.; Stephen, A.; Delagrave, S. Purification of Herpes Virus. US9365832B2, 14 June 2016. [Google Scholar]


{kind=link}
{kind=link}
| Virus Family | Particle Geometry | Genome Size | References | |
|---|---|---|---|---|
| DNA viruses | ||||
| Herpes simplex virus (e.g., HSV-1) | Herpesviridae | 155–240 nm, icosahedral | 152 kb | [14] |
| Orf | Poxviridae | 220–300 × 140–200 nm, ovoid shaped | 140 kb | [15] |
| Vaccinia | Poxviridae | 360 × 270 × 250 nm, brick shaped | 190 kb | [14] |
| Myxoma | Poxviridae | 320 × 235 nm, brick shaped | 162 kb | [16,17] |
| RNA viruses | ||||
| Measle | Paramyxoviridae | 100–300 nm, helical | 15.8 kb | [14] |
| VSV (vesicular stomatitis virus) | Rhabdoviridae | 70 × 200 nm, bullet shaped | 11 kb | [14,18] |
| NDV (Newcastle disease virus) | Paramyxoviridae | 100–500 nm, spherical | 15 kb | [14,19] |
| LCMV (lymphocytic choriomeningitis virus) | Arenaviridae | 78–90 nm, spherical | 10.6 kb (segmented) | [20,21,22] |
| Influenza | Orthomyxoviridae | 50–120 nm, spherical + longer filamentous forms | 13.6 kb | [23,24] |
| Virus | Upstream | Downstream | Final Product | Process Performance | |||||
|---|---|---|---|---|---|---|---|---|---|
| Cell Culture | Virus Harvest and Release | Nucleic Acid Digestion | Clarification (Primary and Secondary) | 1st Purification (Capture) | 2nd Purification (Polish) | Sterile Filtration | Infectivity, Dosage, and Impurities | IU Overall Process Yield | |
| HSV-2 [290], (repl.-def. vaccine production) | Adh. Vero cells, MOI 0.01, TOH 24–72 hpi | Dextran sulfate, 100 µg/mL, up to 24 h, further processing of supernatant | e.g., Benzonase (Merck): 90 U/mL + 5 mM MgCl₂, at 25 °C for 4–6 h | Filtration (e.g., Sartopure PP2, 0.65 µm (Sartorius Stedim)) | AEX (e.g., Mustang Q membrane (Cytiva)): high-salt elution (2 M NaCl) | UF/DF (e.g., hollow fiber PS, 100 kDa, Spectrum Laboratories): TFF conc. (5–10×), DF (3–5×) | n.a. due to VP size, aseptic process suggested | >1 × 107 pfu/mL hcDNA < 10 ng/dose hcP: 30 µg/mL 107 pfu/dose | 10% to 20% |
| Orf [70] | Adh. Vero cells, MOI 0.05, TOH 120 hpi | Intracellular VP release by a FT-cycle, further processing of complete broth | Benzonase (Merck): 250 U/mL, 1 h at RT, after clarification | Filtration 5 µm and 0.65 µm, Sartopure PP3 (Sartorius) or Millistak cellulose with DE (Merck) | SXC binding at 8% PEG8000 on RC membrane stack (pore size 1 μm) | RAM: CC700 (Cytiva) | not discussed | 1.1–4.2 × 106 IU/mL total DNA: ~1 ng/dose total protein < LOD 106 IU/dose | 64% |
| Vaccinia [129], (MVA, batch process) | Avian susp. cells, MOI: 0.05, TOH at cell viability ~70% | Cell culture broth was further processed | Denerase (c-LEcta): 35 U/mL + 3 mM MgCl₂, 4 h 37 °C, after clarification | Primary: Acoustic settler (3 W, 2.1 MHz, 252 mL/h) Secondary: Filtration 0.45 µm, Sartopure PP3 (Sartorius Stedim) | Pre-filtration (0.45 µm, CA SXC: binding at 7.2% PEG6000 on RC membrane stack (pore size 1 µm) | n.a. | not discussed | >5 × 107 TCID50/mL hcDNA < 10 ng/dose total protein: 11–37 μg/dose 1.4 × 108 TCID50/dose | 55% |
| Vaccinia [129], (MVA, continuous perfusion process) | Avian susp. cells, MOI 0.05, TOH start 40 hpi | Continuous harvest of cell culture broth | Denerase (c-LEcta): 37 U/mL + 4 mM MgCl₂, 4 h 37 °C, after clarification | As batch process | As batch process | n.a. | not discussed | Titers as batch process hcDNA < 10 ng/dose total protein: ~10 μg/dose 1.4 × 108 TCID50/dose | 51% |
| Measles [67] | Adh. Vero cells, MOI: 0.001 to 0.01 | Supernatant was further processed | Benzonase (Merck): 50 U/mL + 2 mM MgCl2 for 1 h 37 °C after clarification | Filtration, 3 µm: Sartopure PP3 (Sartorius Stedim) | RAM: CC700 (Cytiva) | UF/DF (cellulose flat sheet membrane, 100 kDa, Merck): TFF conc. (~9×), DF (5×) | n.a. due to VP size, aseptic process suggested | 7.9 × 106 TCID50/mL dsDNA: 354 ng/mL → 18 ng/dose hcP: 18 µg/mL → 1 µg/dose 105 IU/dose | only step yields are disclosed, see reference |
| VSV [154], (rVSV-NDV, fusogenic virus) | Avian susp. cells, MOI 0.0001, TOH at cell viability ~90% (between 48 and 67 hpi) | Cell culture broth was further processed | Denerase (c-LEcta): 20 U/mL + 2 mM MgCl2, 1 h at RT | Primary: Filtration 1–0.4 µm Millistak CE (Cytiva) Secondary: Filtration 0.45 µm Sartopure PP3 (Sartorius Stedim) | AEX membrane Sartobind Q (Sartorius): Elution at 1.2 M NaCl | Pre-dilution (4×), UF/DF (PES hollow fiber, 750 kDa, Cytiva): TFF conc. (4×), DF (6×) | Supor EKV Mini Kleenpak filter 0.2 µm (Cytiva) | 4 × 109 TCID50/mL total DNA: 1.4 ng/mL total protein: 0.3 µg/mL 5 × 107 to 5 × 1010 IU/dose | 65% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schimek, A.; Ng, J.K.M.; Hubbuch, J. Navigating the Purification Process: Maintaining the Integrity of Replication-Competent Enveloped Viruses. Vaccines 2025, 13, 444. https://doi.org/10.3390/vaccines13050444
Schimek A, Ng JKM, Hubbuch J. Navigating the Purification Process: Maintaining the Integrity of Replication-Competent Enveloped Viruses. Vaccines. 2025; 13(5):444. https://doi.org/10.3390/vaccines13050444
Chicago/Turabian StyleSchimek, Adrian, Judy King Man Ng, and Jürgen Hubbuch. 2025. "Navigating the Purification Process: Maintaining the Integrity of Replication-Competent Enveloped Viruses" Vaccines 13, no. 5: 444. https://doi.org/10.3390/vaccines13050444
APA StyleSchimek, A., Ng, J. K. M., & Hubbuch, J. (2025). Navigating the Purification Process: Maintaining the Integrity of Replication-Competent Enveloped Viruses. Vaccines, 13(5), 444. https://doi.org/10.3390/vaccines13050444