Autophagy Promotes Duck Tembusu Virus Replication by Suppressing p62/SQSTM1-Mediated Innate Immune Responses In Vitro

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Duck Embryo Fibroblast (DEF) Primary Cells
2.3. Viral Infection
2.4. Plasmids and Small Interfering RNA (siRNA)
2.5. Transmission Electron Microscopy (TEM)
2.6. Fluorescence Microscopy
2.7. Western Blotting
2.8. Pharmaceutical Treatment
2.9. Quantitative RT-PCR Assay
2.10. Luciferase Reporter Assay
2.11. Median Tissue Culture Infectious Doses (TCID50) Assay
2.12. Statistical Analysis
3. Results
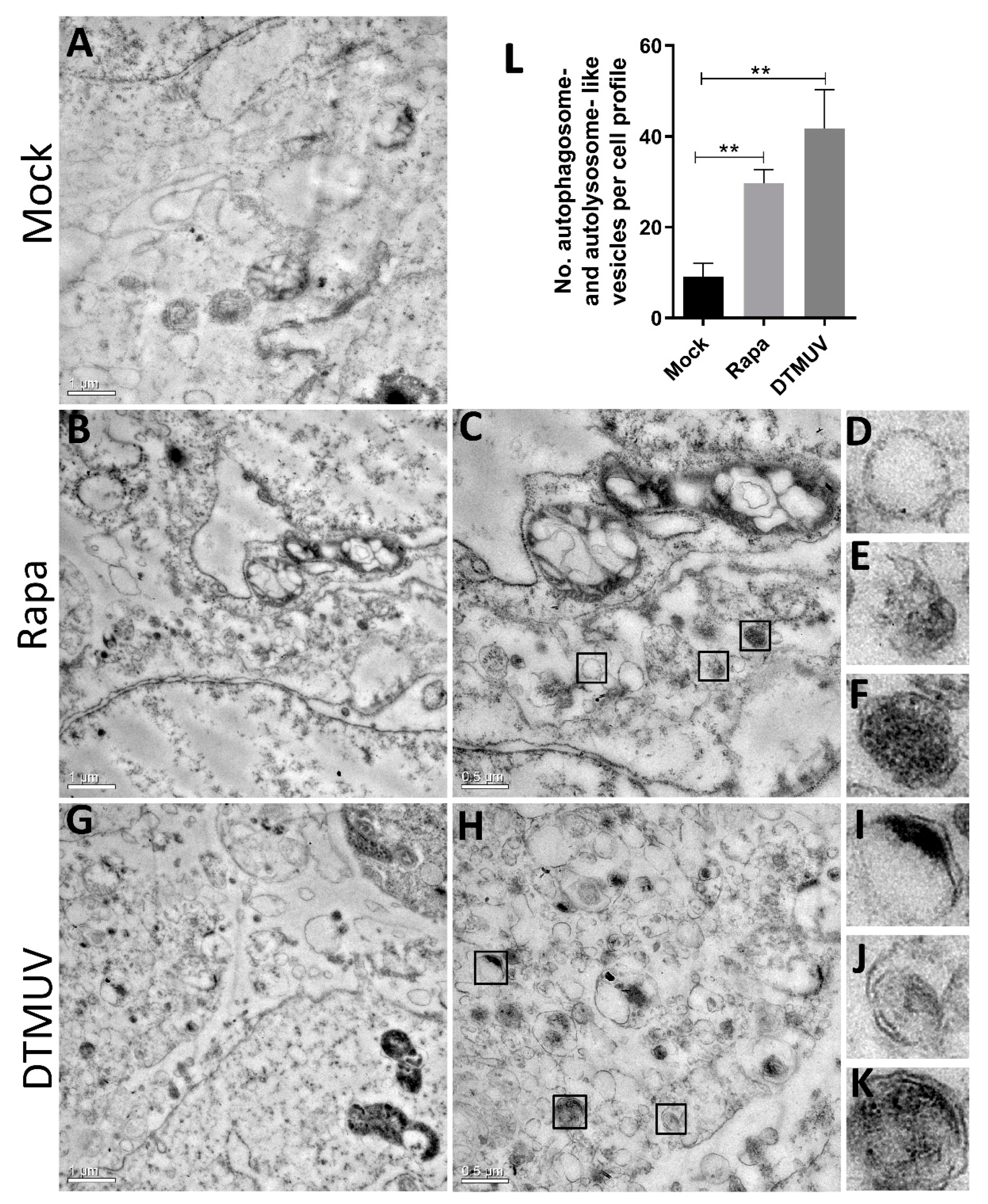
3.1. DTMUV Infection Induces the Formation of Autophagosome-Like Vesicles in DEF Cells
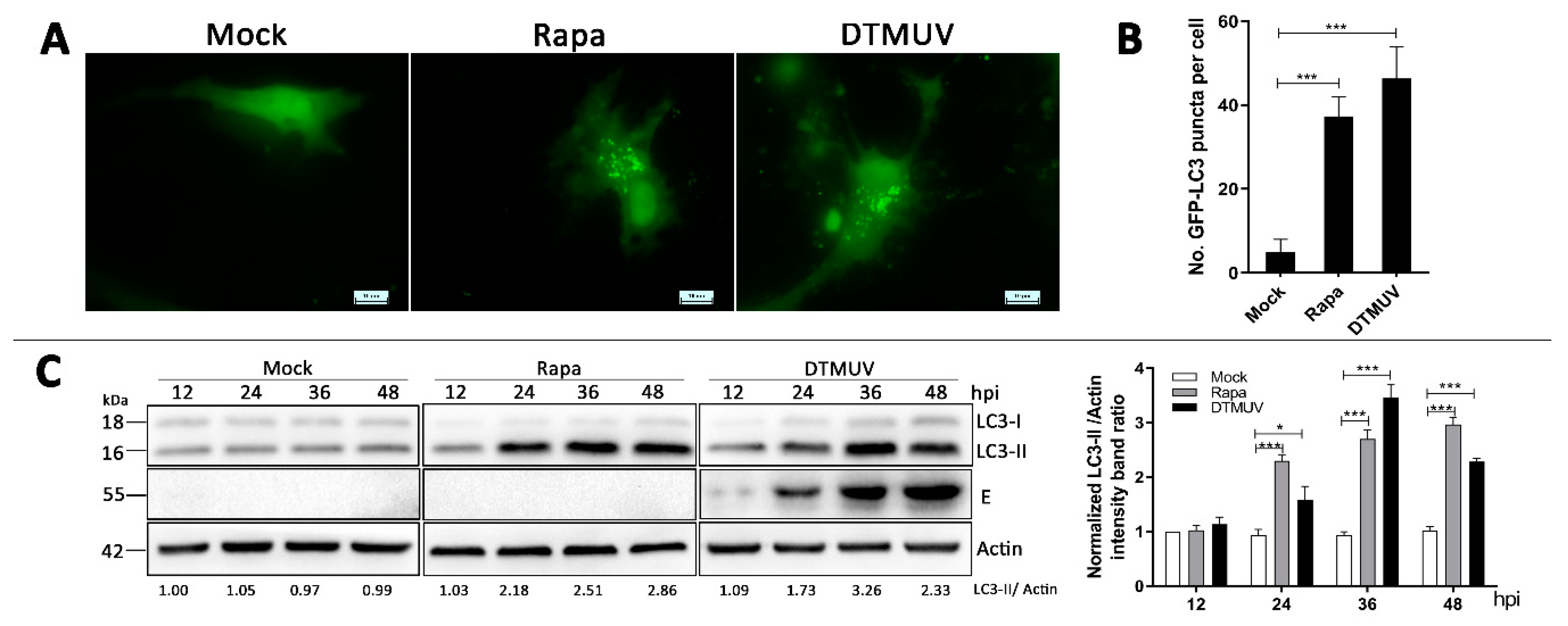
3.2. DTMUV Infection Increases the Levels of Autophagic Markers in DEF Cells
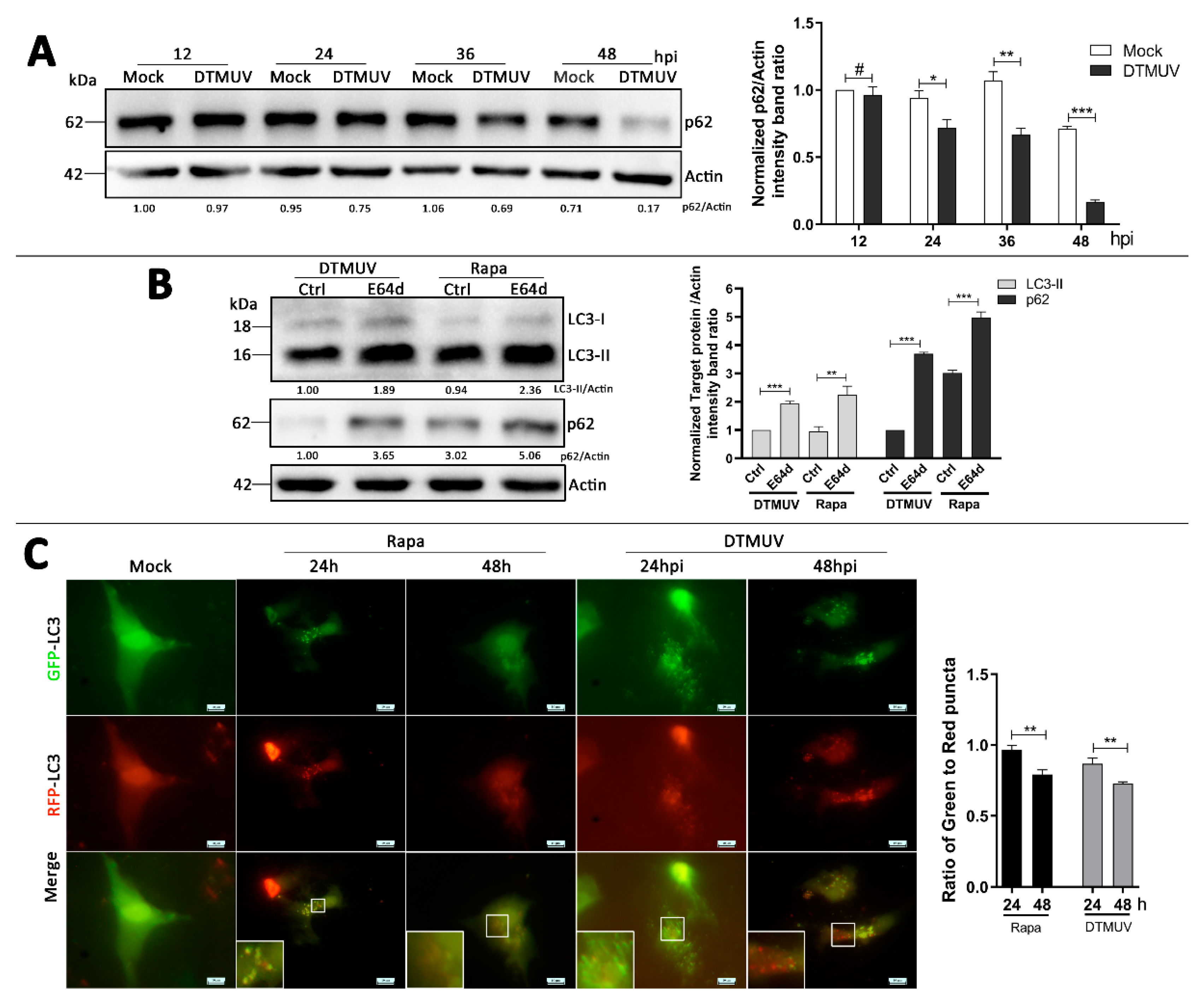
3.3. DTMUV Infection Enhances Autophagic Flux in DEF Cells
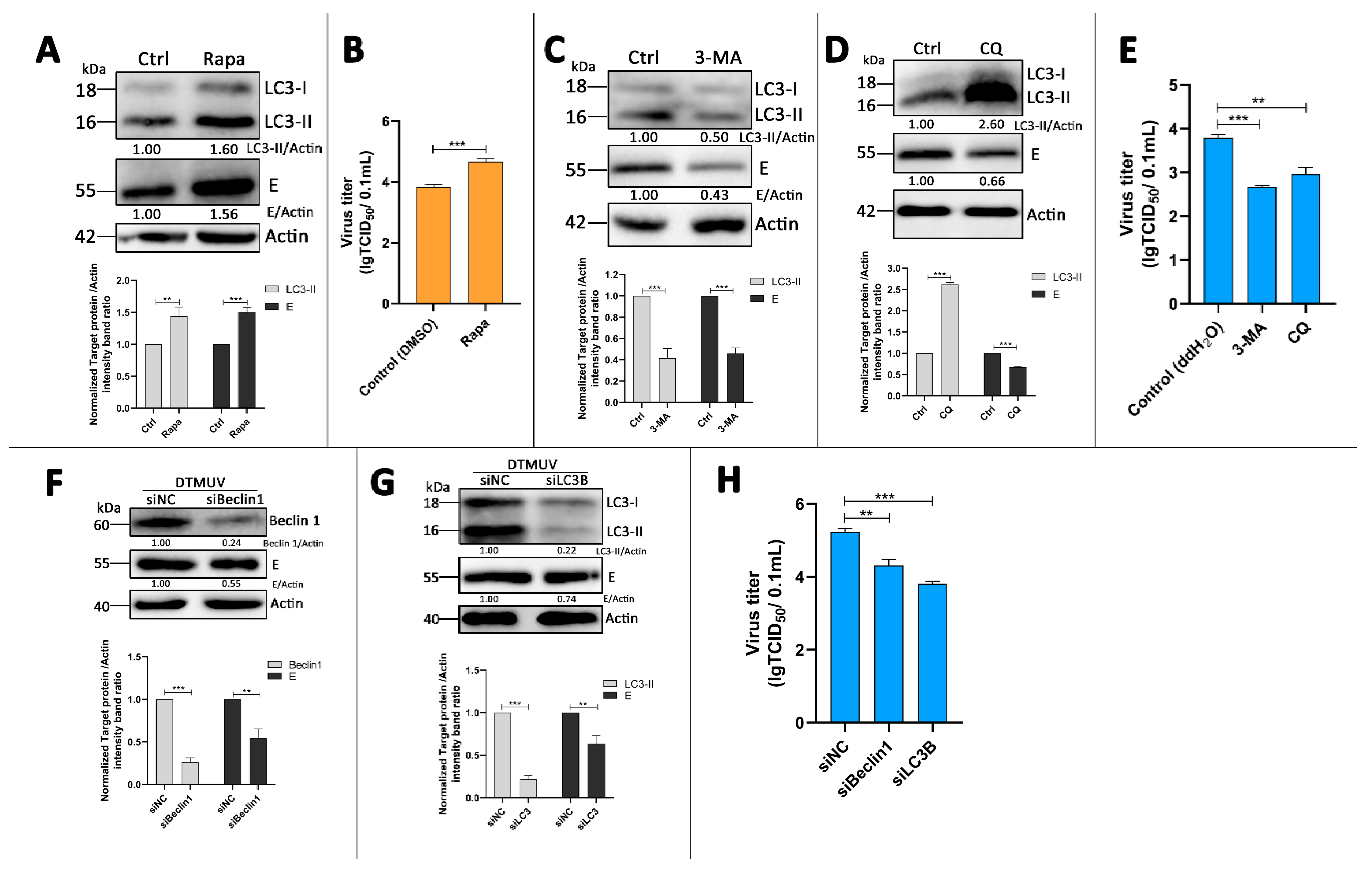
3.4. Autophagy-Altering Treatments Affect the Replication of DTMUV in DEF Cells
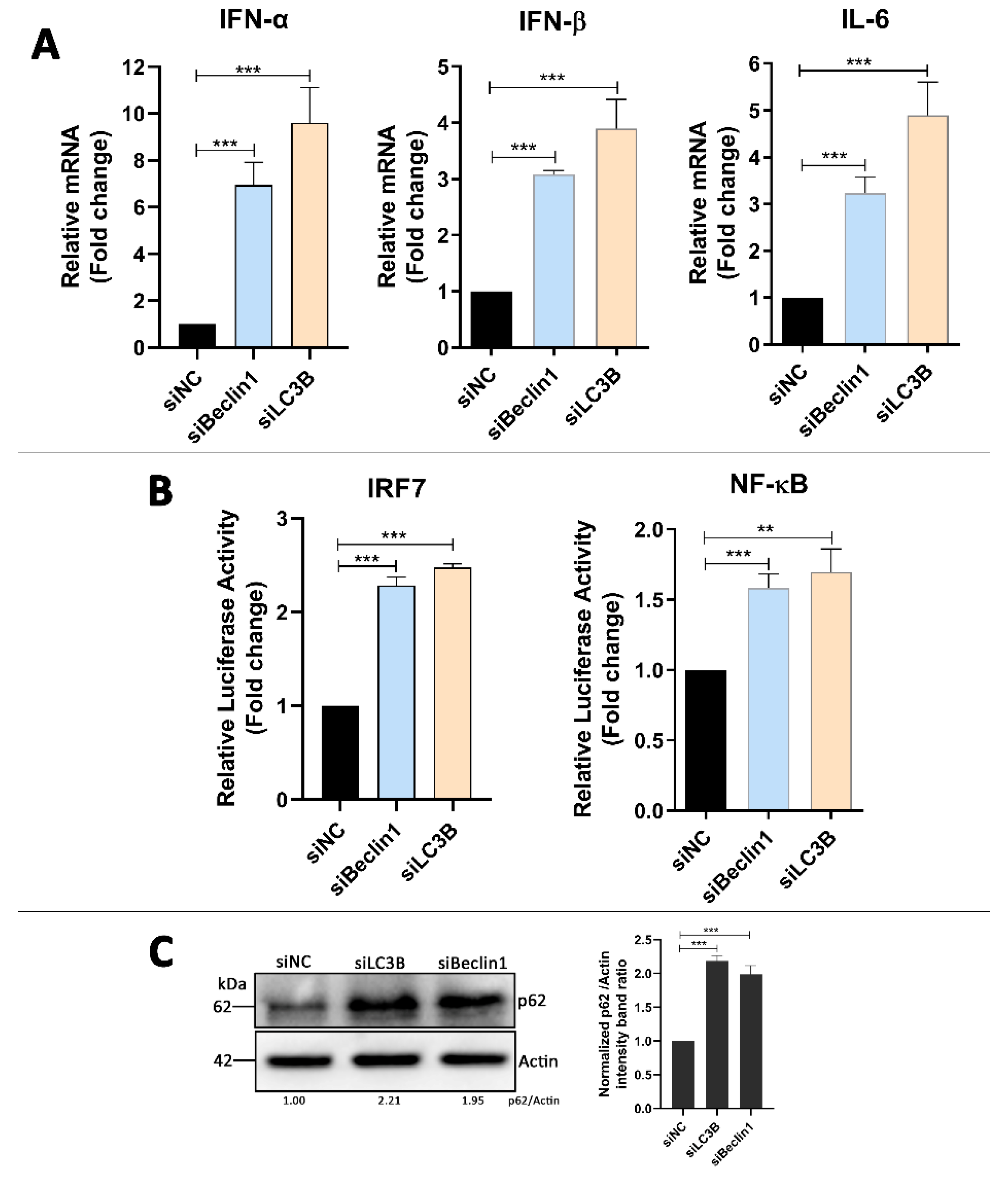
3.5. Absence of Autophagy Enhances Type I Interferons and IL-6 Production in DTMUV-Infected Cells
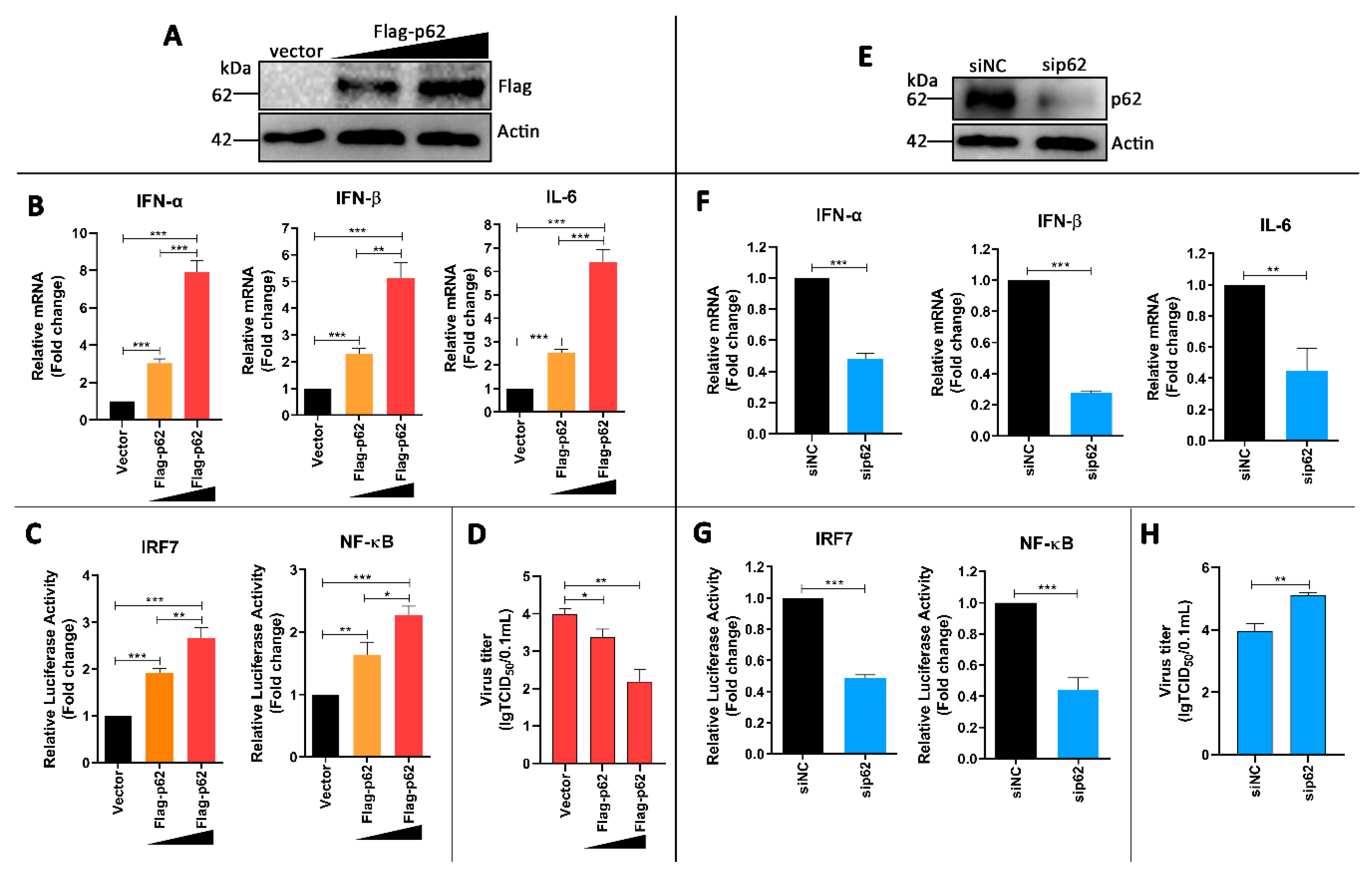
3.6. p62 Regulates IRF7 and NF-kB Pathways and DTMUV Replication in DTMUV-Regulated Cells
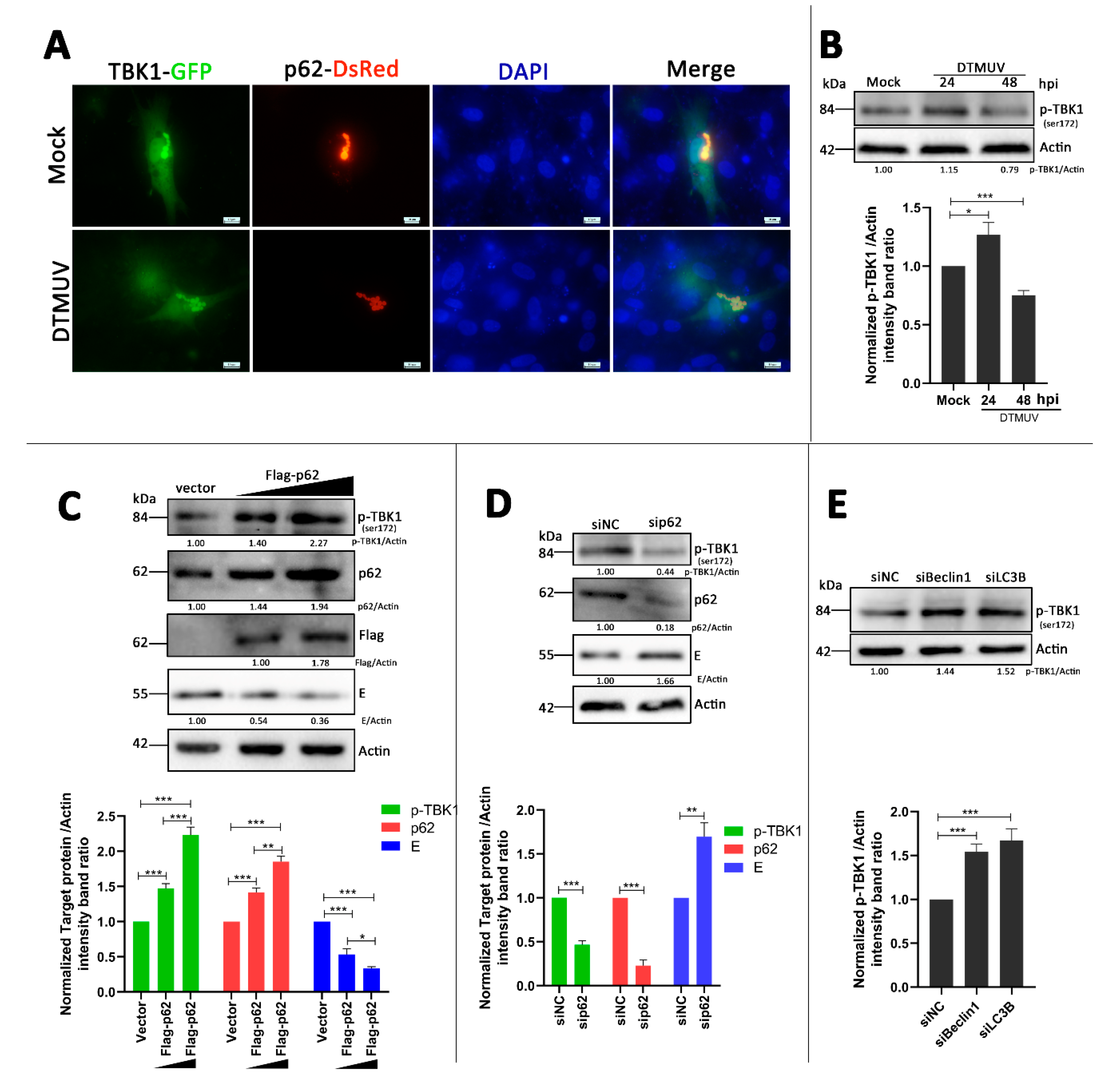
3.7. p62 Regulates the Phosphorylation of TBK1 (p-TBK1) in DTMUV-Infected Cells
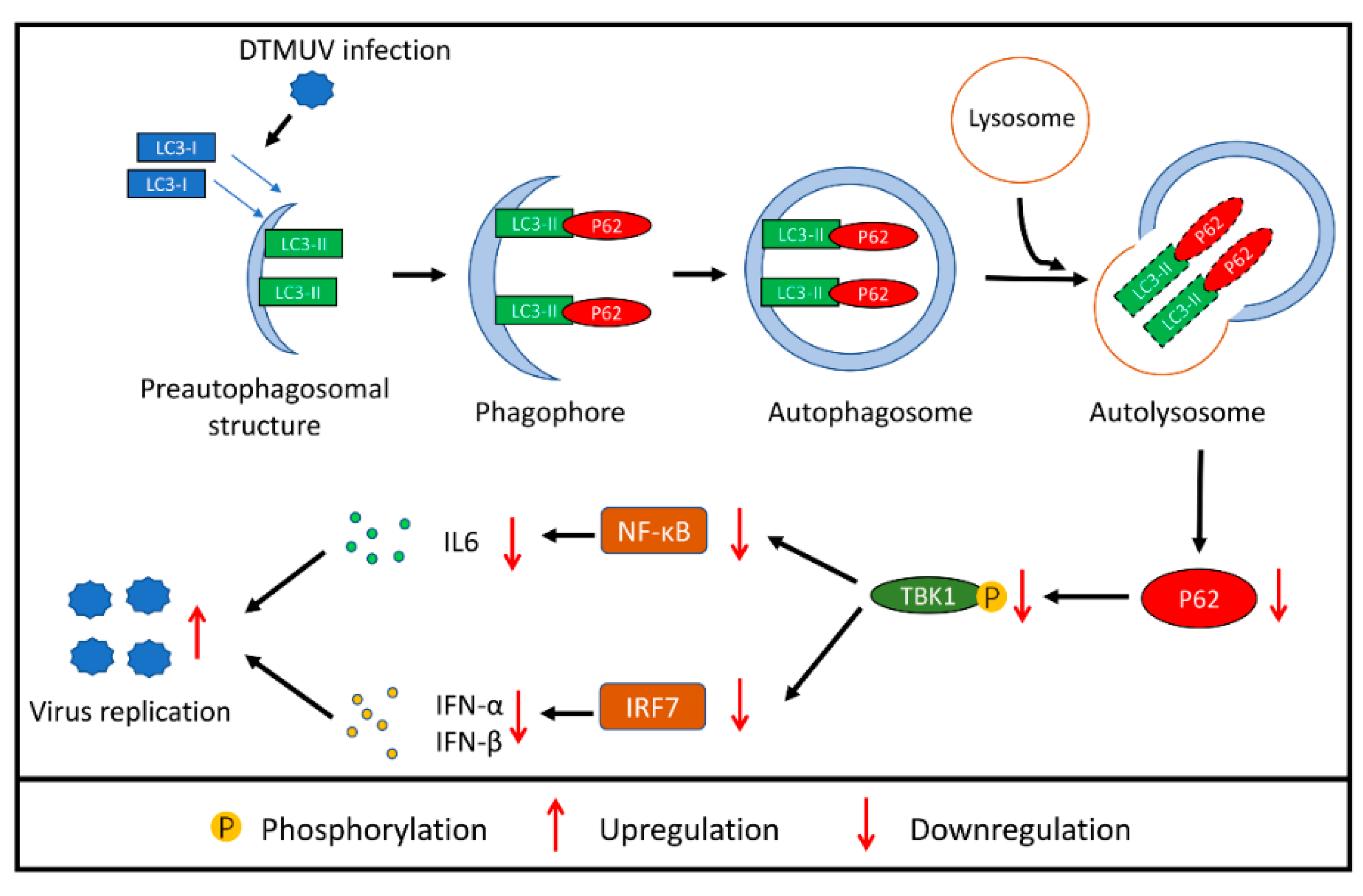
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, P.; Zhao, Y.; Zhang, X.; Xu, D.; Dai, X.; Teng, Q.; Yan, L.; Zhou, J.; Ji, X.; Zhang, S. An infectious disease of ducks caused by a newly emerged Tembusu virus strain in mainland China. Virology 2011, 417, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, S.; Hu, X.; Yu, X.; Wang, Y.; Liu, P.; Lu, X.; Zhang, G.; Hu, X.; Liu, D. Duck egg-drop syndrome caused by BYD virus, a new Tembusu-related flavivirus. PLoS ONE 2011, 6, e18106. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Zhang, C.; Liu, Y.; Ye, W.; Han, J.; Ma, G.; Zhang, D.; Xu, F.; Gao, X.; Tang, Y. Tembusu virus in ducks, China. Emerg. Infect. Dis. 2011, 17, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lv, C.; Yue, R.; Shi, Y.; Wei, L.; Chai, T.; Liu, S. Effect of age on the pathogenesis of duck tembusu virus in Cherry Valley ducks. Front. Microbiol. 2015, 6, 581. [Google Scholar] [CrossRef]
- Ti, J.; Zhang, M.; Li, Z.; Li, X.; Diao, Y. Duck Tembusu virus exhibits pathogenicity to Kunming mice by intracerebral inoculation. Front. Microbiol. 2016, 7, 190. [Google Scholar] [CrossRef]
- Doughty, C.T.; Yawetz, S.; Lyons, J. Emerging causes of arbovirus encephalitis in North America: Powassan, Chikungunya, and Zika viruses. Curr. Neurol. Neurosci. Rep. 2017, 17, 12. [Google Scholar] [CrossRef]
- Peng, B.H.; Wang, T. West Nile Virus Induced Cell Death in the Central Nervous System. Pathogens 2019, 8, 215. [Google Scholar] [CrossRef]
- Yun, S.-I.; Lee, Y.-M. Japanese encephalitis: The virus and vaccines. Hum. Vaccines Immunother. 2014, 10, 263–279. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Zhang, Q.; Sun, M.; Li, S.; Su, W.; Hu, X.; He, W.; Su, J. Efficacy assessment of an inactivated Tembusu virus vaccine candidate in ducks. Res. Vet. Sci. 2017, 110, 72–78. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Fan, X.; Han, S.; Yan, D.; Gao, Y.; Wei, Y.; Liu, X.; Liao, Y.; Guo, H.; Sun, S. Foot-and-mouth disease virus infection suppresses autophagy and NF-κ B antiviral responses via degradation of ATG5-ATG12 by 3C pro. Cell Death Dis. 2018, 8, e2561. [Google Scholar] [CrossRef]
- Pyo, J.O.; Nah, J.; Jung, Y.K. Molecules and their functions in autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jin, S.; Wang, R.-F. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy 2016, 12, 1210–1211. [Google Scholar] [CrossRef]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R.F. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef]
- Prabakaran, T.; Bodda, C.; Krapp, C.; Zhang, B.C.; Christensen, M.H.; Sun, C.; Reinert, L.; Cai, Y.; Jensen, S.B.; Skouboe, M.K. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018, 37, e97858. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM 1 functions as a signaling hub and an autophagy adaptor. EMBO J. 2015, 282, 4672–4678. [Google Scholar]
- Deng, J.; Liu, Y.; Jia, R.; Wang, M.; Chen, S.; Zhu, D.; Liu, M.; Sun, K.; Zhao, X.; Yin, Z. Development of an immunochromatographic strip for detection of antibodies against duck Tembusu virus. J. Virol. Methods 2017, 249, 137–142. [Google Scholar] [CrossRef]
- Vaheri, A.; Ruoslahti, E.; Hovi, T.; Nordling, S. Stimulation of density-inhibited cell cultures by insulin. J. Cell. Physiol. 1973, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Ruoslahti, E.; Nordling, S. Neuraminidase stimulates Division and Sugar Uptake in Density-inhibited Cell Cultures. Nat. New Biol. 1972, 238, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Huang, J.; Jia, R.; Zhang, B.; Wang, M.; Zhu, D.; Chen, S.; Liu, M.; Yin, Z.; Cheng, A. Identification and molecular characterization of a novel duck Tembusu virus isolate from Southwest China. Arch. Virol. 2015, 160, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, S.; Ding, N.; Meng, C.; Meng, S.; Zhang, S.; Zhan, Y.; Qiu, X.; Tan, L.; Chen, H. Autophagy benefits the replication of Newcastle disease virus in chicken cells and tissues. J. Virol. 2014, 88, 525–537. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Perry, A.K.; Chow, E.K.; Goodnough, J.B.; Yeh, W.-C.; Cheng, G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 2004, 199, 1651–1658. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lei, C.-Q.; Ji, Y.; Zhou, H.; Ren, Y.; Peng, Q.; Zeng, Y.; Jia, Y.; Ge, J.; Zhong, B. Duck Tembusu Virus Nonstructural Protein 1 Antagonizes IFN-β Signaling Pathways by Targeting VISA. J. Immunol. 2016, 197, 4704–4713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jia, R.; Pan, Y.; Wang, M.; Chen, S.; Zhu, D.; Liu, M.; Zhao, X.; Yang, Q.; Wu, Y. Therapeutic effects of duck Tembusu virus capsid protein fused with staphylococcal nuclease protein to target Tembusu infection in vitro. Vet. Microbiol. 2019, 235, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.-H.J. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar]
- Metz, P.; Chiramel, A.; Chatel-Chaix, L.; Alvisi, G.; Bankhead, P.; Mora-Rodríguez, R.; Long, G.; Hamacher-Brady, A.; Brady, N.R.; Bartenschlager, R. Dengue virus inhibition of autophagic flux and dependency of viral replication on proteasomal degradation of the autophagy receptor p62. J. Virol. 2015, 89, 8026–8041. [Google Scholar] [CrossRef]
- Pei, J.; Zhao, M.; Ye, Z.; Gou, H.; Wang, J.; Yi, L.; Dong, X.; Liu, W.; Luo, Y.; Liao, M. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy 2014, 10, 93–110. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.l.; Choi, J.; Wakita, T.; Yen, T.B.; Ou, J.h.J. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ou, J.-H.J. Regulation of autophagy by hepatitis C virus for its replication. DNA Cell Biol. 2018, 37, 287–290. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F. Biology of Zika virus infection in human skin cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I-and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS ONE 2012, 7, e45800. [Google Scholar] [CrossRef]
- Ke, P.-Y. The multifaceted roles of autophagy in flavivirus-host interactions. Int. J. Mol. Sci. 2018, 19, 3940. [Google Scholar] [CrossRef]
- Magor, K.E.; Navarro, D.M.; Barber, M.R.; Petkau, K.; Fleming-Canepa, X.; Blyth, G.A.; Blaine, A.H. Defense genes missing from the flight division. Dev. Comp. Immunol. 2013, 41, 377–388. [Google Scholar] [CrossRef]
- Chen, S.; Wang, T.; Liu, P.; Yang, C.; Wang, M.; Jia, R.; Zhu, D.; Liu, M.; Yang, Q.; Wu, Y. Duck interferon regulatory factor 7 (IRF7) can control duck Tembusu virus (DTMUV) infection by triggering type I interferon production and its signal transduction pathway. Cytokine 2019, 113, 31–38. [Google Scholar] [CrossRef]
- Ke, P.-Y.; Chen, S.S.-L. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Yang, S.; Jin, S.; Zhang, Y.; Cui, J. LRRC59 modulates type I interferon signaling by restraining the SQSTM1/p62-mediated autophagic degradation of pattern recognition receptor DDX58/RIG-I. Autophagy 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.-F. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, G.; Zuo, J. Caffeic acid inhibits HCV replication via induction of IFNα antiviral response through p62-mediated Keap1/Nrf2 signaling pathway. Antivir. Res. 2018, 154, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Min, Y.; Kwon, J.; Son, J.; Im, J.S.; Shin, J.; Lee, K.-Y. p62 Negatively Regulates TLR4 Signaling via Functional Regulation of the TRAF6-ECSIT Complex. Immune Netw. 2019, 19, e16. [Google Scholar] [CrossRef] [PubMed]
- Alegre, F.; Moragrega, Á.B.; Polo, M.; Marti-Rodrigo, A.; Esplugues, J.V.; Blas-Garcia, A.; Apostolova, N. Role of p62/SQSTM1 beyond autophagy: A lesson learned from drug-induced toxicity in vitro. Br. J. Pharmacol. 2018, 175, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Waisner, H.; Kalamvoki, M. The ICP0 Protein of Herpes Simplex Virus 1 (HSV-1) Downregulates Major Autophagy Adaptor Proteins Sequestosome 1 and Optineurin during the Early Stages of HSV-1 Infection. J. Virol. 2019, 93, e01258–e01319. [Google Scholar] [CrossRef]
- Zhao, W. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. 2013, 587, 542–548. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Ma, X.; Helgason, E.; Phung, Q.T.; Quan, C.L.; Iyer, R.S.; Lee, M.W.; Bowman, K.K.; Starovasnik, M.A.; Dueber, E.C. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. USA 2012, 109, 9378–9383. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prime Name | Prime Sequence (5′–3′) | Purpose |
|---|---|---|
| Duck-Flag-p62-F | CCGGAATTCATGGATTACAAGGATGACGACGATAAGGCGTTCTCCAGTGA | Gene cloning |
| Duck-Flag-p62-R | CCGCTCGAGAATACATGTGAGGAGGCTG | |
| Duck-DsRed-p62-F | CCGGAATTCATGGCGTTCTCCAGTGACG | Gene cloning |
| Duck-DsRed-p62-R | CGGGGTACCGACATGTGAGGAGGCTG | |
| Duck-siLC3B-F | GGAGCGCAACCUUCCGUUUTT | Gene Knockdown |
| Duck-siLC3B-R | AAACGGAAGGUUGCGCUCCTT | |
| Duck-siBeclin1-F | GCUCAGUACCAGAAGGAAUTT | Gene Knockdown |
| Duck-siBeclin1-R | AUUCCUUCUGGUACUGAGCTT | |
| Duck-sip62-F | GCUGCGGAAGAAGCUUCUATT | Gene Knockdown |
| Duck-sip62-R | UAGAAGCUUCUUCCGCAGCTT | |
| Duck-IFN-α-F | TCCTCCAACACCTCTTCGAC | RT-qPCR |
| Duck-IFN-α-R | GGGCTGTAGGTGTGGTTCTG | |
| Duck-IFN-β-F | AGATGGCTCCCAGCTCTACA | RT-qPCR |
| Duck-IFN-β-R | AGTGGTTGAGCTGGTTGAGG | |
| Duck-IL-6-F | TTCGACGAGGAGAAATGCTT | RT-qPCR |
| Duck-IL-6-R | CCTTATCGTCGTTGCCAGAT | |
| Duck-β-actin-F | GGTATCGGCAGCAGTCTTA | RT-qPCR |
| Duck-β-actin R | TTCACAGAGGCGAGTAACTT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Pan, Y.; Cheng, A.; Zhang, X.; Wang, M.; Chen, S.; Zhu, D.; Liu, M.; Yang, Q.; Wu, Y.; et al. Autophagy Promotes Duck Tembusu Virus Replication by Suppressing p62/SQSTM1-Mediated Innate Immune Responses In Vitro. Vaccines 2020, 8, 22. https://doi.org/10.3390/vaccines8010022
Hu Z, Pan Y, Cheng A, Zhang X, Wang M, Chen S, Zhu D, Liu M, Yang Q, Wu Y, et al. Autophagy Promotes Duck Tembusu Virus Replication by Suppressing p62/SQSTM1-Mediated Innate Immune Responses In Vitro. Vaccines. 2020; 8(1):22. https://doi.org/10.3390/vaccines8010022
Chicago/Turabian StyleHu, Zhiqiang, Yuhong Pan, Anchun Cheng, Xingcui Zhang, Mingshu Wang, Shun Chen, Dekang Zhu, Mafeng Liu, Qiao Yang, Ying Wu, and et al. 2020. "Autophagy Promotes Duck Tembusu Virus Replication by Suppressing p62/SQSTM1-Mediated Innate Immune Responses In Vitro" Vaccines 8, no. 1: 22. https://doi.org/10.3390/vaccines8010022
APA StyleHu, Z., Pan, Y., Cheng, A., Zhang, X., Wang, M., Chen, S., Zhu, D., Liu, M., Yang, Q., Wu, Y., Zhao, X., Huang, J., Zhang, S., Mao, S., Ou, X., Yu, Y., Zhang, L., Liu, Y., Tian, B., ... Jia, R. (2020). Autophagy Promotes Duck Tembusu Virus Replication by Suppressing p62/SQSTM1-Mediated Innate Immune Responses In Vitro. Vaccines, 8(1), 22. https://doi.org/10.3390/vaccines8010022