Novel GMO-Based Vaccines against Tuberculosis: State of the Art and Biosafety Considerations

Abstract
:1. Introduction
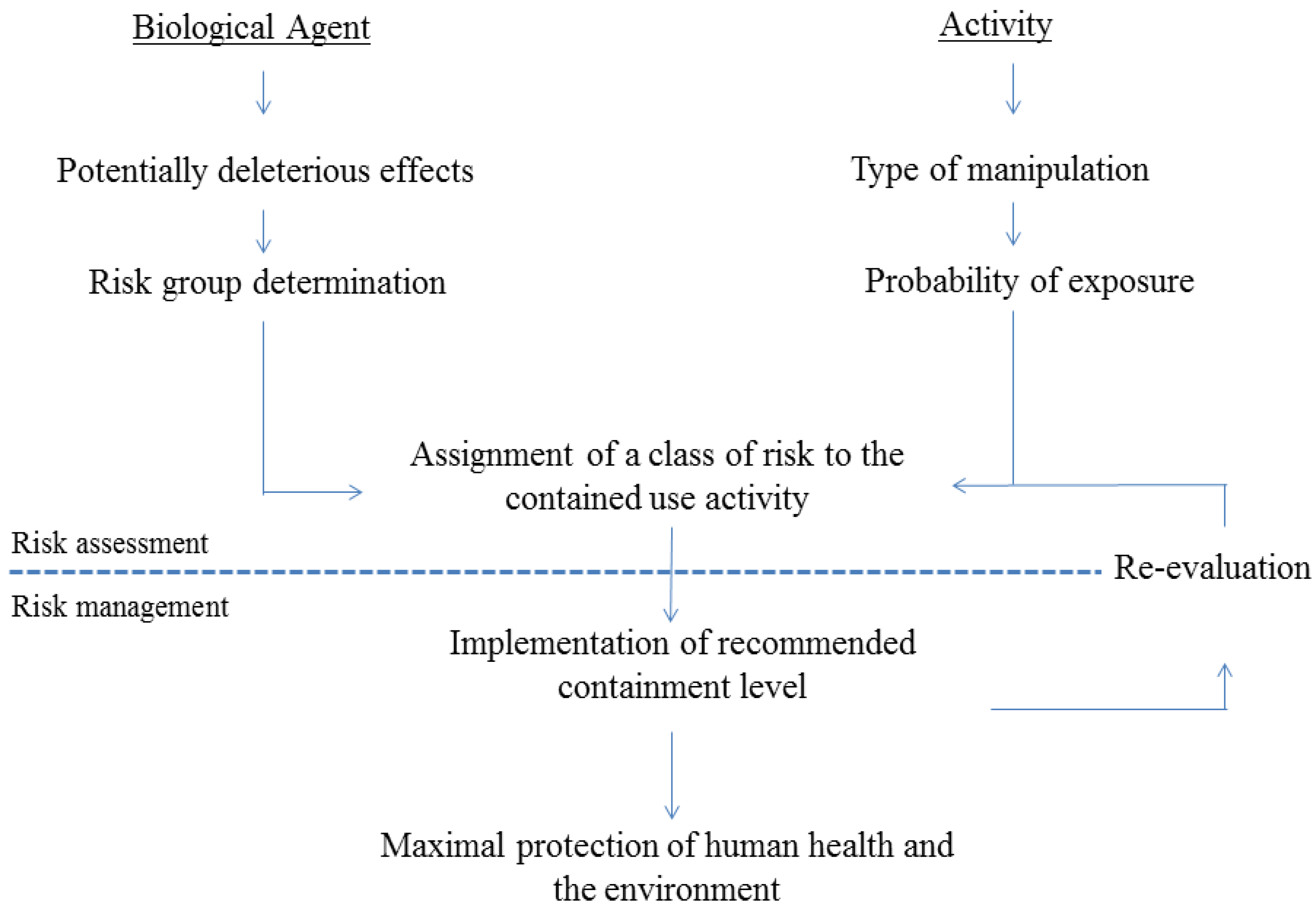
2. Risk Assessment of Activities Involving GMO-Based Vaccines against TB
2.1. General Regulatory Considerations in Europe
2.2. Activities Involving Manipulation of GMO-Based Vaccine Candidates against TB

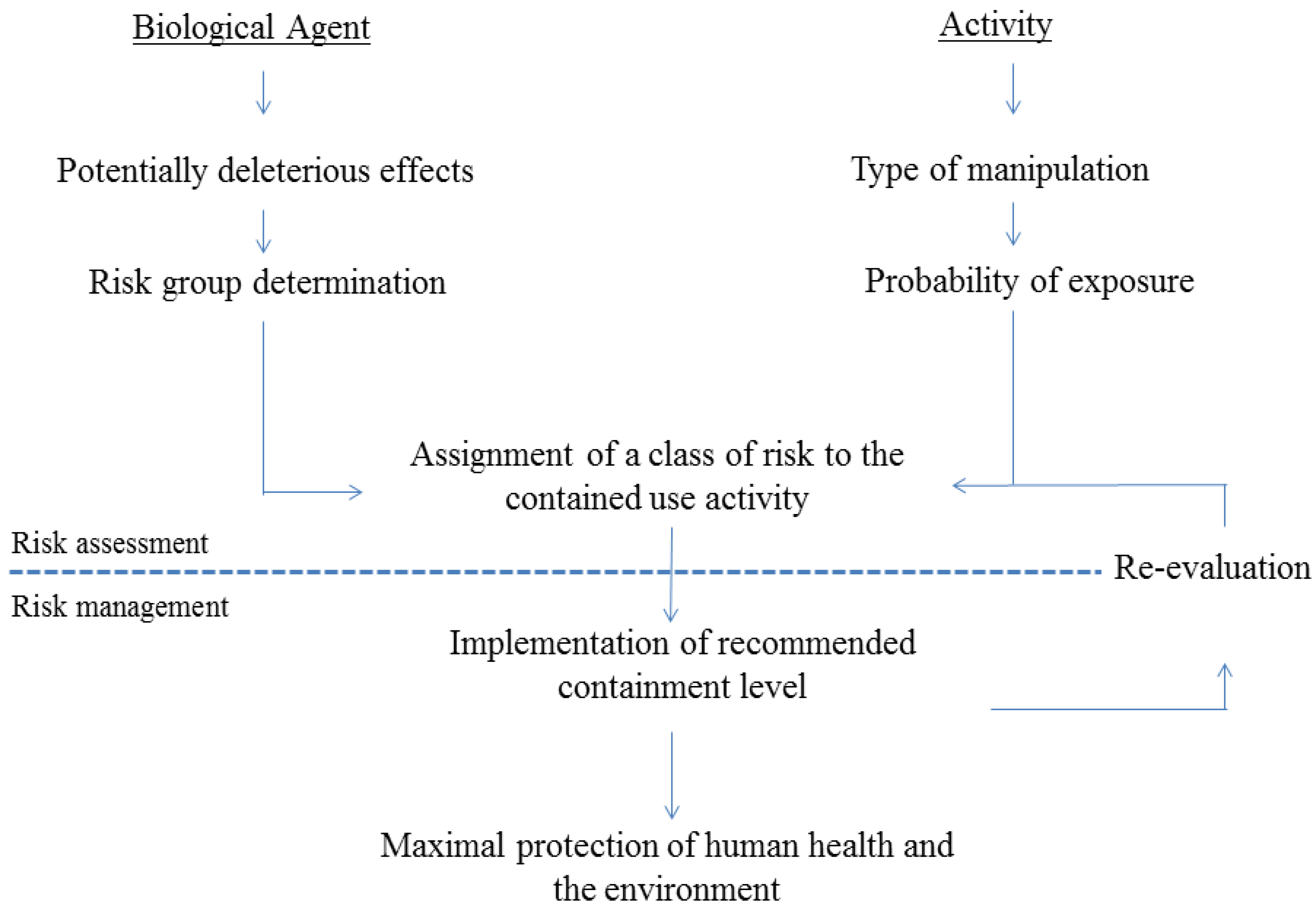{kind=link}
| Legislation | Main elements | Ref. |
|---|---|---|
| Directive 2000/54/EC | This Directive aims at the protection of workers against risks to their health and safety, including the prevention of such risks, arising or likely to arise from exposure to biological agents at work. It requests Member States to determine the nature, degree and duration of worker’s exposure during an activity likely to involve a risk of exposure to biological agents and on the basis of this assessment, to implement adequate protective measures. Biological agents are classified in four risk groups, from the one that is unlikely to cause human disease to a biological agent causing severe disease for which no effective prophylaxis or treatment is available. | [2] |
| Directive 2001/18/EC | This Directive defines the procedure for granting consent for the deliberate release in the environment and placing on the market of GMOs as or in products. It provides for a common methodology to assess on a case-by-case basis the risks for human health and the environment associated with the release of GMOs. It also introduces compulsory monitoring after GMOs have been placed on the market, as well as compulsory public consultation and GMO labeling. | [14] |
| Directive 2009/41/EC | This Directive focuses on the contained use of genetically modified micro-organisms (GMMs), i.e., any activity involving GMMs for which specific containment measures are used to limit their contact with, and to provide a high level of safety for, the general population and the environment. The Directive requests Member States to assess on a case-by-case basis the risks contained uses may pose and to implement appropriate containment and other protective measures to avoid adverse effects on human health and the environment. Contained uses are classified in four classes, from no or negligible risk to activities of high risk. | [15] |
| Regulation (EC) No. 726/2004 | This Regulation lays down procedures for the authorization, supervision and pharmacovigilance of medicinal products for human and veterinary use. For medicinal products derived from biotechnology, it foresees a compulsory centralized authorization procedure in which the European Medicines Agency is responsible for drawing up opinions on any matter concerning the evaluation of the products. | [16] |
| Directive 2001/20/EC | This Directive sets out common rules for the authorization and regulatory follow-up of a clinical trial. It aims at protecting human subjects involved in clinical trials and ensuring that the results are credible, by establishing quality, safety and ethical criteria. Approval of trials is the responsibility of individual EU Member States, who are required to evaluate the products used in clinical studies. | [18] |
3. Update of TB Vaccine Candidates in Clinical Trials
| Vaccine | Backbone | Genetic modification | Current clinical phase of development | Ref. |
|---|---|---|---|---|
| GMO-based vaccine candidates designed to replace BCG | ||||
| VPM1002 | Recombinant BCG | ΔureC::hly Hm(R) deleted in ureC expressing listeriolysin (hly) from Listeria monocytogenes | Phase II | [31,32] |
| MTBVAC | Recombinant Mtb | deleted in phoP and fadD26 without antibiotic-resistance markers | Phase I | [33] |
| Viral vectored sub-unit vaccines designed as booster vaccines | ||||
| MVA-85A (also called AERAS-485) | Recombinant Vaccinia Ankara vector | Expressing Ag85A (Rv3804c) | Phase IIb | [34,35,36,37,38,39,40,41,42,43,44,45,46,47] |
| MVA-85A-IMX313 | Recombinant Vaccinia Ankara vector | Expressing a fusion of Ag85A (Rv3804c) and IMX313 | Phase I | [48] |
| AERAS-402 (also called Crucell Ad35) | Recombinant replication deficient Adenovirus serotype 35 (Ad35) | Expressing Ag85A (Rv3804c); Ag85B (Rv1886c) and TB10.4 (Rv0288) as a fusion protein | Phase II | [49,50] |
| Deleted in E1 | ||||
| AdAg85A | Recombinant replication deficient Adenovirus serotype 5 (Ad5) | Expressing Ag85A (Rv3804c) | Phase I | [51,52] |
| Deleted in E1 and E3 | ||||
| ChAdOx1 85A | Recombinant replication deficient simian Adenovirus | Expressing Ag85A (Rv3804c) | Phase I | [53] |
| Deleted in E1 and E3 | ||||
4. Biosafety Considerations of Clinical Studies with GMO-Based Vaccines
4.1. BCG Replacement with Genetically Modified Mycobacteria
4.1.1. Genetically Modified BCG in Current Live Vaccine Candidates: VPM1002
4.1.1.1. Characteristics of the Parental BCG
4.1.1.2. The Transgene and the Genetic Modification
4.1.1.3. Genetic Stability of VPM1002
4.1.1.4. Safety of VPM1002
4.1.1.5. Transmission of VPM1002
4.1.1.6. VPM1002 Risk Classification
4.1.1.7. Biodistribution of VPM1002 and Environmental Risk Assessment
4.1.1.8. Risk management Measures (Containment, Worker Protection Measures, Waste)
|
4.1.2. Mtb Genetic Background of Current Live Vaccine Candidates: MTBVAC
4.1.2.1. Characteristics of the Parental Mtb
4.1.2.2. Genetic Modifications in MTBVAC
4.1.2.3. Genetic Stability of MTBVAC
4.1.2.4. Safety of MTBVAC
4.1.2.5. Transmission of MTBVAC
4.1.2.6. MTBVAC Risk Classification
4.1.2.7. Biodistribution of MTBVAC and Environmental Risk Assessment
4.1.2.8. Risk Management Measures (Containment, Workers Protection Measures, Waste)
4.2. TB Vaccine Candidates Based on Recombinant Viral Vectors as “Booster” Sub-Unit Vaccines
4.2.1. Intrinsic Characteristics of the Four Viral Strains Currently Used in TB Clinical Trials
4.2.2. The Characteristics of the Transgenes
4.2.3. Biodistribution and Dissemination of the Recombinant Viral Vector Vaccines
4.2.4. Possibility of Recombination with Other Viruses
4.2.5. Risk Classification
4.2.6. Environmental Risk Assessment
4.2.7. Risk Management Measures
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wirth, T.; Hildebrand, F.; Allix-Béguec, C.; Wölbeling, F.; Kubica, T.; Kremer, K.; van Soolingen, D.; Rüsch-Gerdes, S.; Locht, C.; Brisse, S.; et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 2008, 4, e1000160. [Google Scholar] [CrossRef]
- European Commission. Directive 2000/54/EC-biological agents at work of 18 September 2000 on the protection of workers from risks related to exposure to biological agents at work. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:32000L0054:EN:HTML (accessed on 23 April 2014).
- Herman, P.; Fauville-Dufaux, M.; Breyer, D.; van Vaerenbergh, B.; Pauwels, K.; do Thi, C.D.; Sneyers, M.; Wanlin, M.; Snacken, R.; Moens, W. Biosafety recommendations for the contained use of Mycobacterium tuberculosis complex isolates in industrialized countries. Available online: http://www.biosafety.be/CU/PDF/Mtub_Final_DL.pdf (accessed on 23 April 2014).
- WHO. Global Tuberculosis Report 2013. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 23 April 2014).
- Colditz, G.A.; Brewer, T.F.; Berkey, C.S. Efficacy of BCG vaccine in the prevention of tuberculosis: Meta-analysis of the published literature. JAMA 1994, 271, 698–702. [Google Scholar] [CrossRef]
- Colditz, G.A.; Berkey, C.S.; Mosteller, F.; Brewer, T.F.; Wilson, M.E.; Burdick, E.; Fineberg, H.V. The efficacy of bacillus Calmette-Guerin vaccination of newborns and infants in the prevention of tuberculosis: Meta-analyses of the published literature. Pediatrics 1995, 96, 29–35. [Google Scholar]
- Fine, P.E.M. Variation in protection by BCG: Implications of and for heterologous immunity. Lancet 1995, 346, 1339–1345. [Google Scholar] [CrossRef]
- Philips, J.A.; Ernst, J.D. Tuberculosis pathogenesis and immunity. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 353–384. [Google Scholar] [CrossRef]
- Casanova, J.L.; Abel, L. The human model: A genetic dissection of immunity to infection in natural conditions. Nat. Rev. Immunol. 2004, 4, 55–66. [Google Scholar] [CrossRef]
- Corbett, E.L. HIV and tuberculosis: Surveillance revisited [Editorial]. Int. J. Tuberc. Lung Dis. 2003, 7, 709. [Google Scholar]
- Harris, J.; Keane, J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin. Exp. Immunol. 2010, 161, 1–9. [Google Scholar]
- Caccamo, N.; Guggino, G.; Meraviglia, S.; Gelsomino, G.; di Carlo, P.; Titone, L.; Bocchino, M.; Galati, D.; Matarese, A.; Nouta, J.; et al. Analysis of Mycobacterium tuberculosis-specific CD8 T-cells in patients with active tuberculosis and in individuals with latent infection. PLoS One 2009, 4, e5528. [Google Scholar] [CrossRef]
- Bruns, H.; Meinken, C.; Schauenberg, P.; Härter, G.; Kern, P.; Modlin, R.L.; Antoni, C.; Stenger, S. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J. Clin. Invest. 2009, 119, 1167–1177. [Google Scholar] [CrossRef]
- European Commission. Directive 2001/18/EC of the European Parliament and of the Council of 12 March 2001 on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:32001L0018:EN:HTML (accessed on23 April 2014).
- European Commission. Directive 2009/41/EC of the European Parliament and of the Council of 6 May 2009 on the contained use of genetically modified micro-organisms. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:125:0075:01:EN:HTML (accessed on 23 April 2014).
- European Commission. Regulation (EC) N726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:32004R0726:EN:HTML (accessed on 23 April 2014).
- Verheust, C.; Goossens, M.; Pauwels, K.; Breyer, D. Biosafety aspects of modified vaccinia virus Ankara (MVA)-based vectors used for gene therapy or vaccination. Vaccine 2012, 30, 2623–2632. [Google Scholar] [CrossRef]
- European Commission. Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on the medicinal products for human use. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:32001L0020:EN:HTML (accessed on 23 April 2014).
- Laboratory Biosafety Manual, 3rd ed.; WHO: Geneva, Switzerland, 2004.
- Walker, K.B.; Brennan, M.J.; Ho, M.M.; Eskola, J.; Thiry, G.; Sadoff, J.; Dobbelaer, R.; Grode, L.; Liu, M.A.; Fruth, U.; et al. The second Geneva Consensus: Recommendations for novel live TB vaccines. Vaccine 2010, 28, 2259–2270. [Google Scholar] [CrossRef]
- Baldo, A.; van den Akker, E.; Bergmans, H.E.; Lim, F.; Pauwels, K. General considerations on the biosafety of virus-derived vectors used in gene therapy and vaccination. Curr. Gene Ther. 2013, 13, 385–394. [Google Scholar]
- Sterne, J.A.C.; Rodrigues, L.C.; Guedes, I.N. Does the efficacy of BCG decline with time since vaccination? Int. J. Tuber. Lung Dis. 1998, 2, 200–207. [Google Scholar]
- Romano, M.; Huygen, K. An update on vaccines for tuberculosis-there is more to it than just waning of BCG efficacy with time. Expert Opin.Biol.Ther. 2012, 12, 1601–1610. [Google Scholar] [CrossRef]
- Karonga Prevention Trial Group. Randomised controlled trial of single BCG, repeated BCG, or combined BCG and killed Mycobacterium leprae vaccine for prevention of leprosy and tuberculosis in Malawi. Lancet 1996, 348, 17–24. [Google Scholar]
- Brennan, M.J.; Clagett, B.; Fitzgerald, H.; Chen, V.; Williams, A.; Izzo, A.A.; Barker, L.F. Preclinical evidence for implementing a prime-boost vaccine strategy for tuberculosis. Vaccine 2012, 30, 2811–2823. [Google Scholar]
- Bertholet, S.; Ireton, G.C.; Ordway, D.J.; Windish, H.P.; Pine, S.O.; Kahn, M.; Phan, T.; Orme, I.M.; Vedvick, T.S.; Baldwin, S.L.; et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci. Transl. Med. 2012, 2. [Google Scholar] [CrossRef]
- Billeskov, R.; Elvang, T.T.; Andersen, P.L.; Dietrich, J. The HyVac4 subunit vaccine efficiently boosts BCG-primed anti-mycobacterial protective immunity. PLoS One 2012, 7, e39909. [Google Scholar]
- Lin, P.L.; Dietrich, J.; Tan, E.; Abalos, R.M.; Burgos, J.; Bigbee, C.; Bigbee, M.; Milk, L.; Gideon, H.P.; Rodgers, M.; et al. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J. Clin. Invest. 2012, 122, 303–314. [Google Scholar] [CrossRef]
- Montoya, J.; Solon, J.A.; Cunanan, S.R.; Acosta, L.; Bollaerts, A.; Moris, P.; Janssens, M.; Jongert, E.; Demoitié, M.A.; Mettens, P.; et al. A randomized, controlled dose-finding phase II study of the M72/AS01 candidate tuberculosis vaccine in healthy PPD-positive adults. J. Clin. Immunol. 2013, 33, 1360–1375. [Google Scholar] [CrossRef]
- Van Dissel, J.T.; Soonawala, D.; Joosten, S.A.; Prins, C.; Arend, S.M.; Bang, P.; Tingskov, P.N.; Lingnau, K.; Nouta, J.; Hoff, S.T.; et al. Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in volunteers with previous BCG vaccination or tuberculosis infection. Vaccine 2011, 29, 2100–2109. [Google Scholar] [CrossRef]
- Grode, L.; Seiler, P.; Baumann, S.; Hess, J.; Brinkmann, V.; Eddine, A.N.; Mann, P.; Goosmann, C.; Bandermann, S.; Smith, D.; et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. J. Clin. Invest. 2005, 115, 2472–2479. [Google Scholar] [CrossRef]
- Grode, L.; Ganoza, C.A.; Brohm, C.; Weiner, J., III; Eisele, B.; Kaufmann, S.H. Safety and immunogenicity of the recombinant BCG vaccine VPM1002 in a phase 1 open-label randomized clinical trial. Vaccine 2013, 31, 1340–1348. [Google Scholar] [CrossRef]
- Arbues, A.; Aguilo, J.I.; Gonzalo-Asensio, J.; Marinova, D.; Uranga, S.; Puentes, E.; Fernandez, C.; Parra, A.; Cardona, P.J.; Vilaplana, C.; et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine 2013, 31, 4867–4873. [Google Scholar] [CrossRef]
- Brookes, R.H.; Hill, P.C.; Owiafe, P.K.; Ibanga, H.B.; Jeffries, D.J.; Donkor, S.A.; Fletcher, H.A.; Hammond, A.S.; Lienhardt, C.; Adegbola, R.A.; et al. Safety and immunogenicity of the candidate tuberculosis vaccine MVA85A in West Africa. PLoS One 2008, 3, e2921. [Google Scholar] [CrossRef]
- Hawkridge, T.; Scriba, T.J.; Gelderbloem, S.; Smit, E.; Tameris, M.; Moyo, S.; Lang, T.; Veldsman, A.; Hatherill, M.; Merwe, L.; et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J. Infect. Dis. 2008, 198, 544–552. [Google Scholar] [CrossRef]
- Pathan, A.A.; Sander, C.R.; Fletcher, H.A.; Poulton, I.; Alder, N.C.; Beveridge, N.E.; Whelan, K.T.; Hill, A.V.; McShane, H. Boosting BCG with recombinant modified vaccinia ankara expressing antigen 85A: Different boosting intervals and implications for efficacy trials. PLoS One 2007, 2, e1052. [Google Scholar] [CrossRef]
- Sander, C.R.; Pathan, A.A.; Beveridge, N.E.; Poulton, I.; Minassian, A.; Alder, N.; van Wijgerden, J.; Hill, A.V.; Gleeson, F.V.; Davies, R.J.; et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in Mycobacterium tuberculosis-infected individuals. Am. J. Respir. Crit. Care Med. 2009, 179, 724–733. [Google Scholar] [CrossRef]
- Scriba, T.J.; Tameris, M.; Mansoor, N.; Smit, E.; van der Merwe, L.; Isaacs, F.; Keyser, A.; Moyo, S.; Brittain, N.; Lawrie, A.; et al. Modified vaccinia Ankara-expressing Ag85A, a novel tuberculosis vaccine, is safe in adolescents and children, and induces polyfunctional CD4+ T cells. Eur. J. Immunol. 2010, 40, 279–290. [Google Scholar]
- Scriba, T.J.; Tameris, M.; Smit, E.; van der Merwe, L.; Hughes, E.J.; Kadira, B.; Mauff, K.; Moyo, S.; Brittain, N.; Lawrie, A.; et al. A phase IIa trial of the new tuberculosis vaccine, MVA85A, in HIV- and/or Mycobacterium tuberculosis-infected adults. Am. J. Respir. Crit. Care Med. 2012, 185, 769–778. [Google Scholar] [CrossRef]
- Dieye, T.N.; Ndiaye, B.P.; Dieng, A.B.; Fall, M.; Britain, N.; Vermaak, S.; Camara, M.; Diop-Ndiaye, H.; Ngom-Gueye, N.F.; Diaw, P.A.; et al. Two doses of candidate TB vaccine MVA85A in antiretroviral therapy (ART) naive subjects gives comparable immunogenicity to one dose in ART+ subjects. PLoS One 2013, 8, e67177. [Google Scholar]
- Whelan, K.T.; Pathan, A.A.; Sander, C.R.; Fletcher, H.A.; Poulton, I.; Alder, N.C.; Hill, A.V.; McShane, H. Safety and immunogenicity of boosting BCG vaccinated subjects with BCG: Comparison with boosting with a new TB vaccine, MVA85A. PLoS One 2009, 4, e5934. [Google Scholar] [CrossRef]
- Scriba, T.J.; Tameris, M.; Mansoor, N.; Smit, E.; van der Merwe, L.; Mauff, K.; Hughes, E.J.; Moyo, S.; Brittain, N.; Lawrie, A.; Mulenga, H.; et al. Dose-finding study of the novel tuberculosis vaccine, MVA85A, in healthy BCG-vaccinated infants. J. Infect. Dis. 2011, 203, 1832–1843. [Google Scholar] [CrossRef]
- Pathan, A.A.; Minassian, A.M.; Sander, C.R.; Rowland, R.; Porter, D.W.; Poulton, I.; Hill, A.V.; Fletcher, H.A.; McShane, H. Effect of vaccine dose on the safety and immunogenicity of a candidate TB vaccine, MVA85A, in BCG vaccinated UK adults. Vaccine 2012, 30, 5616–5624. [Google Scholar] [CrossRef]
- Minassian, A.M.; Rowland, R.; Beveridge, N.E.; Poulton, I.; Satti, I.; Harris, S.; Poyntz, H.; Hamill, M.; Griffiths, K.; Sander, C.R.; et al. A Phase I study evaluating the safety and immunogenicity of MVA85A, a candidate TB vaccine, in HIV-infected adults. Br. Med. J. Open 2011, 1, e000223. [Google Scholar]
- Tameris, M.; McShane, H.; McClain, J.B.; Landry, B.; Lockhart, S.; Luabeya, A.K.K.; Geldenhuys, H.; Shea, J.; Hussey, G.; van der Merwe, L.; et al. Lessons learnt from the first efficacy trial of a new infant tuberculosis vaccine since BCG. Tuberculosis (Edinb.) 2013, 93, 143–149. [Google Scholar] [CrossRef]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; Shea, J.E.; McClain, J.B.; Hussey, G.D.; Hanekom, W.A.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef]
- Meyer, J.; Harris, S.A.; Satti, I.; Poulton, I.; Poyntz, H.C.; Tanner, R.; Rowland, R.; Griffiths, K.L.; Fletcher, H.A.; McShane, H. Comparing the safety and immunogenicity of a candidate TB vaccineMVA85A administered by intramuscular and intradermal delivery. Vaccine 2013, 31, 1026–1033. [Google Scholar] [CrossRef]
- Spencer, A.J.; Hill, F.; Honeycutt, J.D.; Cottingham, M.G.; Bregu, M.; Rollier, C.S.; Furze, J.; Draper, S.J.; Sogaard, K.C.; Gilbert, S.C.; et al. Fusion of the Mycobacterium tuberculosis antigen 85A to an oligomerization domain enhances its immunogenicity in both mice and non-human primates. PLoS One 2012, 7, e33555. [Google Scholar] [CrossRef]
- Abel, B.; Tameris, M.; Mansoor, N.; Gelderbloem, S.; Hughes, J.; Abrahams, D.; Makhethe, L.; Erasmus, M.; de Kock, M.; van der Merwe, L.; et al. The novel tuberculosis vaccine, AERAS-402, induces robust and polyfunctional CD4+ and CD8+ T cells in adults. Am. J. Respir. Crit. Care Med. 2010, 181, 1407–1417. [Google Scholar] [CrossRef]
- Hoft, D.F.; Blazevic, A.; Stanley, J.; Landry, B.; Sizemore, D.; Kpamegan, E.; Gearhart, J.; Scott, A.; Kik, S.; Pau, M.G.; et al. A recombinant adenovirus expressing immunodominant TB antigens can significantly enhance BCG-induced human immunity. Vaccine 2012, 30, 2098–2108. [Google Scholar] [CrossRef]
- Smaill, F.; Jeyanathan, M.; Smieja, M.; Medina, M.F.; Thanthrige-Don, N.; Zganiacz, A.; Yin, C.; Heriazon, A.; Damjanovic, D.; Puri, L.; et al. A human type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Wang, J.; Thorson, L.; Stokes, R.W.; Santosuosso, M.; Huygen, K.; Zganiacz, A.; Hitt, M.; Xing, Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 2004, 173, 6357–6365. [Google Scholar] [CrossRef]
- Dicks, M.D.; Spencer, A.J.; Edwards, N.J.; Wadell, G.; Bojang, K.; Gilbert, S.C.; Hill, A.V.; Cottingham, M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: Improved systems for vector derivation and comparative immunogenicity. PLoS One 2012, 7, e40385. [Google Scholar] [CrossRef]
- Ho, M.M.; Southern, J.; Kang, H.N.; Knezevic, I. WHO informal consultation on standardization and evaluation of BCG vaccines Geneva, Switzerland 22–23 September 2009. Vaccine 2010, 28, 6945–6950. [Google Scholar] [CrossRef]
- Velmurugan, K.; Grode, L.; Chang, R.; Fitzpatrick, M.; Laddy, D.; Hokey, D.; Derrick, S.; Morris, S.; McCown, D.; Kidd, R.; et al. Nonclinical development of BCG replacement vaccine candidates. Vaccines 2013, 1, 120–138. [Google Scholar] [CrossRef]
- Jackson, M.; Yamamoto, S. Historical Background of Mycobacterium bovis BCG. In BCG-Vaccineand Adjuvant; Takii, T., Maeyama, J., Yamamoto, S., Eds.; Japan Anti-Tuberculosis Association: Tokyo, Japan, 2011; pp. 3–12. [Google Scholar]
- Brosch, R.; Gordon, S.V.; Garnier, T.; Eiglmeier, K.; Frigui, W.; Valenti, P.; dos Santos, S.; Duthoy, S.; Lacroix, C.; Garcia-Pelayo, C.; et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl. Acad. Sci. USA 2007, 104, 5596–5601. [Google Scholar] [CrossRef]
- Horwitz, M.A.; Harth, G.; Dillon, B.J.; Maslesa-Galic, S. Commonly administered BCG strains including an evolutionarily early strain and evolutionarily late strains of disparate genealogy induce comparable protective immunity against tuberculosis. Vaccine 2009, 27, 441–445. [Google Scholar] [CrossRef]
- Horwitz, M.A.; Harth, G.; Dillon, B.J.; Maslesa-Galic, S. Recombinant bacillus Calmette-Guérin (BCG) vaccines expressing the Mycobacterium tuberculosis 30-kDa major secretory protein induce greater protective immunity against tuberculosis than conventional BCG vaccines in a highly susceptible animal model. Proc. Natl. Acad. Sci. USA 2000, 97, 13853–13858. [Google Scholar] [CrossRef]
- Sun, R.; Skeiky, Y.A.; Izzo, A.; Dheenadhayalan, V.; Imam, Z.; Penn, E.; Stagliano, K.; Haddock, S.; Mueller, S.; Fulkerson, J.; et al. Novel recombinant BCG expressing perfringolysin O and the over-expression of key immunodominant antigens; pre-clinical characterization, safety and protection against challenge with Mycobacterium tuberculosis. Vaccine 2009, 27, 4412–4423. [Google Scholar] [CrossRef]
- Festjens, N.; Bogaert, P.; Batni, A.; Houthuys, E.; Plets, E.; Vanderschaeghe, D.; Laukens, B.; Asselbergh, B.; Parthoens, E.; de Rycke, R.; et al. Disruption of the SapM locus in Mycobacterium bovis BCG improves its protective efficacy as a vaccine against M. tuberculosis. EMBO Mol. Med. 2011, 3, 222–234. [Google Scholar] [CrossRef]
- Johansen, P.; Fettelschoss, A.; Amstutz, B.; Selchow, P.; Waeckerle-Men, Y.; Keller, P.; Deretic, V.; Held, L.; Kündig, T.M.; Böttger, E.C.; et al. Relief from Zmp1-mediated arrest of phagosome maturation is associated with facilitated presentation and enhanced immunogenicity of mycobacterial antigens. Clin. Vacc. Immunol. 2011, 18, 907–913. [Google Scholar] [CrossRef]
- Vakzine Projekt Management GmbH. Summary Notification Information Format-B/DE/08/PEI574 Phase I Open Label, Randomized, Controlled, Dose-Escalation Study to Evaluate Safety and Immunogenicity of VPM1002 in Comparison with BCG in Healthy Male Volunteers. Available online: http://gmoinfo.jrc.ec.europa.eu/gmo_report.aspx?CurNot=B/DE/08/PEI574 (accessed on 23 April 2014).
- Frey, J. Classification of Organisms. Part1: Bacteria. Available online: http://www.bafu.admin.ch/publikationen/publikation/01614/index.html?lang=en (accessed on 23 April 2014).
- Health Safety Executive and ACDP. The Approved List of Biological Agents. Available online: http://www.hse.gov.uk/pubns/misc208.pdf (accessed on 23 April 2014).
- Heydebrand von und der Lasa, H.-C. and Federal Ministry of Food and Agriculture. Bekanntmachung der Liste Risikobewerteter Spenderund Empfängerorganismen für gentechnische arbeiten. Available online: http://www.bvl.bund.de/SharedDocs/Downloads/06_Gentechnik/register_datenbanken/organismenliste_pdf.pdf?__blob=publicationFile&v=7 (accessed on 23 April 2014).
- University of Boston and Research Occupational Health Program. Agent Information sheet: Mycobacterium bovis (BCG). Available online: http://www.bu.edu/rohp/files/2012/07/Mycobacterium-bovis-BCG.pdf (accessed on 23 April 2014).
- Norouzi, S.; Aghamohammadi, A.; Mamishi, S.; Rosenzweig, S.D.; Rezaei, N. Bacillus Calmette-Guérin (BCG) complications associated with primary immunodeficiency diseases. J. Infect. 2012, 64, 543–554. [Google Scholar] [CrossRef]
- WHO. Weekly Epidemiological Record Relevé épidémiologique hebdomadaire. Available online: http://www.who.int/wer/2007/wer8221.pdf?ua=1 (accessed on 23 April 2014).
- Hesseling, A.C.; Marais, B.J.; Gie, R.P.; Schaaf, H.S.; Fine, P.E.; Godfrey-Faussett, P.; Beyers, N. The risk of disseminated Bacille Calmette-Guerin (BCG) disease in HIV-infected children. Vaccine 2007, 25, 14–18. [Google Scholar]
- Public Health Agency of Canada. Pathogen Safety Data Sheet: Mycobacterium tuberculosis Complex. Available online: http://www.phac-aspc.gc.ca/lab-bio/res/psds-ftss/tuber-eng.php (accessed on 23 April 2014).
- Centers for Disease Control and Prevention (US). National Institutes of Health. Agent summary statements. In Biosafety in Microbiological and Biomedical Laboratories, 5th ed.; Chosewood, L.C., Wilson, D.E., Eds.; U.S. Government Printing Office: Washington, USA, 2009; pp. 123–289. [Google Scholar]
- Lewis, K.N.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Smith, S.; Behr, M.A.; Sherman, D.R. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille calmette-guérin attenuation. J. Infect. Dis. 2003, 187, 117–123. [Google Scholar] [CrossRef]
- Kozak, R.A.; Alexander, D.C.; Liao, R.; Sherman, D.R.; Behr, M.A. Region of difference 2 contributes to virulence of Mycobacterium tuberculosis. Infect. Immun. 2011, 79, 59–66. [Google Scholar] [CrossRef]
- Hess, J.; Miko, D.; Catic, A.; Lehmensiek, V.; Russell, D.G.; Kaufmann, S.H. Mycobacterium bovis bacille calmette-guerin strains secreting listeriolysin of listeria monocytogenes. Proc. Natl. Acad. Sci. USA 1998, 95, 5299–5304. [Google Scholar]
- Farinacci, M.; Weber, S.; Kaufmann, S.H. The recombinant tuberculosis vaccine rBCG DeltaureC::hly(+) induces apoptotic vesicles for improved priming of CD4+ and CD8+ T cells. Vaccine 2012, 30, 7608–7614. [Google Scholar] [CrossRef]
- Guzman, C.A.; Domann, E.; Ronde, M.; Bruder, D.; Darji, A.; Weiss, S.; Wehland, J.; Chakraborty, T.; Timmis, K.N. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol. Microbiol. 1996, 20, 119–126. [Google Scholar] [CrossRef]
- Rogers, H.W.; Callery, M.P.; Deck, B.; Unanue, E.R. Listeria monocytogenes induces apoptosis of infected hepatocytes. J. Immunol. 1996, 156, 679–684. [Google Scholar]
- Bavdek, A.; Kostanjsek, R.; Antonini, V.; Lakey, J.H.; Dalla-Serra, M.; Gilbert, R.J.C.; Anderluh, G. pH dependence of listeriolysin O aggregation and pore-forming ability. FEBS J. 2012, 279, 126–141. [Google Scholar] [CrossRef]
- Geoffroy, C.; Gaillard, J.L.; Alouf, J.E.; Berche, P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogene. Infect. Immun. 1987, 55, 1641–1646. [Google Scholar]
- Vakzine Projekt Management GmbH. Report on VPM1002 First Results of Clinical Study. Available online: http://www.vakzine-manager.de/en/resources/Produkte/VPM1002_en.pdf (accessed on 23 April 2014).
- Hess, J.; Kaufmann, S.H. Live antigen carriers as tools for improved anti-tuberculosis vaccines. FEMS Immunol. Med. Microbiol. 1999, 23, 165–173. [Google Scholar] [CrossRef]
- TBVI newsletter. First Results Tuberculosis Vaccine Candidate MTBVAC very Promising. Available online: http://www.tbvi.eu/news-agenda/news/news-message/first-results-tuberculosis-vaccine-candidate-very-promising.html (accessed on 23 April 2014).
- Gonzalo-Asensio, J.; Mostowy, S.; Harders-Westerveen, J.; Huygen, K.; Hernandez-Pando, R.; Thole, J.; Behr, M.; Gicquel, B.; Martin, C. PhoP: A missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One 2008, 3, e3496. [Google Scholar] [CrossRef]
- Pérez, E.; Samper, S.; Bordas, Y.; Guilhot, C.; Gicquel, B.; Martin, C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 2001, 41, 179–187. [Google Scholar] [CrossRef]
- Soto, C.Y.; Menéndez, M.C.; Pérez, E.; Samper, S.; Gomez, A.B.; Garcia, M.J.; Martin, C. IS6110 mediates increased transcription of the phoP virulence gene in a multidrug-resistant clinical isolate responsible for tuberculosis outbreaks. J. Clin. Microbiol. 2004, 42, 212–219. [Google Scholar] [CrossRef]
- Martin, C.; Williams, A.; Hernandez-Pando, R.; Cardona, P.J.; Gormley, E.; Bordat, Y.; Soto, C.Y.; Clark, S.O.; Hatch, G.J.; Aguilar, D.; et al. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 2006, 24, 3408–3419. [Google Scholar] [CrossRef]
- Cox, J.S.; Chen, B.; McNeil, M.; Jacobs, W.R. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 1999, 402, 79–83. [Google Scholar] [CrossRef]
- Quintero-Macias, L.; Santos-Mendoza, T.; Donis-Maturano, L.; Silva-Sanchez, A.; Aguilar, D.; Orozco, H.; Gicquel, B.; Estrada-Garcia, I.; Flores-Romo, L.; Hernandez-Pando, R. T-cell responses and in vivo cytotoxicity in the target organ and the regional lymphoid tissue during airborne infection with the virulent Mycobacterium tuberculosis MT103 and its lipid mutant fadD26. Scand. J. Immunol. 2010, 71, 20–28. [Google Scholar] [CrossRef]
- Camacho, L.R.; Ensergueix, D.; Perez, E.; Gicquel, B.; Guilhot, C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol. Microbiol. 1999, 34, 257–267. [Google Scholar] [CrossRef]
- Malaga, W.; Perez, E.; Guilhot, C. Production of unmarked mutations in mycobacteria using site-specific recombination. FEMS Microbiol. Lett. 2003, 219, 261–268. [Google Scholar] [CrossRef]
- Martin, C.; Gicquel, B.; Perez, E.; Gonzalo-Asensio, J.; Arbues, A. European patent specification: Tuberculose vaccine. Available online: http://www.lens.org/images/patent/EP/1997881/B1/EP_1997881_B1.pdf (accessed on 23 April 2014).
- Infante, E.; Aguilar, L.D.; Gicquel, B.; Hernandez Pando, R.H. Immunogenicity and protective efficacy of the Mycobacterium tuberculosis fadD26 mutant. Clin. Exp. Immunol. 2005, 141, 21–28. [Google Scholar] [CrossRef]
- Aronson, N.E.; Santosham, M.; Comstock, G.W. Long-term efficacy of BCG vaccine in american Indians and Alaska natives: A 60-year follow-up study. JAMA 2004, 291, 2086–2091. [Google Scholar] [CrossRef]
- Wold, S.M.; Toth, K. Adenovirus vectors for gene therapy, vaccination, and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Tiesjema, B.; Hermen, H.P.H.; van Eijkeren, J.C.H.; Brandon, E.F.A. Effect of administration route on the biodistribution and shedding of replication-deficient HAdV-5: A qualitative modelling approach. Curr. Gene Ther. 2010, 12, 107–127. [Google Scholar]
- Wold, W.; Ison, M. Adenoviruses. In Fields Virology, 6th ed.; Knipe, D., Howley, P., Eds.; Lippincott Williams & Wilkins: Philidelphia, PA, USA, 2013; pp. 1732–1767. [Google Scholar]
- Goossens, M.; Pauwels, K.; Willemarck, N.; Breyer, D. Environmental risk assessment of clinical trials involving modified vaccinia virus ankara (MVA)-based vectors. Curr. Gene Ther. 2013, 13, 413–420. [Google Scholar]
- Hillis, W.D.; Goodman, R. Serologic classification of chimpanzee adenoviruses by hemagglutination and hemagglutination inhibition. J. Immunol. 1969, 103, 1089–1095. [Google Scholar]
- Warimwe, G.M.; Lorenzo, G.; Lopez-Gil, E.; Reyes-Sandoval, A.; Cottingham, M.G.; Spencer, A.J.; Collins, K.A.; Dicks, M.D.; Milicic, A.; Lall, A.; et al. Immunogenicity and efficacy of a chimpanzee adenovirus-vectored Rift Valley Fever vaccine in mice. Virol. J. 2013, 10. [Google Scholar] [CrossRef]
- Havenga, M.; Vogels, R.; Zuijdgeest, D.; Radosevic, K.; Mueller, S.; Sieuwerts, M.; Weichold, F.; Damen, I.; Kaspers, J.; Lemckert, A.; et al. Novel replication-incompetent adenoviral B-group vectors: High vector stability and yield in PER C6 cells. J. Gen. Virol. 2006, 87, 2135–2143. [Google Scholar] [CrossRef]
- Moss, B. Poxviridae: The virus and their repication. In Field’s Virology, 4th ed.; Knipe, D., Howley, P., Eds.; Lippincot Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2849–2883. [Google Scholar]
- Kennedy, J.S.; Greenberg, R.N. Imvamune: Modified vaccinia Ankara strain as an attenuated smallpox vaccine. Expert Rev. Vaccines 2009, 8, 13–24. [Google Scholar] [CrossRef]
- Drumond, B.P.; Leite, J.A.; da Fonseca, F.G.; Bonjardim, C.A.; Ferreira, P.C.; Kroon, E. Brazilian vaccinia virus strains are genetically divergent and differ from the Lister vaccine strain. Microbes Infect. 2008, 10, 185–197. [Google Scholar] [CrossRef]
- Belisle, J.T.; Vissa, V.D.; Sievert, T.; Takayama, K.; Brennan, P.J.; Besra, G.S. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 1997, 276, 1420–1422. [Google Scholar] [CrossRef]
- Elamin, A.A.; Stehr, M.; Spallek, R.; Rohde, M.; Singh, M. The Mycobacterium tuberculosis Ag85A is a novel diacylglycerol acyltransferase involved in lipid body formation. Mol. Microbiol. 2011, 81, 1577–1592. [Google Scholar] [CrossRef]
- Skjot, R.L.; Brock, I.; Arend, S.M.; Munk, M.E.; Theisen, M.; Ottenhoff, T.H.; Andersen, P. Epitope mapping of the immunodominant antigen TB10.4 and the two homologous proteins TB10.3 and TB12.9, which constitute a subfamily of the esat-6 gene family. Infect. Immun 2002, 70, 5446–5453. [Google Scholar] [CrossRef]
- Kato-Maeda, M.; Bifani, P.J.; Kreiswirth, B.N.; Small, P.M. The nature and consequence of genetic variability within Mycobacterium tuberculosis. J. Clin. Invest. 2001, 107, 533–537. [Google Scholar] [CrossRef]
- Schenk-Braat, E.A.; van Mierlo, M.M.; Wagemaker, G.; Bangma, C.H.; Kaptein, L.C. An inventory of shedding data from clinical gene therapy trials. J. Gene Med. 2007, 9, 910–921. [Google Scholar] [CrossRef]
- Mu, J.; Jeyanathan, M.; Small, C.L.; Zhang, X.; Roediger, E.; Feng, X.; Chong, D.; Gauldie, J.; Xing, Z. Immunization with a bivalent adenovirus-vectored tuberculosis vaccine provides markedly improved protection over its monovalent counterpart against pulmonary tuberculosis. Mol. Ther. 2009, 17, 1093–1100. [Google Scholar] [CrossRef]
- Damjanovic, D.; Zhang, X.; Mu, J.; Medina, M.F.; Xing, Z. Organ distribution of transgene expression following intranasal mucosal delivery of recombinant replication-defective adenovirus gene transfer vector. Genet. Vaccines. Ther. 2008, 6. [Google Scholar] [CrossRef]
- McShane, H.; Pathan, A.A.; Sander, C.R.; Goonetilleke, N.P.; Fletcher, H.A.; Hill, A.V. Boosting BCG with MVA85A: The first candidate subunit vaccine for tuberculosis in clinical trials. Tuberculosis (Edinb.) 2005, 85, 47–52. [Google Scholar] [CrossRef]
- White, A.D.; Sibley, L.; Dennis, M.J.; Gooch, K.; Betts, G.; Edwards, N.; Reyes-Sandoval, A.; Carroll, M.W.; Williams, A.; March, P.D.; et al. Evaluation of the safety and immunogenicity of a candidate tuberculosis vaccine, MVA85A, delivered by aerosol to the lungs of macaques. Clin. Vacc. Immunol. 2013, 20, 663–672. [Google Scholar] [CrossRef]
- Vordermeier, H.M.; Rhodes, S.G.; Dean, G.; Goonetilleke, N.; Huygen, K.; Hill, A.V.; Hewinson, R.G.; Gilbert, S.C. Cellular immune responses induced in cattle by heterologous prime-boost vaccination using recombinant viruses and bacille Calmette-Guerin. Immunology 2004, 112, 461–470. [Google Scholar] [CrossRef]
- Vordermeier, H.M.; Villarreal-Ramos, B.; Cockle, P.J.; McAulay, M.; Rhodes, S.G.; Thacker, T.; Gilbert, S.C.; McShane, H.; Hill, A.V.; Xing, Z.; et al. Viral booster vaccines improve Mycobacterium bovis BCG-induced protection against bovine tuberculosis. Infect.Immun. 2009, 77, 3364–3373. [Google Scholar]
- Hansen, H.; Okeke, M.I.; Nilssen, O.; Traavik, T. Recombinant viruses obtained from co-infection in vitro with a live vaccinia-vectored influenza vaccine and a naturally occurring cowpox virus display different plaque phenotypes and loss of the transgene. Vaccine 2004, 23, 499–506. [Google Scholar] [CrossRef]
- Okeke, M.I.; Nilssen, O.; Moens, U.; Tryland, M.; Traavik, T. In vitro host range, multiplication and virion forms of recombinant viruses obtained from co-infection in vitro with a vaccinia-vectored influenza vaccine and a naturally occurring cowpox virus isolate. Virol. J. 2009, 6. [Google Scholar] [CrossRef]
- Carroll, M.W.; Moss, B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: Propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 1997, 238, 198–211. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Carroll, M.W.; Czerny, C.P.; Merchlinsky, M.; Sisler, J.R.; Moss, B. Marker rescue of the host range restriction defects of modified vaccinia virus Ankara. Virology 1998, 251, 334–342. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; Kuiken, T.; de Swart, R.L.; van Amerongen, G.; Vos, H.W.; Niesters, H.G.; van Schalkwijk, P.; van der Kwast, T.; Wyatt, L.S.; Moss, B.; et al. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 2001, 19, 3700–3709. [Google Scholar] [CrossRef]
- Suter, M.; Meisinger-Henschel, C.; Tzatzaris, M.; Hulsemann, V.; Lukassen, S.; Wulff, N.H.; Hausmann, J.; Howley, P.; Chaplin, P. Modified vaccinia Ankara strains with identical coding sequences actually represent complex mixtures of viruses that determine the biological properties of each strain. Vaccine 2009, 27, 7442–7450. [Google Scholar] [CrossRef]
- Goonetilleke, N.P.; McShane, H.; Hannan, C.M.; Anderson, R.J.; Brookes, R.H.; Hill, A.V. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J. Immunol. 2003, 171, 1602–1609. [Google Scholar] [CrossRef]
- Dye, C.; Fine, P.E. A major event for new tuberculosis vaccines. Lancet 2013, 381, 972–974. [Google Scholar] [CrossRef]
- Chauhan, P.; Reddy, P.V.; Singh, R.; Jaisinghani, N.; Gandotra, S.; Tyagi, A.K. Secretory phosphatases deficient mutant of Mycobacterium tuberculosis imparts protection at the primary site of infection in guinea pigs. PLoS One 2013, 8, e77930. [Google Scholar]
- Sampson, S.L.; Mansfield, K.G.; Carville, A.; Magee, D.M.; Quitugua, T.; Howerth, E.W.; Bloom, B.R.; Hondalus, M.K. Extended safety and efficacy studies of a live attenuated double leucine and pantothenate auxotroph of Mycobacterium tuberculosis as a vaccine candidate. Vaccine 2011, 29, 4839–4847. [Google Scholar] [CrossRef]
- Hedhli, D.; Denis, O.; Barkan, D.; Daffé, M.; Glickman, M.S.; Huygen, K. M. tuberculosis mutants lacking oxygenated mycolates show increased immunogenicity and protective efficacy as compared to M. bovis BCG vaccine in an experimental mouse model. PLoS One 2013, 8, e76442. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leunda, A.; Baldo, A.; Goossens, M.; Huygen, K.; Herman, P.; Romano, M. Novel GMO-Based Vaccines against Tuberculosis: State of the Art and Biosafety Considerations. Vaccines 2014, 2, 463-499. https://doi.org/10.3390/vaccines2020463
Leunda A, Baldo A, Goossens M, Huygen K, Herman P, Romano M. Novel GMO-Based Vaccines against Tuberculosis: State of the Art and Biosafety Considerations. Vaccines. 2014; 2(2):463-499. https://doi.org/10.3390/vaccines2020463
Chicago/Turabian StyleLeunda, Amaya, Aline Baldo, Martine Goossens, Kris Huygen, Philippe Herman, and Marta Romano. 2014. "Novel GMO-Based Vaccines against Tuberculosis: State of the Art and Biosafety Considerations" Vaccines 2, no. 2: 463-499. https://doi.org/10.3390/vaccines2020463
APA StyleLeunda, A., Baldo, A., Goossens, M., Huygen, K., Herman, P., & Romano, M. (2014). Novel GMO-Based Vaccines against Tuberculosis: State of the Art and Biosafety Considerations. Vaccines, 2(2), 463-499. https://doi.org/10.3390/vaccines2020463