Interaction of Viral Capsid-Derived Virus-Like Particles (VLPs) with the Innate Immune System



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Virus-Like Particles (VLPs)—Brief Overview
2. VLPs Interaction with the Innate Immune System
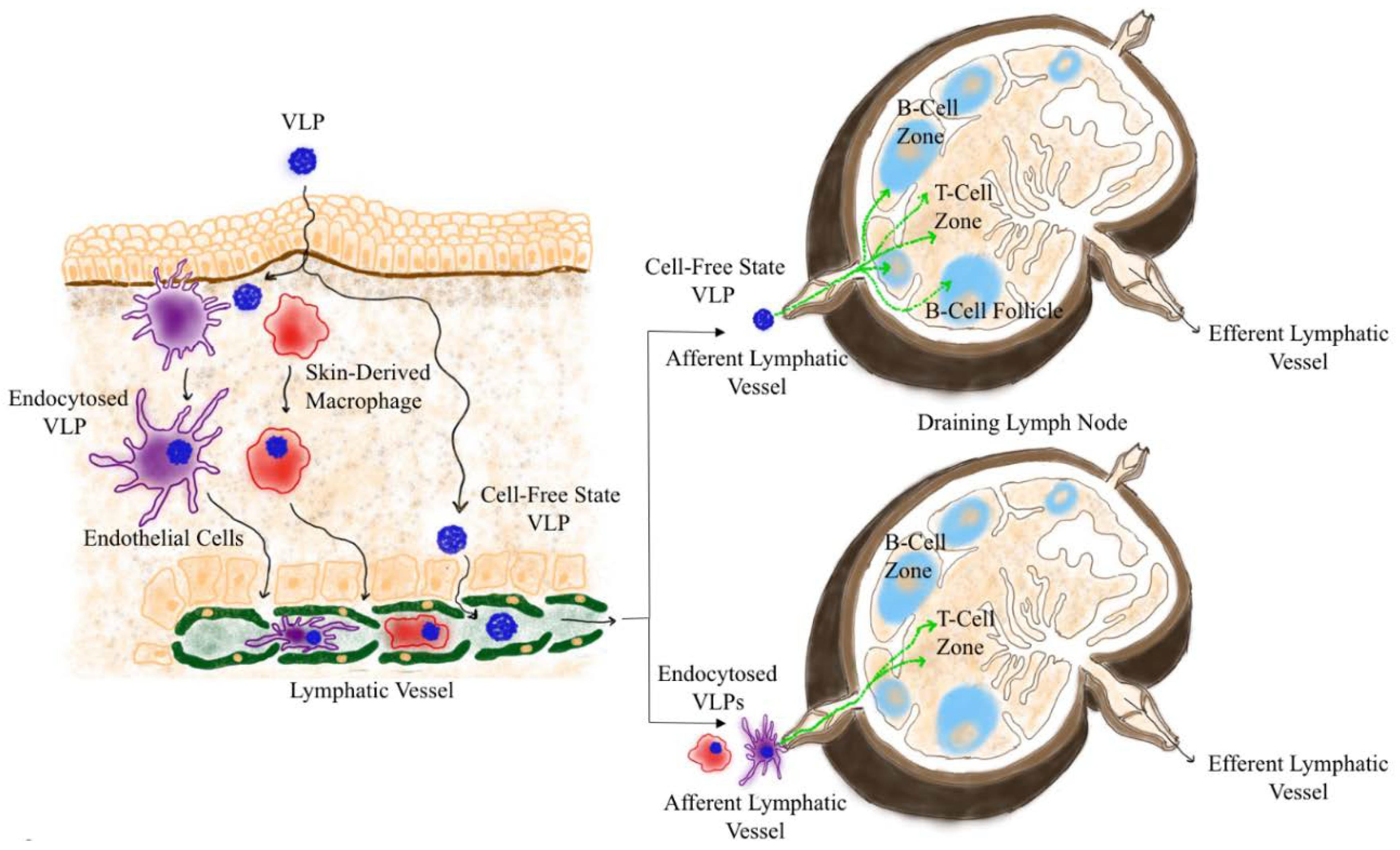
2.1. Drainage of VLP into the Lymphatoid Organs
2.2. Trafficking of VLPs Within Draining LN
2.3. VLPs and Innate Humoral Immune Response
2.4. Efficient Presentation of VLPs by Both MHC Pathways
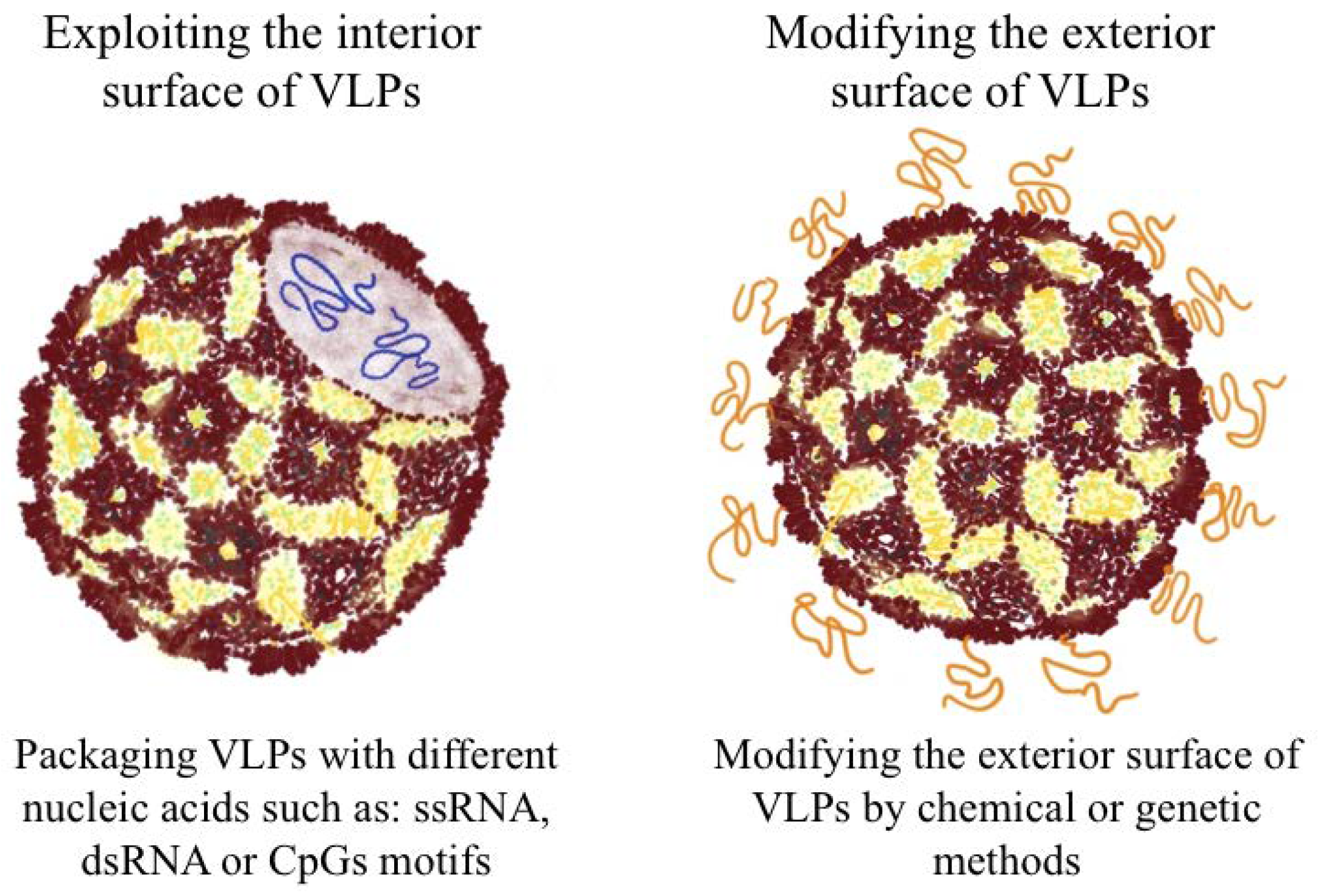
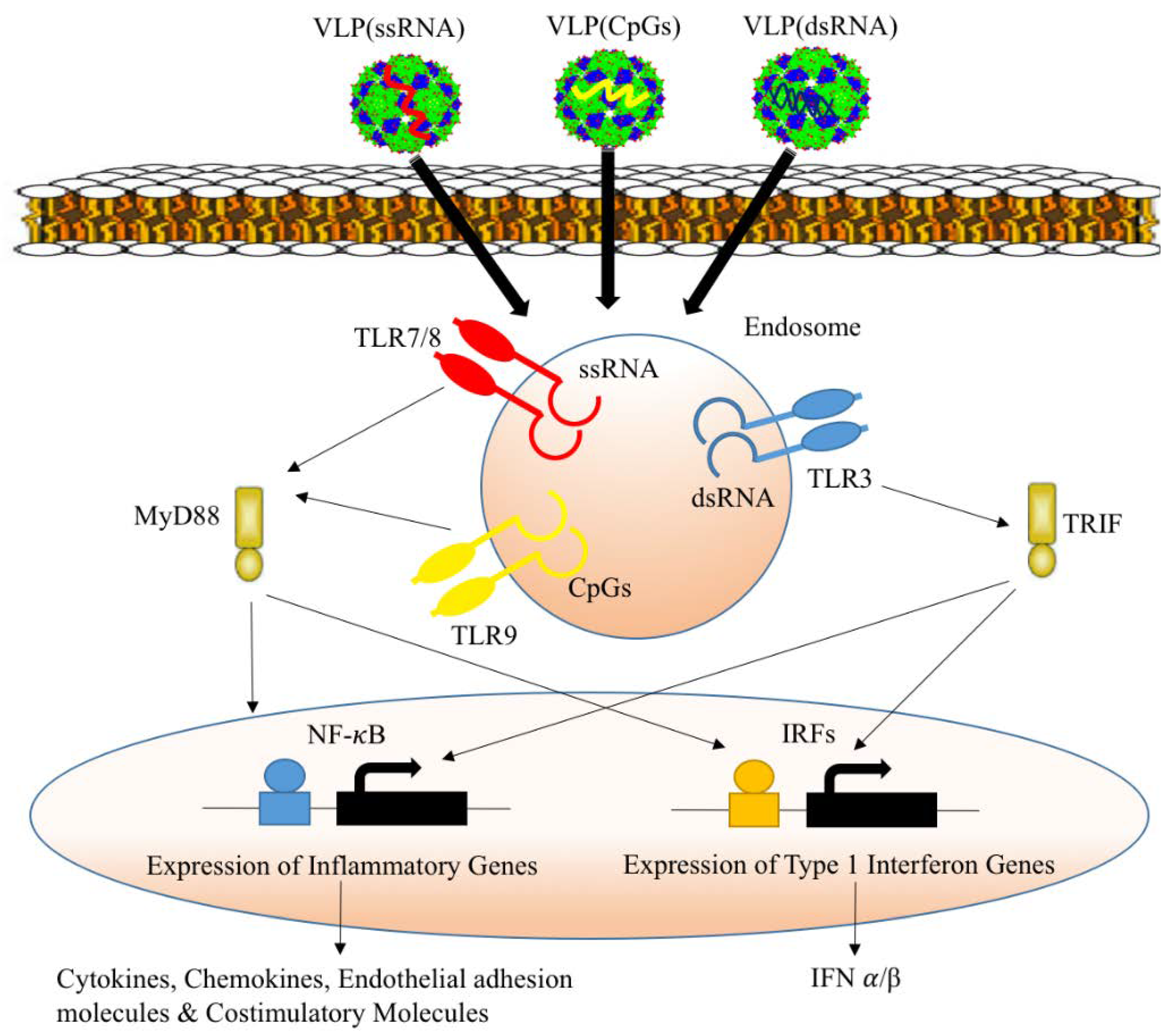
2.5. Packaging VLPs with Innate Immune-Modulators
2.6. Important Considerations on VLP Based Vaccines
2.7. Challenges for VLP-Based Vaccine Development
- Even though several VLP-based vaccines are on the market, some more recent candidates struggle with stability. In addition, no vaccine that displays foreign epitopes has made it to the market so far. Hence, real-life, market PoC for such vaccines is missing. While there is no a priori reason that this should not be possible, it may still be perceived as a potential risk.
- Most if not all nucleocapsid VLPs derived from RNA viruses package RNA from the production host cells. This may need an additional Quality Control effort.
- If epitopes are to be fused into VLPs, this can create substantial problems, as VLPs may not assemble anymore.
- The selected epitope may not be protective
- Induced immune responses may be too low
- The selected indication may sound interesting but does not attract interest from industry and/or the end-customer
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| VLPs | Virus-Like Particles |
| PRR | Pattern Recognition Receptor |
| PASP | Pathogen-Associated Structural Pattern |
| TLR | Toll-Like Receptor |
| LN | Lymph Node |
| SCS | Sub-Capsular Sinus |
| DC | Dendritic Cell |
| cDC | Conventional Dendritic Cell |
| pDC | Plasmacytoid Dendritic Cell |
| dDC | Dermal Dendritic Cell |
| fDC | Follicular Dendritic Cell |
| LC | Langerhans Cell |
| APC | Antigen-Presenting Cell |
| CTL | Cytotoxic T-Lymphocyte |
| CRP | C Reactive Protein |
| PTX3 | Pentraxin-Related Protein |
| MHC-I | Major Histocompatability Class I |
| MHC-II | Major Histocompatibility Class II |
| LCMV | Lymphocytic Choriomeningitis Virus |
| dsRNA | Double Stranded RNA |
| ssRNA | Single Stranded RNA |
| TNF | Tumor Necrosis Factor |
| IFN | Interferon |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of Activated B-Cells |
| AP-1 | Activator Protein 1 |
| IRF | Interferon Regulatory factor |
| CCL2 | Chemokine (C-C motif) Ligand 2 |
| CXCL8 | Chemokine (C-X-C motif) Ligand 8 |
| MyD88 | Myeloid Differentiation Primary Response Gene 88 |
| TRIF | TIR-Domain-Containing Adapter-Inducing Interferon-β |
References
- Schwarz, B.; Uchida, M.; Douglas, T. Biomedical and Catalytic Opportunities of Virus-Like Particles in Nanotechnology. Adv. Virus Res. 2017, 97, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Goldinger, S.M.; Dummer, R.; Baumgaertner, P.; Mihic-Probst, D.; Schwarz, K.; Hammann-Haenni, A.; Willers, J.; Geldhof, C.; Prior, J.O.; Kundig, T.M.; et al. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8(+) T-cell responses in melanoma patients. Eur. J. Immunol. 2012, 42, 3049–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, M.B.; Kim, S.Y.; Yun, W.S.; Lim, Y.T. Sequential delivery of an anticancer drug and combined immunomodulatory nanoparticles for efficient chemoimmunotherapy. Int. J. Nanomed. 2015, 10, 5981–5992. [Google Scholar] [CrossRef]
- Schwarz, K.; Meijerink, E.; Speiser, D.E.; Tissot, A.C.; Cielens, I.; Renhof, R.; Dishlers, A.; Pumpens, P.; Bachmann, M.F. Efficient homologous prime-boost strategies for T cell vaccination based on virus-like particles. Eur. J. Immunol. 2005, 35, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-Display: Decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Argenio, D.A.; Wilson, C.B. A decade of vaccines: Integrating immunology and vaccinology for rational vaccine design. Immunity 2010, 33, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Guo, J.H.; Fan, M.W. The effect of antigen size on the immunogenicity of antigen presenting cell targeted DNA vaccine. Int. Immunopharmacol. 2012, 12, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsen, M.O.; Gomes, A.C.; Cabral-Miranda, G.; Krueger, C.C.; Leoratti, F.M.; Stein, J.V.; Bachmann, M.F. Delivering adjuvants and antigens in separate nanoparticles eliminates the need of physical linkage for effective vaccination. J. Control. Release 2017, 251, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ruedl, C.; Storni, T.; Lechner, F.; Bachi, T.; Bachmann, M.F. Cross-presentation of virus-like particles by skin-derived CD8(-) dendritic cells: A dispensable role for TAP. Eur. J. Immunol. 2002, 32, 818–825. [Google Scholar] [CrossRef]
- Keller, S.A.; Bauer, M.; Manolova, V.; Muntwiler, S.; Saudan, P.; Bachmann, M.F. Cutting edge: Limited specialization of dendritic cell subsets for MHC class II-associated presentation of viral particles. J. Immunol. 2010, 184, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Egen, J.G.; Huang, A.Y.; Germain, R.N. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science 2006, 312, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, Y.R.; Batista, F.D. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 2007, 27, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.F.; Degn, S.E.; Pitcher, L.A.; Woodruff, M.; Heesters, B.A.; Carroll, M.C. Trafficking of B cell antigen in lymph nodes. Annu. Rev. Immunol. 2011, 29, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Link, A.; Zabel, F.; Schnetzler, Y.; Titz, A.; Brombacher, F.; Bachmann, M.F. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J. Immunol. 2012, 188, 3724–3733. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.; Pombo, A.; Jenkins, C.; MacPherson, G.G. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 1998, 161, 1313–1319. [Google Scholar] [PubMed]
- Kissenpfennig, A.; Henri, S.; Dubois, B.; Laplace-Builhe, C.; Perrin, P.; Romani, N.; Tripp, C.H.; Douillard, P.; Leserman, L.; Kaiserlian, D.; et al. Dynamics and function of Langerhans cells in vivo: Dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 2005, 22, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.E.; Langlois, M.P.; Daudelin, J.F.; Tarrab, E.; Savard, P.; Leclerc, D.; Lamarre, A. Complement Component 3 Regulates IFN-alpha Production by Plasmacytoid Dendritic Cells following TLR7 Activation by a Plant Virus-like Nanoparticle. J. Immunol. 2017, 198, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.; Vilsgaard Ravn, K.; Juul Sorensen, I.; Jonson, G.; Holm Nielsen, E.; Svehag, S.E. Serum amyloid P component binds to influenza A virus haemagglutinin and inhibits the virus infection in vitro. Scand. J. Immunol. 1997, 46, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Clingan, J.M.; Matloubian, M. B Cell-intrinsic TLR7 signaling is required for optimal B cell responses during chronic viral infection. J. Immunol. 2013, 191, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Jegerlehner, A.; Maurer, P.; Bessa, J.; Hinton, H.J.; Kopf, M.; Bachmann, M.F. TLR9 signaling in B cells determines class switch recombination to IgG2a. J. Immunol. 2007, 178, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Bessa, J.; Zabel, F.; Link, A.; Jegerlehner, A.; Hinton, H.J.; Schmitz, N.; Bauer, M.; Kundig, T.M.; Saudan, P.; Bachmann, M.F. Low-affinity B cells transport viral particles from the lung to the spleen to initiate antibody responses. Proc. Natl. Acad. Sci. USA 2012, 109, 20566–20571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, D.; Young, J.W.; Banchereau, J. Dendritic cells. Adv. Immunol. 1999, 72, 255–324. [Google Scholar] [PubMed]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.Y.; Bruce, A.T.; Pardoll, D.M.; Levitsky, H.I. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity 1996, 4, 349–355. [Google Scholar] [CrossRef]
- Huang, A.Y.; Golumbek, P.; Ahmadzadeh, M.; Jaffee, E.; Pardoll, D.; Levitsky, H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science 1994, 264, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Storni, T.; Bachmann, M.F. Loading of MHC class I and II presentation pathways by exogenous antigens: A quantitative in vivo comparison. J. Immunol. 2004, 172, 6129–6135. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.V.; Song, R. Phagocytic processing of exogenous particulate antigens by macrophages for presentation by class I MHC molecules. J. Immunol. 1994, 153, 4925–4933. [Google Scholar] [PubMed]
- Fang, H.; Tan, M.; Xia, M.; Wang, L.; Jiang, X. Norovirus P particle efficiently elicits innate, humoral and cellular immunity. PLoS ONE 2013, 8, e63269. [Google Scholar] [CrossRef] [PubMed]
- Subklewe, M.; Paludan, C.; Tsang, M.L.; Mahnke, K.; Steinman, R.M.; Munz, C. Dendritic cells cross-present latency gene products from Epstein-Barr virus-transformed B cells and expand tumor-reactive CD8(+) killer T cells. J. Exp. Med. 2001, 193, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Clark, K.; Benacerraf, B.; Rock, K.L. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. USA 1993, 90, 4942–4946. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Dudziak, D.; Kamphorst, A.O.; Heidkamp, G.F.; Buchholz, V.R.; Trumpfheller, C.; Yamazaki, S.; Cheong, C.; Liu, K.; Lee, H.W.; Park, C.G.; et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007, 315, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Jennings, G.T.; Martina, B.E.; Keller, I.; Beck, M.; Pumpens, P.; Osterhaus, A.D.; Bachmann, M.F. A VLP-based vaccine targeting domain III of the West Nile virus E protein protects from lethal infection in mice. Virol. J. 2010, 7, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Storni, T.; Ruedl, C.; Schwarz, K.; Schwendener, R.A.; Renner, W.A.; Bachmann, M.F. Nonmethylated CG motifs packaged into virus-like particles induce protective cytotoxic T cell responses in the absence of systemic side effects. J. Immunol. 2004, 172, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3516–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Utaisincharoen, P.; Kespichayawattana, W.; Anuntagool, N.; Chaisuriya, P.; Pichyangkul, S.; Krieg, A.M.; Sirisinha, S. CpG ODN enhances uptake of bacteria by mouse macrophages. Clin. Exp. Immunol. 2003, 132, 70–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.B.; Moltedo, B.; Alexopoulou, L.; Bonifaz, L.; Flavell, R.A.; Moran, T.M. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J. Immunol. 2004, 173, 6882–6889. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Storni, T.; Manolova, V.; Didierlaurent, A.; Sirard, J.C.; Rothlisberger, P.; Bachmann, M.F. Role of Toll-like receptors in costimulating cytotoxic T cell responses. Eur. J. Immunol. 2003, 33, 1465–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Hiscott, J.; Lin, R. STING-ing the antiviral pathway. J. Mol. Cell Biol. 2010, 2, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Vance, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Fitzgerald, K.A.; Paludan, S.R. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulen, M.F.; Koch, U.; Haag, S.M.; Schuler, F.; Apetoh, L.; Villunger, A.; Radtke, F.; Ablasser, A. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 2017, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Krugman, S. The newly licensed hepatitis B vaccine. Characteristics and indications for use. JAMA 1982, 247, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Kozlovska, T.M.; Cielens, I.; Dreilinna, D.; Dislers, A.; Baumanis, V.; Ose, V.; Pumpens, P. Recombinant RNA phage Q beta capsid particles synthesized and self-assembled in Escherichia coli. Gene 1993, 137, 133–137. [Google Scholar] [CrossRef]
- Galaway, F.A.; Stockley, P.G. MS2 viruslike particles: A robust, semisynthetic targeted drug delivery platform. Mol. Pharm. 2013, 10, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Maurer, P.; Jennings, G.T.; Willers, J.; Rohner, F.; Lindman, Y.; Roubicek, K.; Renner, W.A.; Muller, P.; Bachmann, M.F. A therapeutic vaccine for nicotine dependence: Preclinical efficacy, and Phase I safety and immunogenicity. Eur. J. Immunol. 2005, 35, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Akache, B.; Weeratna, R.D.; Deora, A.; Thorn, J.M.; Champion, B.; Merson, J.R.; Davis, H.L.; McCluskie, M.J. Anti-IgE Qb-VLP Conjugate Vaccine Self-Adjuvants through Activation of TLR7. Vaccines (Basel) 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Ambuhl, P.M.; Tissot, A.C.; Fulurija, A.; Maurer, P.; Nussberger, J.; Sabat, R.; Nief, V.; Schellekens, C.; Sladko, K.; Roubicek, K.; et al. A vaccine for hypertension based on virus-like particles: Preclinical efficacy and phase I safety and immunogenicity. J. Hypertens. 2007, 25, 63–72. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohsen, M.O.; Gomes, A.C.; Vogel, M.; Bachmann, M.F. Interaction of Viral Capsid-Derived Virus-Like Particles (VLPs) with the Innate Immune System. Vaccines 2018, 6, 37. https://doi.org/10.3390/vaccines6030037
Mohsen MO, Gomes AC, Vogel M, Bachmann MF. Interaction of Viral Capsid-Derived Virus-Like Particles (VLPs) with the Innate Immune System. Vaccines. 2018; 6(3):37. https://doi.org/10.3390/vaccines6030037
Chicago/Turabian StyleMohsen, Mona O., Ariane C. Gomes, Monique Vogel, and Martin F. Bachmann. 2018. "Interaction of Viral Capsid-Derived Virus-Like Particles (VLPs) with the Innate Immune System" Vaccines 6, no. 3: 37. https://doi.org/10.3390/vaccines6030037
APA StyleMohsen, M. O., Gomes, A. C., Vogel, M., & Bachmann, M. F. (2018). Interaction of Viral Capsid-Derived Virus-Like Particles (VLPs) with the Innate Immune System. Vaccines, 6(3), 37. https://doi.org/10.3390/vaccines6030037