Tauopathy Analysis in P301S Mouse Model of Alzheimer Disease Immunized with DNA and MVA Poxvirus-Based Vaccines Expressing Human Full-Length 4R2N or 3RC Tau Proteins

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells
2.3. Viruses
2.4. Human Tau Antigens
2.5. Construction of Plasmid Transfer Vectors pCyA-Tau4R2N and pCyA-Tau3RC
2.6. Generation of Recombinant Viruses MVA-Tau4R2N and MVA-Tau3RC
2.7. Characterization of MVA-Tau4R2N and MVA-Tau3RC
2.7.1. PCR
2.7.2. Western Blot
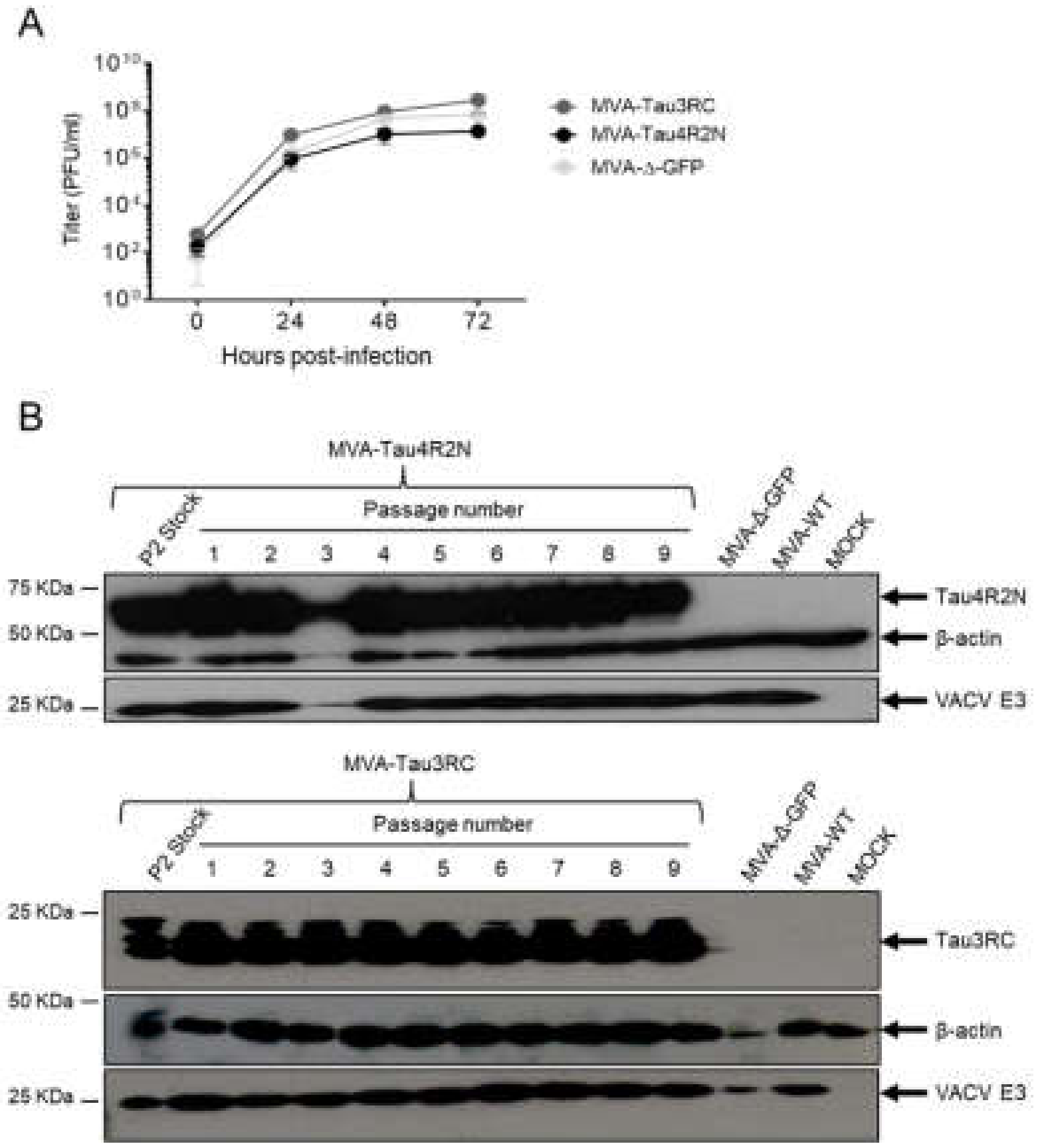
2.7.3. Virus Growth
2.8. Genetic Stability of MVA-Tau4R2N and MVA-Tau3RC
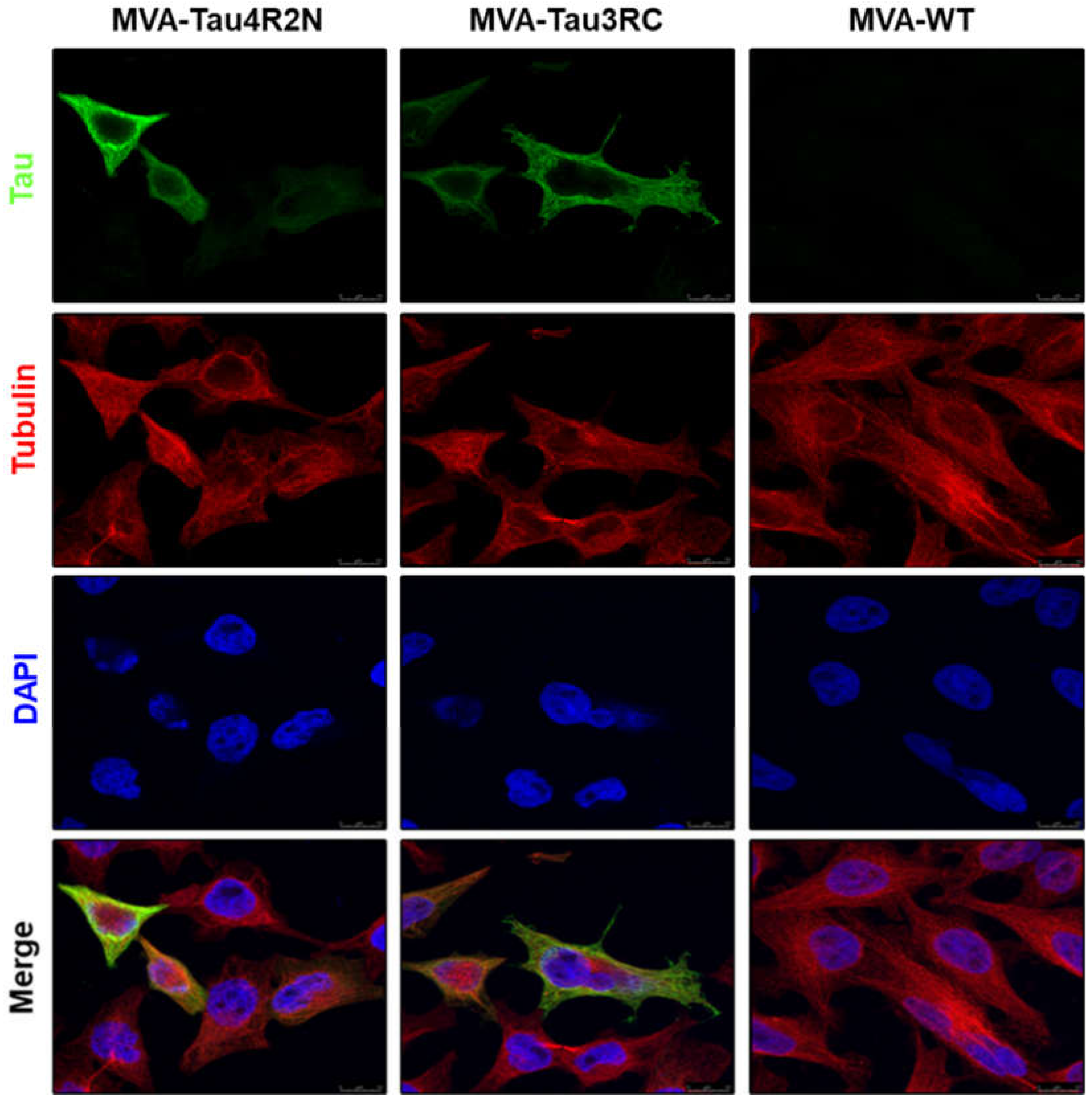
2.9. Analysis of the Expression of Human Tau4R2N and Tau3RC Proteins by Confocal Immunofluorescence Microscopy
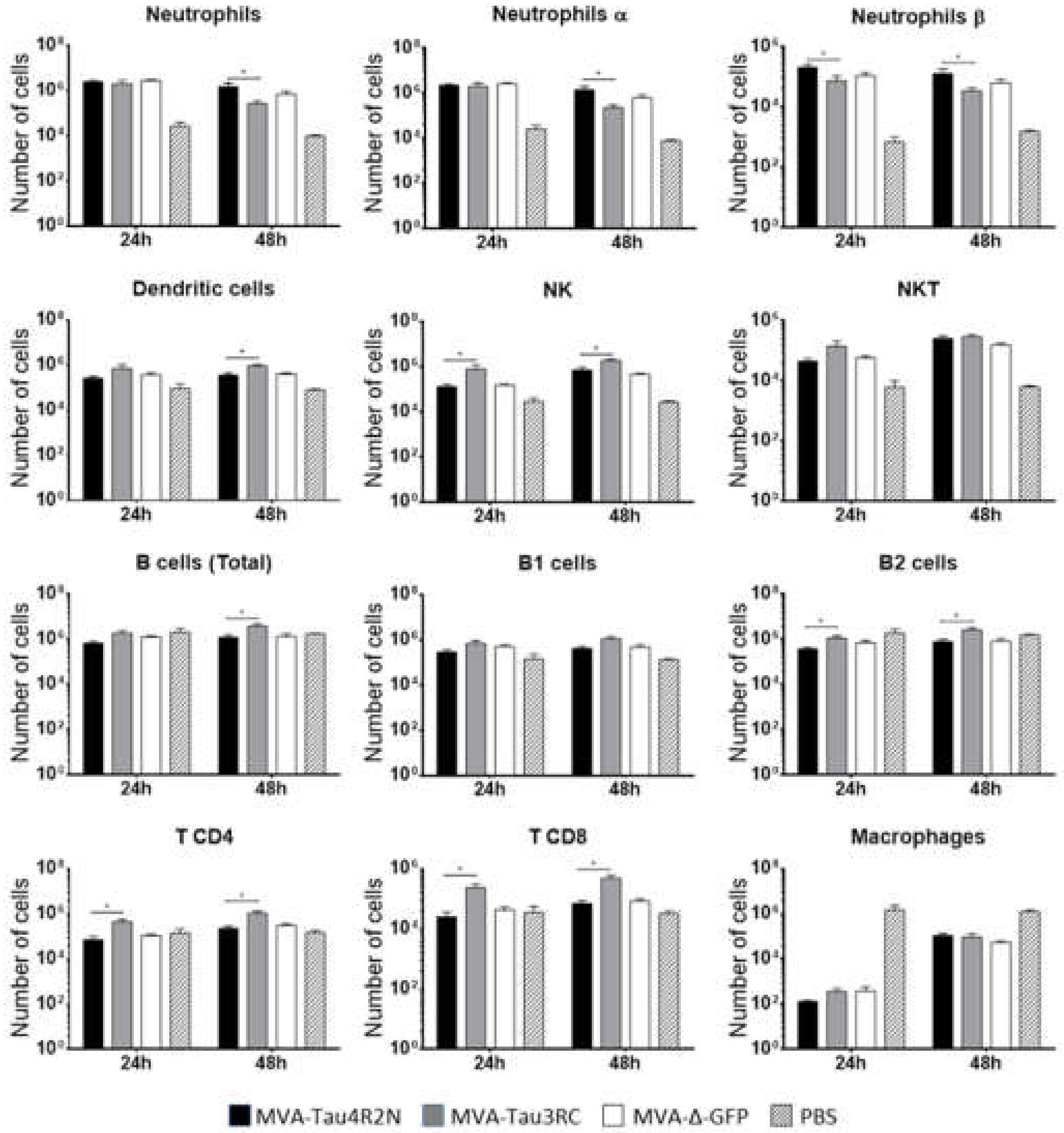
2.10. Recruitment of Immune Cells in the Peritoneal Cavity of C57BL/6 Mice Inoculated with MVA-Tau4R2N and MVA-Tau3RC
2.11. P301S Transgenic Mice Immunization Schedule
2.12. Study of Tau Phosphorylation in Brain Samples by Western Blot or Immunohistochemistry
2.13. Rotarod Test
2.14. Data Analysis and Statistical Procedures
3. Results
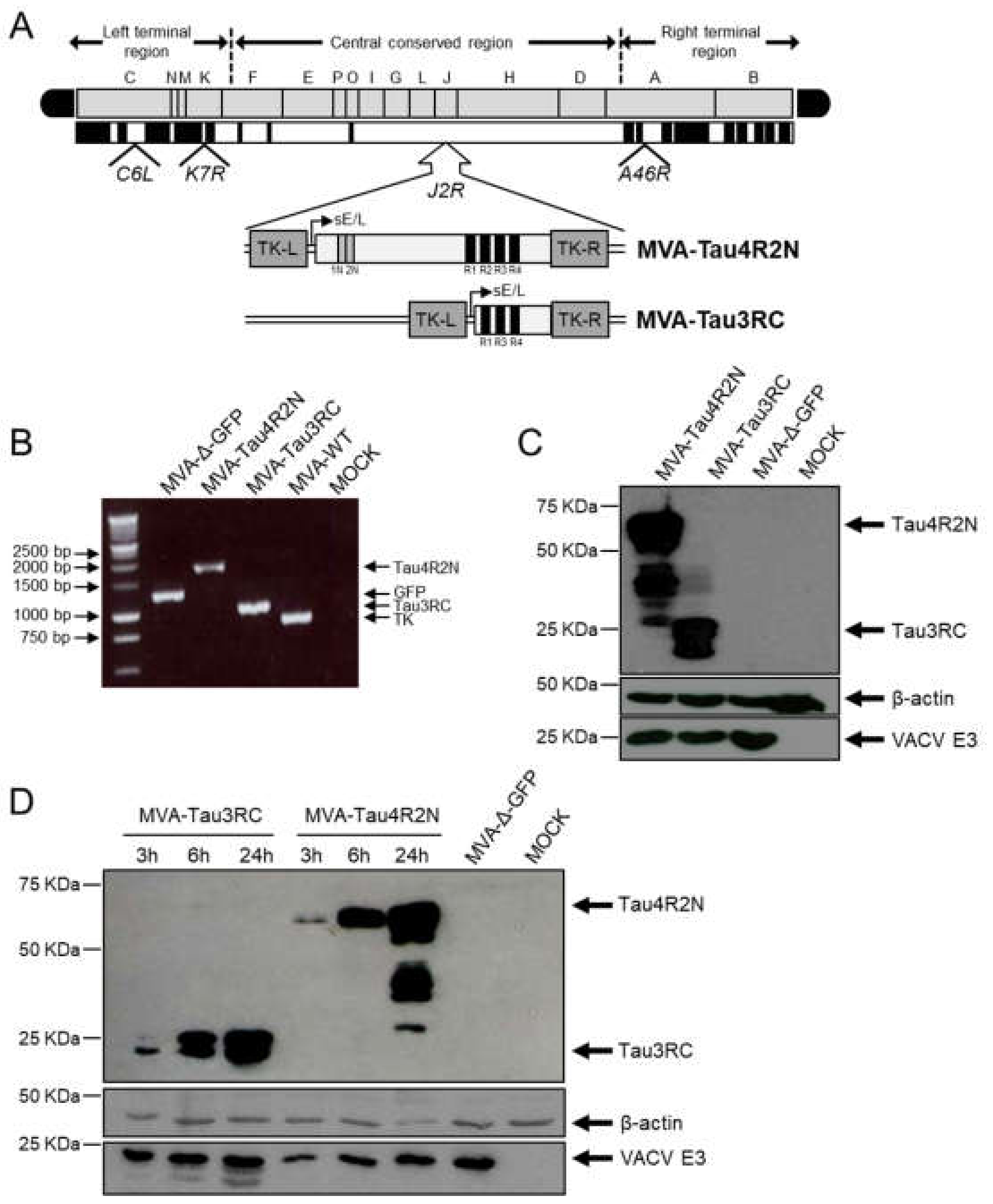
3.1. Generation and In Vitro Characterization of MVA-Tau4R2N and MVA-Tau3RC
3.2. The Human Tau4R2N and Tau3RC Proteins Expressed by MVA-Tau Vaccine Candidates are Located in the Cytoplasm and Co-Localize with Tubulin
3.3. MVA-Tau Vaccine Candidates Impact on the Recruitment of Immune Cells to the Peritoneal Cavity of Infected Mice
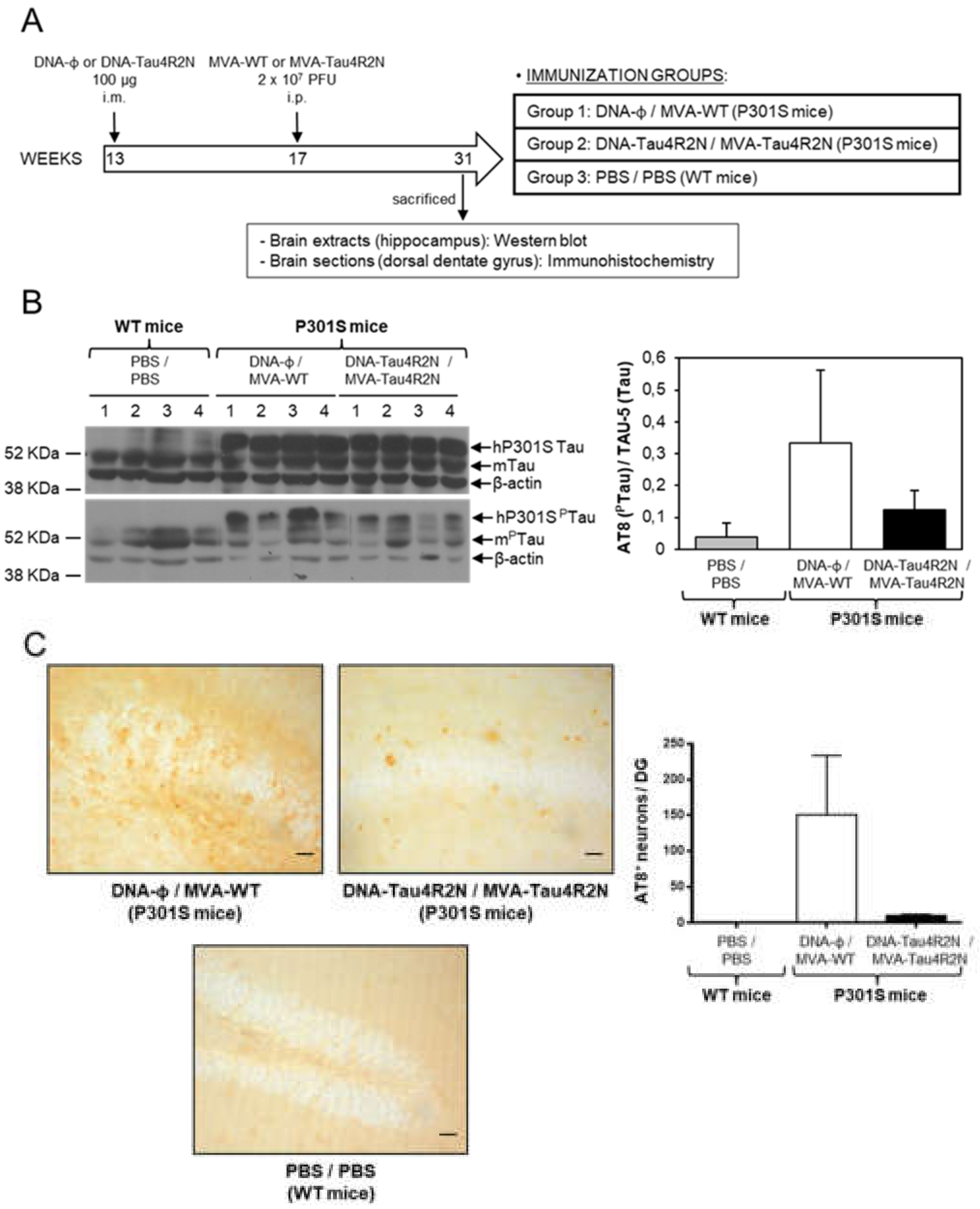
3.4. MVA-Tau4R2N does not Reduce Significantly Hyperphosphorylated Tau in the Brains of Vaccinated Transgenic P301S Mice
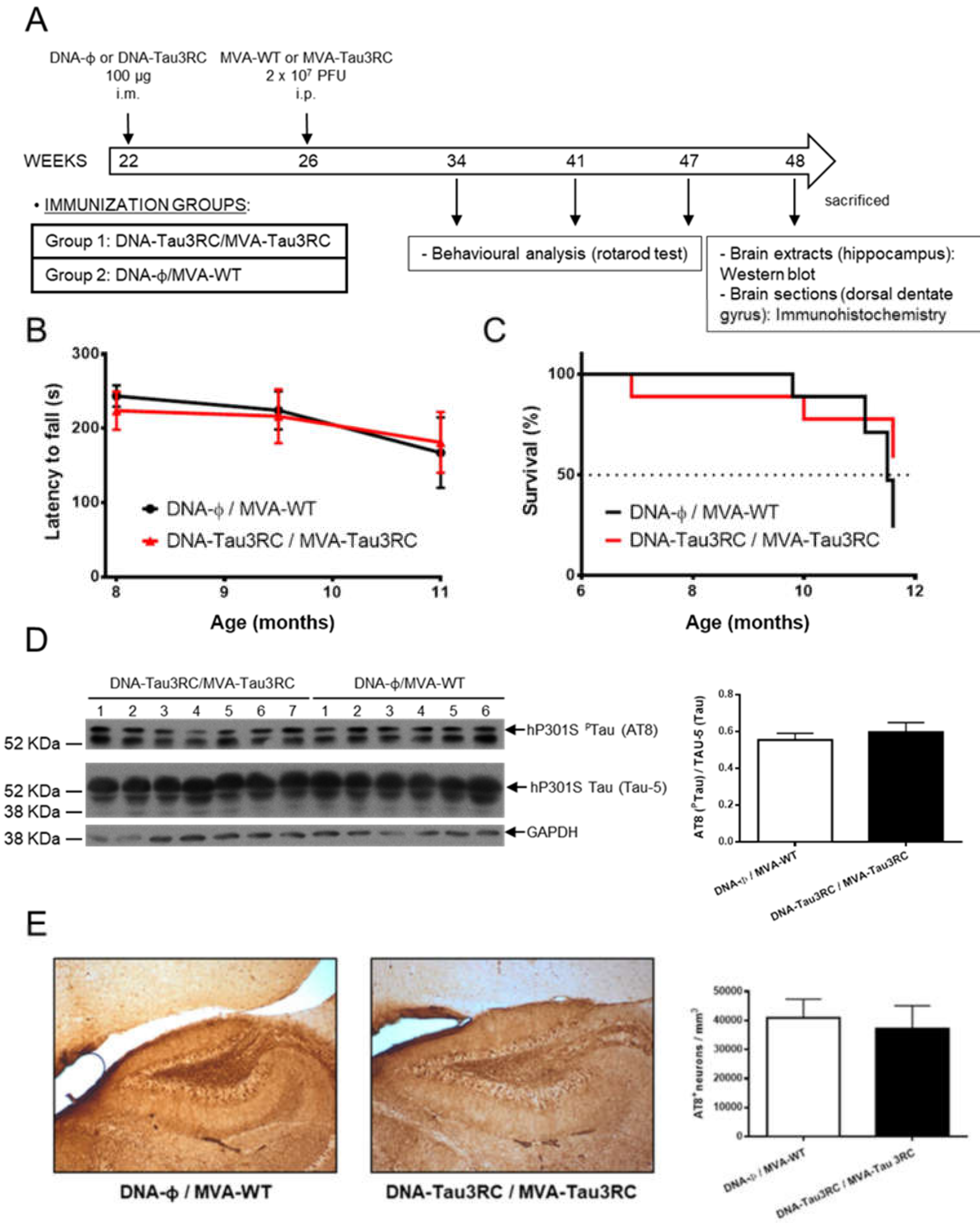
3.5. MVA-Tau3RC does not Control Motor Failure and Mortality of Vaccinated Transgenic P301S Mice, Neither Decrease the Levels of Hyperphosphorylated Tau
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Yankner, B.A. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron 1996, 16, 921–932. [Google Scholar] [CrossRef]
- Alzheimer, A. Über eigenartige Krankheitsfälle des späteren Alters. Gesanipte Neurol. Psychiatr. 1911, 4, 356–385. [Google Scholar] [CrossRef]
- Medina, M.; Avila, J. New perspectives on the role of tau in Alzheimer’s disease. Implications for therapy. Biochem. Pharmacol. 2014, 88, 540–547. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Drummond, E. Developing therapeutic vaccines against Alzheimer’s disease. Expert Rev. Vaccines 2016, 15, 401–415. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Sigurdsson, E.M.; Knudsen, E.; Asuni, A.; Fitzer-Attas, C.; Sage, D.; Quartermain, D.; Goni, F.; Frangione, B.; Wisniewski, T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-beta derivatives. J. Neurosci. 2004, 24, 6277–6282. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef]
- Lemere, C.A.; Masliah, E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat. Rev. Neurol 2010, 6, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Vaccination to Alzheimer Disease. Is it a Promising Tool or a Blind Way? Curr. Med. Chem. 2016, 23, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley-Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T.; Bouras, C.; Kovari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.; Miller, C.A.; Poon, W.W.; Vinters, H.V.; Corrada, M.; Kawas, C.; Hayden, E.Y.; Teplow, D.B.; Glabe, C.; Albay, R., 3rd.; et al. Synaptic Amyloid-beta Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. Am. J. Pathol. 2016, 186, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ramos, A.; Diaz-Hernandez, M.; Cuadros, R.; Hernandez, F.; Avila, J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006, 580, 4842–4850. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.T.; Sigurdsson, E.M. Tau immunotherapy for Alzheimer’s disease. Trends Mol. Med. 2015, 21, 394–402. [Google Scholar] [CrossRef]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; Lopez-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef]
- Novak, P.; Zilka, N.; Zilkova, M.; Kovacech, B.; Skrabana, R.; Ondrus, M.; Fialova, L.; Kontsekova, E.; Otto, M.; Novak, M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimers Dis 2019, 6, 63–69. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 2018, 4, 195–214. [Google Scholar] [CrossRef]
- Asuni, A.A.; Boutajangout, A.; Quartermain, D.; Sigurdsson, E.M. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007, 27, 9115–9129. [Google Scholar] [CrossRef]
- Maphis, N.M.; Peabody, J.; Crossey, E.; Jiang, S.; Jamaleddin Ahmad, F.A.; Alvarez, M.; Mansoor, S.K.; Yaney, A.; Yang, Y.; Sillerud, L.O.; et al. Qss Virus-like particle-based vaccine induces robust immunity and protects against tauopathy. NPJ Vaccines 2019, 4, 26. [Google Scholar] [CrossRef]
- Congdon, E.E.; Gu, J.; Sait, H.B.; Sigurdsson, E.M. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcgamma receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 2013, 288, 35452–35465. [Google Scholar] [CrossRef]
- Kudrna, J.J.; Ugen, K.E. Gene-based vaccines and immunotherapeutic strategies against neurodegenerative diseases: Potential utility and limitations. Hum. Vaccin Immunother 2015, 11, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.E.; Perdiguero, B.; Garcia-Arriaza, J.; Esteban, M. Clinical applications of attenuated MVA poxvirus strain. Expert Rev. Vaccines 2013, 12, 1395–1416. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Protective efficacy of Modified Vaccinia virus Ankara in preclinical studies. Vaccine 2013, 31, 4235–4240. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.C.; Gherardi, M.M.; Esteban, M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: Virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Cepeda, V.; Hallengard, D.; Sorzano, C.O.; Kummerer, B.M.; Liljestrom, P.; Esteban, M. A novel poxvirus-based vaccine, MVA-CHIKV, is highly immunogenic and protects mice against chikungunya infection. J. Virol. 2014, 88, 3527–3547. [Google Scholar] [CrossRef] [PubMed]
- Lazaro-Frias, A.; Gomez-Medina, S.; Sanchez-Sampedro, L.; Ljungberg, K.; Ustav, M.; Liljestrom, P.; Munoz-Fontela, C.; Esteban, M.; Garcia-Arriaza, J. Distinct Immunogenicity and Efficacy of Poxvirus-Based Vaccine Candidates against Ebola Virus Expressing GP and VP40 Proteins. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Perez, P.; Marín, Q.M.; Lazaro-Frias, A.; Jimenez de Oya, N.; Blazquez, A.B.; Escribano-Romero, E.; Sorzano, C.Ó.S.; Ortego, J.; Saiz, J.C.; Esteban, M.; et al. A Vaccine Based on a Modified Vaccinia Virus Ankara Vector Expressing Zika Virus Structural Proteins Controls Zika Virus Replication in Mice. Sci. Rep. 2018, 8, 17385. [Google Scholar] [CrossRef]
- Perez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of tau into filaments in the presence of heparin: The minimal sequence required for tau-tau interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef]
- Montejo de Garcini, E.; de la Luna, S.; Dominguez, J.E.; Avila, J. Overexpression of tau protein in COS-1 cells results in the stabilization of centrosome-independent microtubules and extension of cytoplasmic processes. Mol. Cell Biochem. 1994, 130, 187–196. [Google Scholar] [CrossRef]
- Medina, M.; Montejo de Garcini, E.; Avila, J. The role of tau phosphorylation in transfected COS-1 cells. Mol. Cell Biochem. 1995, 148, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.E.; Najera, J.L.; Jimenez, E.P.; Jimenez, V.; Wagner, R.; Graf, M.; Frachette, M.J.; Liljestrom, P.; Pantaleo, G.; Esteban, M. Head-to-head comparison on the immunogenicity of two HIV/AIDS vaccine candidates based on the attenuated poxvirus strains MVA and NYVAC co-expressing in a single locus the HIV-1BX08 gp120 and HIV-1(IIIB) Gag-Pol-Nef proteins of clade B. Vaccine 2007, 25, 2863–2885. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Arnaez, P.; Gomez, C.E.; Sorzano, C.O.; Esteban, M. Improving Adaptive and Memory Immune Responses of an HIV/AIDS Vaccine Candidate MVA-B by Deletion of Vaccinia Virus Genes (C6L and K7R) Blocking Interferon Signaling Pathways. PLoS ONE 2013, 8, e66894. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Najera, J.L.; Gomez, C.E.; Tewabe, N.; Sorzano, C.O.; Calandra, T.; Roger, T.; Esteban, M. A candidate HIV/AIDS vaccine (MVA-B) lacking vaccinia virus gene C6L enhances memory HIV-1-specific T-cell responses. PLoS ONE 2011, 6, e24244. [Google Scholar] [CrossRef]
- Gomez, C.E.; Perdiguero, B.; Cepeda, M.V.; Mingorance, L.; Garcia-Arriaza, J.; Vandermeeren, A.; Sorzano, C.O.; Esteban, M. High, broad, polyfunctional, and durable T cell immune responses induced in mice by a novel hepatitis C virus (HCV) vaccine candidate (MVA-HCV) based on modified vaccinia virus Ankara expressing the nearly full-length HCV genome. J. Virol. 2013, 87, 7282–7300. [Google Scholar] [CrossRef]
- Di Pilato, M.; Mejias-Perez, E.; Zonca, M.; Perdiguero, B.; Gomez, C.E.; Trakala, M.; Nieto, J.; Najera, J.L.; Sorzano, C.O.; Combadiere, C.; et al. NFkappaB activation by modified vaccinia virus as a novel strategy to enhance neutrophil migration and HIV-specific T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, E1333–E1342. [Google Scholar] [CrossRef]
- Marin, M.Q.; Perez, P.; Gomez, C.E.; Sorzano, C.O.S.; Esteban, M.; Garcia-Arriaza, J. Removal of the C6 Vaccinia Virus Interferon-beta Inhibitor in the Hepatitis C Vaccine Candidate MVA-HCV Elicited in Mice High Immunogenicity in Spite of Reduced Host Gene Expression. Viruses 2018, 10, 414. [Google Scholar] [CrossRef]
- Fernandez-Nogales, M.; Santos-Galindo, M.; Merchan-Rubira, J.; Hoozemans, J.J.M.; Rabano, A.; Ferrer, I.; Avila, J.; Hernandez, F.; Lucas, J.J. Tau-positive nuclear indentations in P301S tauopathy mice. Brain Pathol. 2017, 27, 314–322. [Google Scholar] [CrossRef]
- Engel, T.; Goni-Oliver, P.; Lucas, J.J.; Avila, J.; Hernandez, F. Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 2006, 99, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arriaza, J.; Esteban, M. Enhancing poxvirus vectors vaccine immunogenicity. Hum. Vaccin. Immunother. 2014, 10, 2235–2244. [Google Scholar] [CrossRef]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buee, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.L.; Tung, Y.C.; Liu, F.; Gong, C.X.; Iqbal, K. Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res. Ther. 2017, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M. Tau Immunotherapy. Neurodegener. Dis. 2016, 16, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Scholtzova, H.; Chianchiano, P.; Pan, J.; Sun, Y.; Goni, F.; Mehta, P.D.; Wisniewski, T. Amyloid beta and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol. Commun. 2014, 2, 101. [Google Scholar] [CrossRef]
- Scholtzova, H.; Kascsak, R.J.; Bates, K.A.; Boutajangout, A.; Kerr, D.J.; Meeker, H.C.; Mehta, P.D.; Spinner, D.S.; Wisniewski, T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J. Neurosci. 2009, 29, 1846–1854. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, X.; Pascual, G.; Wadia, J.S.; Keogh, E.; Hoozemans, J.J.; Siregar, B.; Inganas, H.; Stoop, E.J.M.; Goudsmit, J.; et al. Structural Basis for Recognition of a Unique Epitope by a Human Anti-tau Antibody. Structure 2018, 26, 1626–1634. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Arriaza, J.; Marín, M.Q.; Merchán-Rubira, J.; Mascaraque, S.M.; Medina, M.; Ávila, J.; Hernández, F.; Esteban, M. Tauopathy Analysis in P301S Mouse Model of Alzheimer Disease Immunized with DNA and MVA Poxvirus-Based Vaccines Expressing Human Full-Length 4R2N or 3RC Tau Proteins. Vaccines 2020, 8, 127. https://doi.org/10.3390/vaccines8010127
García-Arriaza J, Marín MQ, Merchán-Rubira J, Mascaraque SM, Medina M, Ávila J, Hernández F, Esteban M. Tauopathy Analysis in P301S Mouse Model of Alzheimer Disease Immunized with DNA and MVA Poxvirus-Based Vaccines Expressing Human Full-Length 4R2N or 3RC Tau Proteins. Vaccines. 2020; 8(1):127. https://doi.org/10.3390/vaccines8010127
Chicago/Turabian StyleGarcía-Arriaza, Juan, María Q. Marín, Jesús Merchán-Rubira, Sara M. Mascaraque, Miguel Medina, Jesús Ávila, Félix Hernández, and Mariano Esteban. 2020. "Tauopathy Analysis in P301S Mouse Model of Alzheimer Disease Immunized with DNA and MVA Poxvirus-Based Vaccines Expressing Human Full-Length 4R2N or 3RC Tau Proteins" Vaccines 8, no. 1: 127. https://doi.org/10.3390/vaccines8010127
APA StyleGarcía-Arriaza, J., Marín, M. Q., Merchán-Rubira, J., Mascaraque, S. M., Medina, M., Ávila, J., Hernández, F., & Esteban, M. (2020). Tauopathy Analysis in P301S Mouse Model of Alzheimer Disease Immunized with DNA and MVA Poxvirus-Based Vaccines Expressing Human Full-Length 4R2N or 3RC Tau Proteins. Vaccines, 8(1), 127. https://doi.org/10.3390/vaccines8010127